Co3O4–C3N4 p–n nano-heterojunctions for the simultaneous degradation of a mixture of pollutants under solar irradiation†
Received
18th September 2016
, Accepted 4th November 2016
First published on 7th November 2016
Abstract
Environmental remediation employing sunlight-active semiconductor nano-heterostructures provides effective solutions for handling emerging contaminants through a greener approach. Herein, we report the creation of ultrafine dispersions of Co3O4 nanoparticles in a g-C3N4 matrix by a simple one-pot synthetic strategy involving the co-pyrolysis of constituent raw materials. Calcination of a homogeneous mixture of melamine and cobalt nitrate at 550 °C/2 h leads to the formation of Co3O4–C3N4 p–n nano-heterojunctions that displayed extended absorption in the visible wavelength region owing to the synergistic role of Co3O4 particles. Moreover, the surface area values of the composites reached 90 m2 g−1, a tenfold increase from the value of 8 m2 g−1 obtained for the pristine C3N4. The band bending, induced by the p–n nano-heterojunctions, leads to the formation of intimate interfaces having enhanced photophysical properties. The mass normalized photoluminescence spectra of the heterojunctions indicated reduced exciton recombinations that are validated further by the enhanced sunlight-induced photocatalytic degradation of a mixture of methylene blue and tetracycline organic pollutants.
Environmental significance
The widespread industrial use of organic compounds like pharmaceuticals, antibiotics, and textile dyes has, in recent times, posed a major threat to the environment due to issues associated with their degradation and safe disposal. Consequently, we are faced with the emergence of newer hazards, like drug-resistant bacteria, that warrant immediate remedial measures. The greener approach for the alleviation of such issues is the use of sunlight-active photocatalysts with appreciable efficiencies and recyclability. This technique is superior to adsorption as photocatalysis does not necessitate secondary remediation measures for the disposal of pollutants. In the present work, Co3O4–C3N4 p–n nano-heterojunctions, obtained by a simple one-pot synthetic strategy for the first time, are demonstrated to be efficient photocatalysts for the simultaneous degradation of a mixture of environmental pollutants like the tetracycline antibiotic and methylene blue dye. The ultrafine dispersions of nanosized Co3O4 particles in C3N4 sheets lead to the formation of intimate interfaces that induced synergy in visible light absorption and reduced exciton combinations for enhanced photophysical properties. This template-free approach, eliminating the use of fluorinated acids, also provided a tenfold increase in surface area values affording good photocatalytic efficiencies.
|
Introduction
The uncontrolled disposal of organic effluents like pharmaceuticals, dyes, pesticides, organic compounds, etc. into aquatic sources is a clear cause of environmental pollution that warrants immediate remedial measures.1,2 Of the various strategies emerging, photocatalytic degradation of organic pollutants by semiconductor nanoparticles offers a green and environmentally friendly solution.3,4 Consequently, a significant amount of work has been carried out utilizing metal oxide nanoparticles like titania, ZnO, NiO, BiOX, and halloysite nanotubes for photocatalytic applications in the past decade.5–10 However, owing to band gap constraints and lower efficiencies, the widespread use of such photocatalysts could not be realized for a larger application like environmental remediation. More recently, the emergence of visible light-active photocatalysts has infused renewed interest in the quest for clean and economical solutions for pollution control. Graphitic carbon nitride (g-C3N4), an n-type semiconductor with a medium band gap of about 2.7 eV, is relatively a new entrant in the domain of visible light-active photocatalysts.11,12 The ease of synthesis from inexpensive precursors, high chemical and thermal stability, and band gap compatibility with other semiconductors are the primary attributes that attracted researchers to pursue the use of C3N4 for diverse applications ranging from photocatalytic water splitting to sensors.13,14 Despite such appealing potential, C3N4 suffers from the major shortcomings of low visible light utilization, fast recombination of photogenerated excitons and low specific surface area.15 Various methods are therefore attempted to address these deficiencies and strategies like surface modification, morphological advancements, doping of metal species, composite formation, sensitization with organic dyes, etc. are currently explored.16–19 Amongst these methods, composite formation leading to generation of heterostructures through intimate interfaces of g-C3N4 with metal oxides, sulphides, halides and noble metals has shown promise through improvements in photophysical properties.12,20 Subsequently, g-C3N4 has been coupled with several metal oxides such as TiO2, ZnO, WO3, Cu2O, In2O3, Fe2O3, BiVO4, etc. to form g-C3N4/metal oxide heterojunctions.21–27
Cobalt oxide (Co3O4) is a visible light-active p-type semiconductor with attractive electronic and structural properties. Several p–n heterojunctions of Co3O4 with other semiconductors like TiO2, BiVO4, etc. are reported to have enhanced photocatalytic activity due to reduced electron hole recombinations at the p–n junction interfaces.28–30 The creation of p–n heterojunctions leads to band bending and generation of an internal electric field due to which the exciton recombinations are reduced.31–34 Due to its favorable band alignment, Co3O4 is a potential metal oxide to form a p–n heterostructure with g-C3N4. Co3O4 loaded on the mesoporous g-C3N4 surface was evaluated for the photocatalytic evolution of oxygen from water as well as for the photocatalytic degradation of organic compounds.35,36 However, the synthesis of mesoporous layers of g-C3N4 sheets involved a silica templating method and employing fluorinated solvents for the post-synthesis etching process made it time-consuming and environmentally hazardous. Guo et al. synthesized Co3O4–C3N4 by a two-step calcination technique using a mechanical mixture of Co3O4 and g-C3N4 wherein the Co3O4 content was too low (below 2 wt%).37 Hitherto, the reported works are based on processing strategies that are cumbersome and less efficient. It is therefore imperative to develop simple synthetic approaches that lead to dispersions of nano-Co3O4 on C3N4 sheets. In a recent and notable contribution, Dontsova et al. synthesized a C3N4–Co3O4–Co(III) complex by the one-step thermal condensation of dicyandiamide with Co(II) chloride-containing salt melts at 550 °C in flowing N2 atmosphere. The non-porous solids thus obtained are demonstrated to be useful for water oxidation and Rhodamine B degradation.38 Here in this study, we further supplement the one-step synthesis strategy to arrive at phase-pure Co3O4–C3N4 systems, for the first time, by a suitable choice of precursors and pyrolysis conditions. Co3O4–C3N4 (CC) p–n heterojunctions containing ultrafine dispersions of Co3O4 in C3N4 sheets are prepared by co-pyrolysing a homogeneous mixture of the precursors, melamine, and cobalt nitrate in air at 550 °C. The composite formed showed improved sunlight-driven photocatalytic activity and the efficiency was evaluated by analyzing the degradation of a mixture of organic compounds (antibiotic + dye) as a case study on water pollution.
Experimental
Synthesis
Pristine g-C3N4 is prepared by the thermal condensation and evaporation of melamine and pure Co3O4 is obtained by the thermal decomposition of cobalt nitrate at 550 °C at a ramping rate of 3 °C min−1 for 2 h in air atmosphere. In a typical synthesis, Co3O4–C3N4 composites are prepared using a one-pot synthetic approach by mixing 4 g of melamine with different amounts of cobalt nitrate (0.1 g, 0.4 g, and 0.7 g) and heated at 550 °C in air at a ramping rate of 3 °C min−1 for 2 h. The as-synthesised composites are correspondingly designated as CC0.1, CC0.4, and CC0.7 and used as such for characterization and photocatalytic application. For a comparative evaluation, a mechanical mixture of the Co3O4–C3N4 (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :96 by weight) composite is prepared by homogeneously mixing the constituent powders in an agate mortar and denoted hereafter as CCM.
:96 by weight) composite is prepared by homogeneously mixing the constituent powders in an agate mortar and denoted hereafter as CCM.
Photocatalytic degradation studies
The photocatalytic activity of the prepared samples is evaluated by the sunlight-induced degradation of a mixture of the organic effluents methylene blue (MB) and tetracycline (TC). About 500 mg L−1 photocatalyst is added to a mixture containing 10−5 M MB and 10−4 M TC and the resulting mixture is stirred in the dark for 30 minutes, prior to irradiation, to attain the adsorption–desorption equilibrium. The reaction mixture is then irradiated under sunlight and the suspension is collected at regular intervals of 30 minutes for measuring the changes in the concentrations of MB (664 nm) and TC (357 nm) using a UV-vis spectrophotometer. The individual degradation of MB and TC is also studied for comparison.
Results and discussion
The composition and thermal stability of g-C3N4 and the different compositions of Co3O4–C3N4 composites are evaluated by post-synthesis thermogravimetric analysis (TGA) in the temperature range of 50 °C to 850 °C in air atmosphere at a rate of 3 °C min−1. Fig. 1, providing the decomposition profiles of the samples, indicates that the pure g-C3N4 decomposes completely at T > 600 °C while nearly 4 wt% residue (Co3O4) is left for the composite CC0.1 sample. The other two composites (CC0.4 and CC0.7) leave behind 15 wt% and 97 wt% Co3O4 residue, respectively. The weight retained indicates the Co3O4 content in the composite samples and the prepared composites are therefore estimated to have C3N4–4 wt% Co3O4 (CC0.1), C3N4–15 wt% Co3O4 (CC0.4), and C3N4–97 wt% Co3O4 (CC0.7) compositions. The most notable aspect of the thermal analysis is the early thermal degradation (at T < 400 °C) observed in CC0.1 and CC0.4 composite samples. Incorporation of Co3O4 nanoparticles into C3N4 sheets improves the surface area by tenfold (presented in detail on the results of BET) and is believed to induce oxidative etching of C3N4 sheets leading to an early weight loss compared to the bulk C3N4.
 |
| | Fig. 1 Thermal decomposition patterns of g-C3N4 and Co3O4–C3N4 composites. | |
The powder XRD patterns of the as-prepared g-C3N4, Co3O4, mechanical mixture CCM and Co3O4–C3N4 composite (CC0.1, CC0.4 and CC0.7) samples are shown in Fig. 2. The pristine g-C3N4 is characterised by well-defined peaks at 27.4° and 13.1° indexed to the (002) and (100) diffraction planes corresponding to the interplanar stacking of aromatic systems and the interlayer structural packing, respectively.11 The peaks of pure Co3O4 are of the cubic spinel-type structure at 2θ values of 18.90°, 31.29°, 36.81°, 38.54°, 44.80°, 59.37° and 65.27° corresponding to (111), (220), (311), (222), (400), (511) and (440) planes, respectively (JCPDS 00-042-1467).28 The XRD pattern of CC0.1 (C3N4–4 wt% Co3O4) shows peaks of both g-C3N4 and Co3O4. The increase in Co3O4 content reduces the peak intensities of g-C3N4 in CC0.4 (C3N4–15 wt% Co3O4). The CC0.7 (C3N4–97 wt% Co3O4) composite is dominantly the Co3O4 phase with no peaks for the g-C3N4 phase as has been substantiated by the TGA analysis. It is presumed that the increase in cobalt nitrate content in the precursor mix can lead to significant release of nitrate fumes due to which there is considerable loss of C3N4 from the composition of CC0.7. The mechanical mixture CCM shows predominant peaks of C3N4 and a minor peak at 36.81° representing the (311) plane of Co3O4, indicating inhomogeneous distribution of the Co3O4 phase and presumably a lack of heterostructure formation.
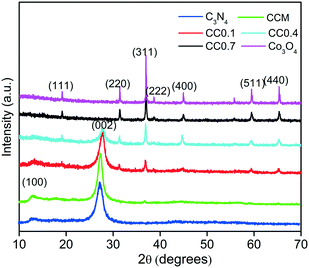 |
| | Fig. 2 XRD patterns of the as-prepared Co3O4–C3N4 (CC) composites, g-C3N4, Co3O4, and mechanical mixture CCM. | |
The TEM images of pristine C3N4 and Co3O4 are shown in Fig. 3(a and b). Thick planar aggregates of g-C3N4 sheets are observed in Fig. 3(a) while Fig. 3(b) reveals irregularly shaped Co3O4 particles of above 50 nm size when synthesized separately without C3N4. In Fig. 3(c and d), exfoliated planar sheets of g-C3N4 are seen decorated with dark dots, corresponding to Co3O4 nanoparticles of 10–15 nm size, throughout the sheets and are distinct from the pristine C3N4 and Co3O4. The size of the Co3O4 nanoparticles was reduced significantly (10–15 nm) when incorporation within the C3N4 sheets through the one-pot co-pyrolysis method was adopted. The evolution of volatile products like NO2 and NH3 during the pyrolysis and condensation processes of composite preparation can presumably restrict the growth of nanoparticles and can exfoliate the aggregated sheets of g-C3N4. Moreover, incorporation of ultrafine dispersions of Co3O4 nanoparticles in the matrix also leads to enhanced surface area. The TEM images of CC0.7 shown in Fig. S1† show the presence of aggregated, micron-sized Co3O4 particles on C3N4 sheets.
 |
| | Fig. 3 TEM images of (a) g-C3N4, (b) Co3O4, and (c and d) the Co3O4–C3N4 (CC0.1) composite. | |
The HRTEM image in Fig. 4(a–c) shows Co3O4 nanoparticles of size 10 to 20 nm forming an intimate interface with the g-C3N4 sheets. The lattice of Co3O4 nanoparticles is clearly visible in the HRTEM images. However, the lattice of C3N4 sheets is not visible due to indistinct in-plane diffraction (100) plane. The close interface between the C3N4 sheets and Co3O4 nanoparticles revealed the nano-heterojunction formation. Fig. 4(c) and (d) shows the lattice d spacing of uniformly dispersed Co3O4 nanoparticles on C3N4 sheets. The interplanar spacing of 0.23 nm, 0.24 nm, and 0.46 nm corresponded to the (222), (311), and (111) respectively and are consistent with the XRD results.39,40
 |
| | Fig. 4 (a) and (b) HRTEM images; (c) lattice fringes (d) SAED pattern of Co3O4–C3N4 (CC) composites. | |
The EDAX spectra of g-C3N4, Co3O4 and the CC0.1 composite are shown in Fig. 5(a–c) which confirm the elemental composition of the synthesized samples. Energy-dispersive X-ray spectrum (EDS) mapping analyses from SEM of the CC composite presented in Fig. 5(d–h) illustrate the homogeneous distribution of the four elements (C, N, O, and Co) throughout the sample. This analysis substantiates that the composite is composed of only C, N, Co and O elements without any impurities. The lower content of Co3O4 in the composite and its near-homogeneous distribution is vivid from the elemental mapping analysis.
 |
| | Fig. 5 EDAX spectra of (a) g-C3N4, (b) Co3O4 and (c) the Co3O4–C3N4 (CC) composite, (d) SEM image showing the selected area of elemental mapping and EDS mapping of elements (e) C K, (f) N K, (g) Co K and (h) O K. | |
The N2 adsorption–desorption isotherms of the CC composites and g-C3N4 are shown in Fig. 6. The adsorption isotherms are of typical type II b (BDDT Classification) behavior and indicate adsorption in sheet-like structures. The change in volume of adsorbed nitrogen (in other words porosity) between the samples is a clear indication of the influence of second phase (Co3O4) addition on the porous structure of C3N4. The porosity of the C3N4-rich sample CC0.1 has been clearly enhanced by the addition of 4 wt% Co3O4. With further increase in the amount of the second phase, the adsorbed volume drops obviously indicating that the enhancement in adsorption volume was not driven solely by the very small second phase particles. The surface area values of the different CC composites and the pure g-C3N4 and Co3O4 were calculated for a better understanding of the composite nanostructure (Table 1). The CC0.1 composite had a surface area of 90 m2 g−1 while pure g-C3N4 showed only a surface area value of 8 m2 g−1. The surface area of the CC0.1 composite was, therefore, enhanced 10-fold due to the dispersion of ultrafine Co3O4 nanoparticles in the C3N4 sheets preventing aggregation. The increase in specific surface area is expected to benefit the photocatalytic degradation process. The release of volatile decomposition products (NO2 and NH3) during the pyrolysis process can also induce the formation of mesopores and exfoliation of sheets contributing to the enhanced surface area in the composite samples.41 However, as the Co3O4 content increased, the surface area values declined and reached 57 m2 g−1 for the CC0.4 sample and 4 m2 g−1 for the CC0.7 composite sample. As has been seen from TGA studies, the CC0.7 composite is 97% by weight Co3O4 and the formation of larger sized Co3O4 particles, in the absence of C3N4 sheets, should have lead to the drastic reduction in surface area values. The individually synthesized Co3O4 particles exhibited negligible surface area. The mechanical mixture (CCM) showed a surface area of only 7 m2 g−1, confirming its aggregated structure devoid of any ultrafine dispersions of nano-Co3O4.
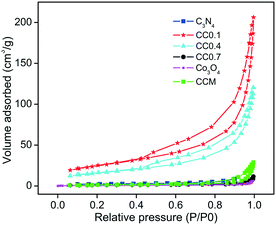 |
| | Fig. 6 N2 adsorption–desorption isotherms of Co3O4–C3N4 (CC) composites, g-C3N4, Co3O4, and mechanical mixture CCM. | |
Table 1 Surface area values of C3N4, Co3O4, and Co3O4–C3N4 composites (CC)
| Sample |
Surface area (m2 g−1) |
| C3N4 |
8 |
| Co3O4 |
2 |
| CC0.1 |
90 |
| CC0.4 |
57 |
Hence, based on the TGA, XRD, TEM and BET surface area analysis, the CC0.1 composite with a composition of C3N4–4 wt% Co3O4 is chosen as the ideal candidate for application studies and is hereafter denoted as CC unless otherwise stated.
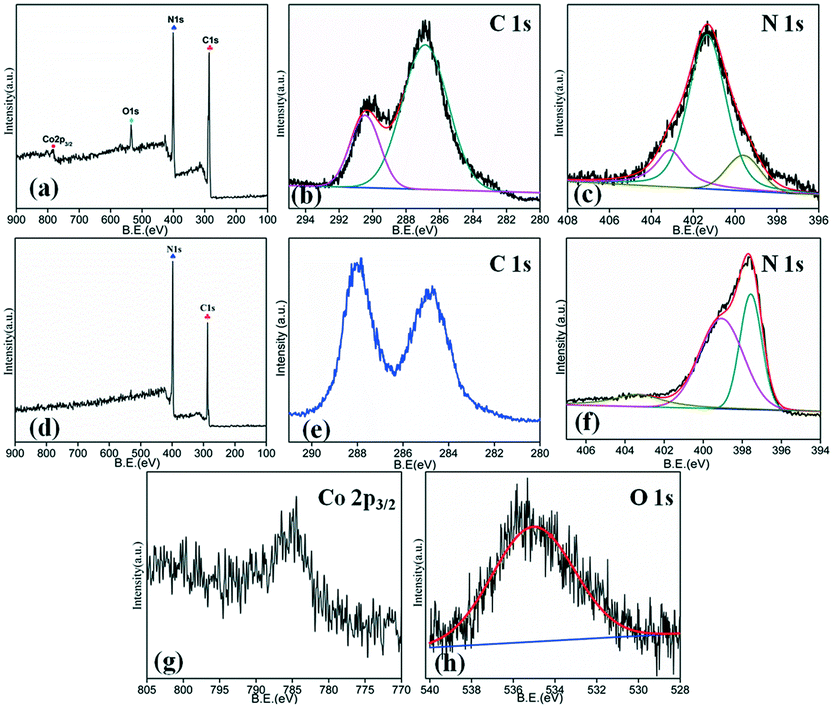
The surface elemental analysis of each sample is carried out by XPS measurements. The survey spectrum of the composite shown in Fig. 7(a) indicates the presence of carbon, nitrogen, cobalt and oxygen. The C1s [Fig. 7(b)] spectrum shows two dominant peaks around 286.86 eV and 290.45 eV corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N sp2 bonds and the interaction of Co3O4 with g-C3N4 in the CC composite, respectively.37,42 The N1s spectrum [Fig. 7(c)] of g-C3N4 shows three impressive peaks around 399.6, 401.3 and 403.1 eV ascribed to the C–N sp3 bonds, CN sp2 bonds and the interaction of g-C3N4 sheets with Co3O4 particles.42 For comparison, the C1s and N1s survey spectra of pristine g-C3N4 are shown in Fig. 7(d)–(f). The C1s spectrum [Fig. 7(e)] of pristine g-C3N4 showed two dominant peaks around 284.4 and 288.1 eV. The peak at 284.4 eV is ascribed to the adventitious carbon and the peaks at 288.1 eV to N–CN bonds, which is the major carbon species. The high resolution spectrum of N1s [Fig. 7(f)] showed two dominant peaks at 398.3 and 400.8 eV. The peak at 398.3 eV is ascribed to the C–NC bond in the triazine rings and the peak at 400.8 eV is ascribed to tertiary nitrogen N–(C)3 bonds. The high resolution Co2p spectrum shown in Fig. 7(g) indicates that Co3O4 contains the mixed oxidation states of Co2+ and Co3+ at 784.3 eV and 797.1 eV, respectively.43,44 The O1s spectrum in Fig. 7(h) shows a distinct peak at 534.9 eV, which is ascribed to surface adsorbed hydroxyl or oxygen atoms.45
N sp2 bonds and the interaction of Co3O4 with g-C3N4 in the CC composite, respectively.37,42 The N1s spectrum [Fig. 7(c)] of g-C3N4 shows three impressive peaks around 399.6, 401.3 and 403.1 eV ascribed to the C–N sp3 bonds, CN sp2 bonds and the interaction of g-C3N4 sheets with Co3O4 particles.42 For comparison, the C1s and N1s survey spectra of pristine g-C3N4 are shown in Fig. 7(d)–(f). The C1s spectrum [Fig. 7(e)] of pristine g-C3N4 showed two dominant peaks around 284.4 and 288.1 eV. The peak at 284.4 eV is ascribed to the adventitious carbon and the peaks at 288.1 eV to N–CN bonds, which is the major carbon species. The high resolution spectrum of N1s [Fig. 7(f)] showed two dominant peaks at 398.3 and 400.8 eV. The peak at 398.3 eV is ascribed to the C–NC bond in the triazine rings and the peak at 400.8 eV is ascribed to tertiary nitrogen N–(C)3 bonds. The high resolution Co2p spectrum shown in Fig. 7(g) indicates that Co3O4 contains the mixed oxidation states of Co2+ and Co3+ at 784.3 eV and 797.1 eV, respectively.43,44 The O1s spectrum in Fig. 7(h) shows a distinct peak at 534.9 eV, which is ascribed to surface adsorbed hydroxyl or oxygen atoms.45
 |
| | Fig. 7 (a) Survey spectrum and high-resolution XPS patterns of (b) C 1s and (c) N 1s of the g-Co3O4–C3N4 (CC) composite; (d) survey spectrum and high-resolution XPS patterns of (e) C 1s and (f) N 1s of g-C3N4 and of (g) Co 2p and (h) O 1s of Co3O4. | |
The optical absorption of the as-prepared samples is measured using UV-vis absorption spectroscopy and is presented in Fig. 8. The pristine g-C3N4 showed absorption from UV to visible light with the main absorption edge at 440 nm and a weaker absorption tail in the visible light region.46 The characteristic absorption edge of C3N4 in the CC composite showed a slight blue shift due to a quantum confinement effect of the exfoliated nanosheets of C3N4.47 Moreover, incorporation of Co3O4 improved the optical absorption with a peak at around 732 nm in the visible region. This is attributed to the small band gap of Co3O4, which has direct transitions at 1.45 and 2.07 eV, corresponding to O2− to Co3+ excitation and O2− to Co2+ charge transfer, respectively.48 The enhanced visible light absorption is observed also from the change in colour of pure g-C3N4 from yellow to light grayish for the composite. The improved visible light absorption signifies the efficient coupling of the constituents and formation of effective heterostructure nanocomposites. On the contrary, the mechanical mixture CCM indicated a pattern resembling that of C3N4 without any absorption beyond 450 nm (Fig. S2†). The processing advantages of the one-pot synthesis are thus categorically established where the formation of intimate interfaces of Co3O4–C3N4 led to the creation of p–n nano-heterojunctions with improved photophysical properties.
 |
| | Fig. 8 UV-vis absorption spectra of Co3O4–C3N4 (CC), Co3O4 and g-C3N4. | |
The FTIR spectra of CC and pristine g-C3N4 are shown in Fig. S3.† The composite preparative conditions did not affect the signature peaks of g-C3N4. The functional groups of g-C3N4 are concentrated in the 1100–1650 cm−1 corresponding to the stretching vibration of C–N and CN. The absorption at 810 cm−1 is attributed to the breathing modes of triazine rings.49
Photocatalytic degradation of pollutant mixture (antibiotic + dye)
The tetracycline degradation studies on all the prepared composites in comparison with pristine C3N4 and Co3O4 are shown in Fig. S4.† The CC0.1 (i.e. CC) composite exhibited maximum degradation efficiency within 180 minutes of irradiation. Further, the practical application of the efficient composite (CC) is evaluated by the degradation of a mixture of organic pollutants (antibiotic + dye) under sunlight irradiation. The degradation profiles of the two pollutants in the mixture (MB + TC) using the CC composite are elucidated from UV-vis measurements as presented in Fig. S5.† The intensity reduction at 360 nm corresponds to TC degradation while that at 668 nm represents MB degradation. The photocatalyst CC effectively degraded both the pollutants from the mixture. Fig. 9(a) shows the TC degradation profile of the composite in comparison with pristine g-C3N4 and Co3O4. The prepared composite degrades more than 97% of TC within 180 min of sunlight irradiation compared to 55% by bare g-C3N4 and 35% by Co3O4. Co3O4 being a p-type visible light-active semiconductor shows little effectiveness primarily due to its negligible specific surface area. Fig. 9(b) shows the MB degradation profile of the samples and it is seen that more than 90% of MB is degraded by CC within 30 min of sunlight irradiation and 100% of MB is degraded within 90 min of exposure. Bare g-C3N4 also shows 70% MB degradation within 180 min of sunlight irradiation. The Co3O4 sample indicates only 15% MB degradation. The degradation efficiency of the composite (CC) is significantly higher due primarily to increased surface area as observed in BET SA measurements. The reduced exciton recombination in the Co3O4–C3N4 p–n heterojunctions is also a vital factor aiding the increase in photocatalytic efficiency (substantiated by PL measurements discussed below). Fig. 9(c) and (d) show the C/C0vs. time profiles of the mixture of organic pollutants (antibiotic + dye). TC and MB show maxima at 360 nm and 668 nm, respectively, which can thus be used to evaluate the concentration of these pollutants within the sample mixture. Fig. 9(c) shows the change in C/C0 of TC in the organic mixture when irradiated for 180 min. The composite degraded 78% of TC from its mixture with MB in the provided span of irradiation. The degradation activity of TC is slightly reduced in the mixture when compared to its individual activity, as shown in Fig. 9(a). Similarly, Fig. 9(d) shows the change in C/C0 of MB in the organic mixture when irradiated for 180 min. The composite degraded 100% of MB within 120 min of irradiation of the mixture. The summary of % degradation efficiency of CC, C3N4, and Co3O4 is shown in Table S1.† Enhanced performances are obtained for the CC heterostructures in all cases.
 |
| | Fig. 9 (a) TC photocatalytic degradation profile, (b) MB photocatalytic degradation profile, (c) TC photocatalytic degradation profile in the mixture of MB + TC and (d) MB photocatalytic degradation profile in the mixture of MB + TC. | |
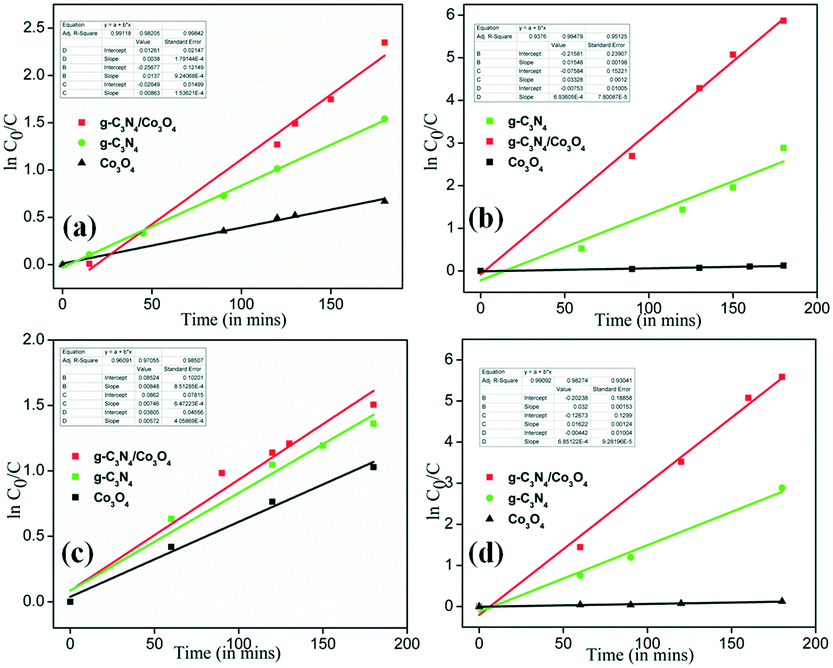
The photocatalytic degradation of an organic pollutant generally is regarded as pseudo-first-order kinetics and follows eqn (1) with C0 in millimolar units:
where
C0 is the concentration of TC and MB after the initial adsorption–desorption equilibrium,
C is the concentration after visible light irradiation and
K is the first-order kinetics rate constant.
Fig. 10(a) shows the rate of photocatalytic degradation of TC using the composite in comparison with pristine g-C
3N
4 and Co
3O
4.
Fig. 10(b) shows the rate of photocatalytic degradation of MB using the composite. Similarly,
Fig. 10(c) and (d) show the ln(
C0/
C)
vs. time plots of the mixture of organic pollutants (antibiotic + dye) using CC, compared with pristine g-C
3N
4 and Co
3O
4. The slope gives the rate constant of the degradation process and
Table 2 summarizes the rate constants for the different samples. The maximum rate constant in all the processes is obtained for the composite prepared (CC) which affirms its improved photocatalytic behavior. To understand the mineralization efficiency of the prepared CC heterostructure, chemical oxygen demand (COD) analysis is performed using MB and it is observed that CC displayed better mineralization efficiency compared to pristine C
3N
4 (Fig. S6
†). We have selected MB for the COD analysis as degradation of TC yields small amounts of carbonaceous products as reported earlier.
50
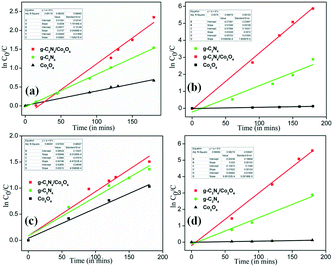 |
| | Fig. 10 (a) Rates of photocatalytic degradation of TC, (b) rates of photocatalytic degradation of MB, (c) rates of photocatalytic degradation of TC in the organic mixture and (d) rates of photocatalytic degradation of MB in the organic mixture. | |
Table 2 Rate constants of the photocatalysts for different photocatalytic degradation experiments
| Sample |
Rate constant (min−1) |
| TC alone |
TC in mixture |
MB alone |
MB in mixture |
| C3N4 |
0.0086 |
0.0074 |
0.0155 |
0.0161 |
| Co3O4 |
0.0038 |
0.0057 |
0.0006 |
0.0006 |
| CC |
0.0137 |
0.0084 |
0.0333 |
0.0333 |
The recyclability studies on CC are performed to ascertain its stability and determine its practical viability. Fig. 11 shows 5 cycles of photocatalytic degradation tests performed on a mixture of TC + MB solutions. The composite shows very little change in activity even after five cycles, suggesting excellent recyclability of the photocatalyst. Both the pollutants are effectively degraded suggesting the practical use of such heterojunctions for the treatment of discharges from pharmaceutical waste, etc. The XRD and FTIR patterns of Co3O4–C3N4 (CC) after the cycling studies, presented in Fig. S7(a) and (b),† indicated no changes in phase and functional groups, respectively.
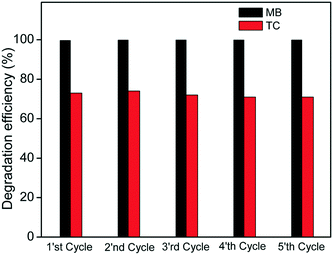 |
| | Fig. 11 Five cyclic runs of the Co3O4–C3N4 (CC) photocatalyst for the degradation of TC and MB in the organic mixture. | |
Active species formed during the photocatalytic reaction of the Co3O4–C3N4 (CC) composite are presented in Fig. 12. Different scavengers are employed to detect the ROS species. 10 mM isopropanol (IPA) (as a quencher of OH−), 6 mM AgNO3 (as a quencher of an electron), 6 mM benzoquinone (BQ) (as a quencher of O2−) and 10 mM triethanolamine (TEA) (as a quencher of holes) are respectively added to the photocatalytic system and irradiated under sunlight.33,51 The photocatalytic degradation of TC is not affected by the addition of isopropanol (IPA) while the degradation is quenched drastically in the presence of benzoquinone (BQ), triethanolamine (TEA) and partially due to AgNO3. Therefore, it can be concluded that the superoxide anions (O2−), electrons and holes are the main reactive species generated by the photocatalysts for TC degradation under sunlight irradiation.
 |
| | Fig. 12 Results of species trapping experiments performed using IPA, TEA, BQ, and AgNO3. | |
The mass normalized photoluminescence spectra of g-C3N4 and the Co3O4–C3N4 composite presented in Fig. 13 are used to elucidate the effect of heterojunction formation on the exciton recombination rates. The main emission peak of g-C3N4 and Co3O4–C3N4 appears at 454 nm and the intensity of the latter is considerably low compared to that of g-C3N4. The lower PL intensity is an indication of the lower recombination of electron–hole pairs.51,52 Pristine C3N4 showed higher PL intensity and therefore lower photocatalytic activity. The recombination of photogenerated charge carriers was inhibited in the heterostructure prepared, contributing to the enhancement of photocatalytic efficiency through a plausible mechanism as discussed below. The mechanical mixture indicated a slight decrease in the PL intensity compared to C3N4 due to the incorporation of 4% Co3O4.
 |
| | Fig. 13 PL spectra of g-C3N4, Co3O4–C3N4 (CC) and mechanical mixture CCM. | |
Photocatalytic mechanism
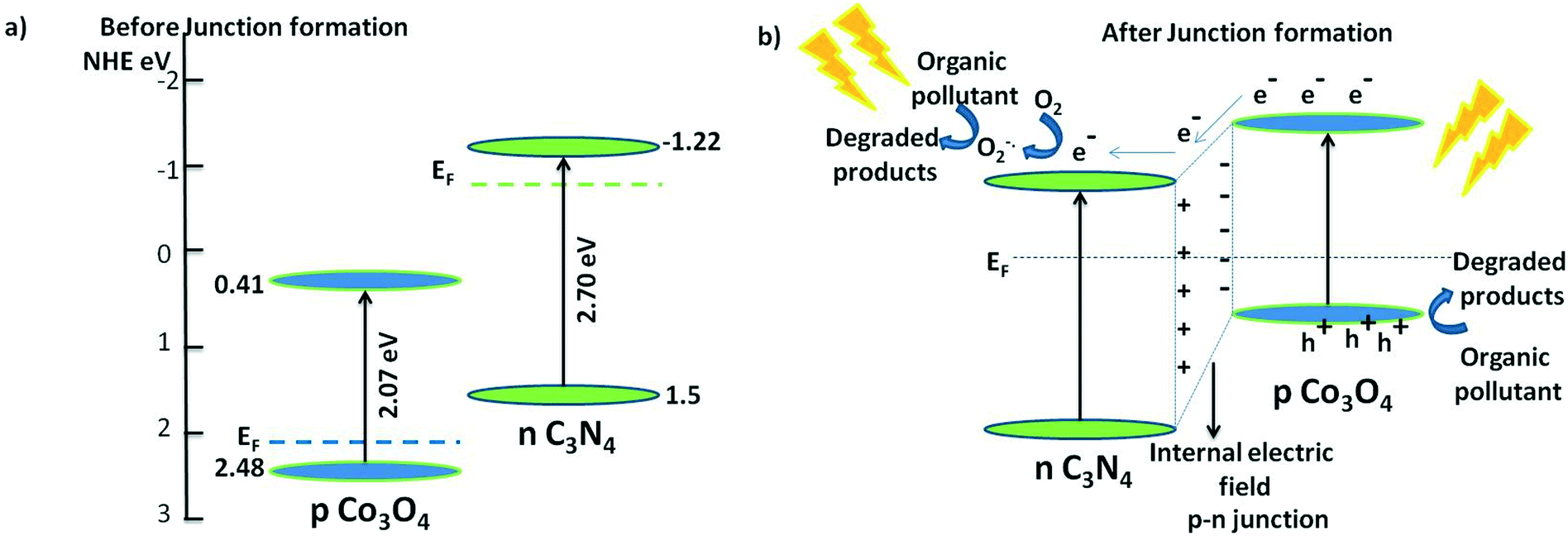
Based on the results of active species trapping and PL measurements, a possible mechanism of the degradation is shown in Fig. 14. g-C3N4 is an n-type semiconductor and Co3O4 is ascertained to be a p-type semiconductor. The interface of the Co3O4–C3N4 composite thus works as a p–n heterojunction. The valence band edge potential and conduction band edge potential of a semiconductor are calculated using Mulliken's electronegativity theory as shown in the following equations:32
 |
| | Fig. 14 a) Band alignment of p-type Co3O4 and n-type C3N4 before junction formation and b) band alignment and the photocatalytic mechanism of Co3O4–C3N4 p–n nano-heterojunctions. | |
Table 3 The absolute electronegativity, band gap energy, valence and conduction band edge potentials of Co3O4 and C3N4
| Semiconductor |
Absolute electronegativity (χ) |
Bandgap energy (eV) |
Valence band potential EVB (eV) |
Conduction band potential ECB (eV) |
| Co3O4 |
5.903 |
2.07 |
2.48 |
0.41 |
| C3N4 |
4.64 |
2.70 |
1.5 |
−1.22 |
According to the above results, the band positions of C3N4 and Co3N4 before having any contact are shown in Fig. 14(a). The Fermi level of the n-type C3N4 is located near the conduction band (CB) while that of the p-type Co3O4 is located near the valence band (VB). In the p–n junction the CB potential of C3N4 is more negative and the, electrons will therefore diffuse from the CB to the CB of Co3O4 leaving a negative charge accumulation in Co3O4. On the other hand, the diffusion of holes from the VB of Co3O4 to the VB of C3N4 provides positive charge accumulation near the vicinity of the junction. As reported earlier, the Fermi level equilibration of the two semiconductors leads to generation of an internal electric field and hence charge diffusion in either side, which in turn shifts the band positions of the two semiconductors.28,31–33,53 The band positions of the n-type C3N4 are down-shifted along with the Femi level; whereas, the band positions of the p-type Co3O4 are up-shifted along with the Fermi levels allowing the CB of Co3O4 to be more negative. Upon sunlight irradiation, electrons in both the visible light-active semiconductors are photoexcited to their respective CB, leaving holes in the VB. At the junction interface, since the CB of Co3O4 is more negative, electrons are diffused to the CB of C3N4, leaving holes in the CB of Co3O4 (Fig. 14(b)). Thus, the formation of the p–n heterojunction serves to separate the charge carriers as much as possible through the formation of the internal electric field and hence the charge recombinations are prohibited in the junction. As shown in the ROS scavenging trials above (Fig. 12), the electrons diffused to the CB of C3N4 react with oxygen, producing superoxide anion radicals, which degrade the organic pollutants efficiently while holes in the VB directly degrade the organic pollutants. Overall, the high surface area, increased visible light absorption, and formation of an effective p–n nano-junction in Co3O4–C3N4 served to outperform the photocatalytic degradation of the individual components. Additionally, the nanodimensions and highly dispersed states of Co3O4 suppressed the interfacial charge recombination, thereby enhancing the charge separation.
A survey of the published works employing Co3O4 as the second phase in the C3N4 matrix, as presented in Table S2,† indicates an advantage for the current synthesis methodology over the reported ones. The facile one-pot approach of co-pyrolysing the constituent raw materials in air atmosphere results in ultrafine dispersions of Co3O4 nanoparticles in C3N4 sheets leading to a tenfold increase in surface area and a nearly homogeneous distribution of the Co3O4 phase. The silica templating method commonly employed for the preparation of mesoporous g-C3N4 necessitates an acid etching step to remove silica, making the process environmentally non-friendly. Moreover, the formation of intimate interfaces of p–n heterojunctions favours enhanced photophysical properties, thereby reducing exciton recombinations and improving the photocatalytic behavior. It is also to be noted that the developed photocatalysts are capable of simultaneously degrading multiple pollutants from a mixture of contaminants that are believed to be emerging environmental threats. A direct comparison of the photocatalytic efficiencies is however not feasible owing to differences (type of pollutant, concentration of catalyst, etc.) in the degradation studies.
Conclusions
Co3O4–C3N4, a p–n heterojunction composite, prepared using a one-pot synthesis methodology resulted in a 10-fold enhancement of surface area compared to pristine C3N4 and improved visible light absorption. Ultrafine Co3O4 (10–15 nm in size) particles are seen dispersed in g-C3N4 sheets leading to the formation of p–n nano-heterojunctions with appropriate band bending. This has resulted in reduced exciton recombinations as confirmed by mass normalised PL measurements. The radical scavenging experiments revealed that the active species employed in the photo-induced degradation processes are superoxide anions, holes and electrons. The C3N4–4 wt% Co3O4 composite exhibited enhanced photocatalytic activity by effectively degrading a mixture of tetracycline and methylene blue under sunlight irradiation.
Acknowledgements
Mr. Kiran Mohan and Mrs. Soumya Valsalam are thankfully acknowledged for the HRTEM micrographs and SEM EDS mapping, respectively. Ms. Athira A. S. is gratefully acknowledged for the COD analysis. The authors are grateful to the Council of Scientific and Industrial Research (CSIR, Government of India) for the 12th five year plan project on “IntelCoat” (CSC0114). Author S. P. thanks the CSIR for the research fellowship.
References
- N. Bhandary, A. P. Singh, S. Kumar, P. P. Ingole, G. S. Thakur, A. K. Ganguli and S. Basu, ChemSusChem, 2016, 9, 2816–2823 CrossRef CAS PubMed.
- M. Zhang, W. Jiang, D. Liu, J. Wang, Y. Liu, Y. Zhu and Y. Zhu, Appl. Catal., B, 2016, 183, 263–268 CrossRef CAS.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- V. Kalarivalappil, C. M. Divya, W. Wunderlich, S. C. Pillai, S. J. Hinder, M. Nageri, V. Kumar and B. K. Vijayan, Catal. Lett., 2016, 146, 474–482 CrossRef CAS.
- V. S. Smitha, K. V. Baiju, P. Perumal, S. Ghosh and K. G. Warrier, Eur. J. Inorg. Chem., 2012, 2012, 226–233 CrossRef CAS.
- Y. Wang, R. Shi, J. Lin and Y. Zhu, Energy Environ. Sci., 2011, 4, 2922–2929 CAS.
- D. Malwal and P. Gopinath, Environ. Sci.: Nano, 2015, 2, 78–85 RSC.
- Y. Bai, L. Ye, L. Wang, X. Shi, P. Wang and W. Bai, Environ. Sci.: Nano, 2016, 3, 902–909 RSC.
- L. Ye, Y. Su, X. Jin, H. Xie and C. Zhang, Environ. Sci.: Nano, 2014, 1, 90–112 RSC.
- L. Yu, H. Wang, Y. Zhang, B. Zhang and J. Liu, Environ. Sci.: Nano, 2016, 3, 28–44 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- J. Liu, H. Wang and M. Antonietti, Chem. Soc. Rev., 2016, 45, 2308–2326 RSC.
- B. Wang, H. Wang, X. Zhong, Y. Chai, S. Chen and R. Yuan, Chem. Commun., 2016, 52, 5049–5052 RSC.
- G. Dong, Y. Zhang, Q. Pan and J. Qiu, J. Photochem. Photobiol., C, 2014, 20, 33–50 CrossRef CAS.
- Z. Zhang, K. Liu, Z. Feng, Y. Bao and B. Dong, Sci. Rep., 2016, 6, 19221 CrossRef CAS PubMed.
- F. Guo, J. Chen, M. Zhang, B. Gao, B. Lin and Y. Chen, J. Mater. Chem. A, 2016, 4, 10806–10809 CAS.
- A. A. S. Nair and R. Sundara, J. Phys. Chem. C, 2016, 120, 9612–9618 CAS.
- K. Takanabe, K. Kamata, X. Wang, M. Antonietti, J. Kubota and K. Domen, Phys. Chem. Chem. Phys., 2010, 12, 13020–13025 RSC.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- B. Zeng, L. Zhang, X. Wan, H. Song and Y. Lv, Sens. Actuators, B, 2015, 211, 370–376 CrossRef CAS.
- S.-W. Cao, X.-F. Liu, Y.-P. Yuan, Z.-Y. Zhang, Y.-S. Liao, J. Fang, S. C. J. Loo, T. C. Sum and C. Xue, Appl. Catal., B, 2014, 147, 940–946 CrossRef CAS.
- J. Chen, S. Shen, P. Guo, M. Wang, P. Wu, X. Wang and L. Guo, Appl. Catal., B, 2014, 152–153, 335–341 CrossRef CAS.
- L. Huang, H. Xu, Y. Li, H. Li, X. Cheng, J. Xia, Y. Xu and G. Cai, Dalton Trans., 2013, 42, 8606–8616 RSC.
- J.-X. Sun, Y.-P. Yuan, L.-G. Qiu, X. Jiang, A.-J. Xie, Y.-H. Shen and J.-F. Zhu, Dalton Trans., 2012, 41, 6756–6763 RSC.
- X. Xiao, J. Wei, Y. Yang, R. Xiong, C. Pan and J. Shi, ACS Sustainable Chem. Eng., 2016, 4, 3017–3023 CrossRef CAS.
- K. Yang, C. Meng, L. Lin, X. Peng, X. Chen, X. Wang, W. Dai and X. Fu, Catal. Sci. Technol., 2016, 6, 829–839 CAS.
- X. Chang, T. Wang, P. Zhang, J. Zhang, A. Li and J. Gong, J. Am. Chem. Soc., 2015, 137, 8356–8359 CrossRef CAS PubMed.
- L. Zhang, Z. Gao, C. Liu, Y. Zhang, Z. Tu, X. Yang, F. Yang, Z. Wen, L. Zhu, R. Liu, Y. Li and L. Cui, J. Mater. Chem. A, 2015, 3, 2794–2801 CAS.
- J. Wang and F. E. Osterloh, J. Mater. Chem. A, 2014, 2, 9405–9411 CAS.
- Z. He, Y. Shi, C. Gao, L. Wen, J. Chen and S. Song, J. Phys. Chem. C, 2014, 118, 389–398 CAS.
- J. Jiang, X. Zhang, P. Sun and L. Zhang, J. Phys. Chem. C, 2011, 115, 20555–20564 CAS.
- D. Jiang, L. Chen, J. Zhu, M. Chen, W. Shi and J. Xie, Dalton Trans., 2013, 42, 15726–15734 RSC.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- P. Qiu, H. Chen and F. Jiang, RSC Adv., 2014, 4, 39969–39977 RSC.
- J. Zhang, M. Grzelczak, Y. Hou, K. Maeda, K. Domen, X. Fu, M. Antonietti and X. Wang, Chem. Sci., 2012, 3, 443–446 RSC.
- C. Han, L. Ge, C. Chen, Y. Li, X. Xiao, Y. Zhang and L. Guo, Appl. Catal., B, 2014, 147, 546–553 CrossRef CAS.
- C. Fettkenhauer, X. Wang, K. Kailasam, M. Antonietti and D. Dontsova, J. Mater. Chem. A, 2015, 3, 21227–21232 CAS.
- D. Su, S. Dou and G. Wang, Sci. Rep., 2014, 4, 5767 CrossRef PubMed.
- H. Wang, C. Chen, Y. Zhang, L. Peng, S. Ma, T. Yang, H. Guo, Z. Zhang, D. S. Su and J. Zhang, Nat. Commun., 2015, 6 Search PubMed.
- G. Zhang, J. Zhang, M. Zhang and X. Wang, J. Mater. Chem., 2012, 22, 8083–8091 RSC.
- W. Jiang, W. Luo, R. Zong, W. Yao, Z. Li and Y. Zhu, Small, 2016, 12, 4370–4378 CrossRef CAS PubMed.
- G. Zhang, S. Zang, L. Lin, Z.-A. Lan, G. Li and X. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 2287–2296 CAS.
- Y. Li and N. Chopra, J. Catal., 2015, 329, 514–521 CrossRef CAS.
- D. K. Mishra, J. Mohapatra, M. K. Sharma, R. Chattarjee, S. K. Singh, S. Varma, S. N. Behera, S. K. Nayak and P. Entel, J. Magn. Magn. Mater., 2013, 329, 146–152 CrossRef CAS.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- P. Niu, L. Zhang, G. Liu and H.-M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- M. Long, W. Cai, J. Cai, B. Zhou, X. Chai and Y. Wu, J. Phys. Chem. B, 2006, 110, 20211–20216 CrossRef CAS PubMed.
- S. Samanta, S. Martha and K. Parida, ChemCatChem, 2014, 6, 1453–1462 CAS.
- C. Liu, G. Wu, J. Chen, K. Huang and W. Shi, New J. Chem., 2016, 40, 5198–5208 RSC.
- P. Suyana, K. R. Sneha, B. N. Nair, V. Karunakaran, A. P. Mohamed, K. G. K. Warrier and U. S. Hareesh, RSC Adv., 2016, 6, 17800–17809 RSC.
- Y. Wang, X. Bai, H. Qin, F. Wang, Y. Li, X. Li, S. Kang, Y. Zuo and L. Cui, ACS Appl. Mater. Interfaces, 2016, 8, 17212–17219 CAS.
- Y. Ao, K. Wang, P. Wang, C. Wang and J. Hou, Dalton Trans., 2016, 45, 7986–7997 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6en00410e |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab
*ab