DOI:
10.1039/C6RA23342B
(Paper)
RSC Adv., 2016,
6, 107256-107262
Chlorination of florfenicol (FF): reaction kinetics, influencing factors and by-products formation†
Received
20th September 2016
, Accepted 1st November 2016
First published on 2nd November 2016
Abstract
Florfenicol (FF) is a widely used antibiotic, which is commonly found in natural waters. In this study, we investigated the removal fate of FF in two different drinking water treatment plants (DWTPs), which suggest that FF was easily transformed by free available chlorine (FAC) and the potential reactions of FF with FAC was the focus of this study. The oxidation kinetics of FF by FAC (7 × 10−4 mol) are very rapid with large pseudo-first-order rate constants kobs = 0.31 min−1, while FF (5 mg L−1) can be completely transformed in 30 min. The results showed that high Cl− (the dominant seawater constituent), Br−, and lower humic acid (HA, main constituents in freshwater) favor the FF oxidation. 21 degradation products were identified by liquid chromatography-tandems mass spectrometry (LC-MS/MS) and the possible routes for FF chlorination were proposed. These results are of importance toward the goal of assessing the persistence of FF in water chlorination.
1. Introduction
Antibiotics as emerging environmental contaminants have been linked to the promotion of bacterial resistance and may cause immune and metabolism diseases, which have received increasing attention.1,2 Florfenicol (FF) is a broad-spectrum antibiotic permitted for use as veterinary drugs in animals used for food production, which belongs to a group of agents used in veterinary medicine named amphenicols.3,4 Since chloramphenicol application in foods and animals was prohibited by the European Union in 1994, FF has been suggested as a potential substitute.5–7 It is used to treat respiratory diseases of pigs and cattle, and has been approved for use in China, Japan, Europe, Norway, Canada, and South Korea for the treatment of various diseases.8–11 In recent years, since it has been administered to farm animals and released into the environment, either in leaching from uneaten medicated pelleted feed, or through urinary, branchial and faecal excretions, research has verified a growing amount of FF in the aquatic environment. FF can persist in the environment for a long period, and is commonly found in rivers, ponds, treatment effluents and even in tap waters and bottled waters. According to Wang et al., FF was found in tap water with the median concentration of 8.9 ng L−1.12 In addition, up to 2.84 μg L−1 for florfenicol was detected in pond water of Southern Jiangsu in China.13 The possible health consequences of FF exposure also include direct side effects, such as the effects on growth and hematopoietic function of children, and promote the development of antimicrobial-resistant bacteria related to the human microbiome. In addition, the application of florfenicol may cause adverse effects on the cardiovascular system.14 Therefore, it is of great importance to investigate the fate and effect of FF by pertinent environmental transformation processes.
In this study, we investigated the fate of FF in two different drinking water treatment plants (DWTPs), which suggested the free available chlorine (FAC) plays an important role. Prior studies indicated that various antibiotics were highly susceptible toward chlorine oxidation and were readily transformed, however, the persistence, influence factors, and transformation for FF chlorination was less well known.15 The purpose of this study was to determine the reaction kinetics of FF with different dosages of free chlorine first. Then, the influencing factors on the removal of FF were explored at initial concentrations of FF, Cl−, Br− and the concentration of humic acid. Since aqueous chlorine is not a strong enough oxidant to mineralize antibiotics, numerous transformation products may be formed due to oxidation or substitution reactions. The transformation products and the possible pathways of FF by chlorination were identified and inferred. In addition, efforts have been made to determine the identities of disinfection byproducts (DBPs) species, containing C-DBPs and N-DBPs, which is essential for understanding the transformation of FF during chlorination.
2. Materials and methods
2.1. Materials
White crystalline powder of florfenicol (C12H14Cl2FNO4S, CAS no. 73231-34-2, MW 358.21) was supplied by of Sigma Company and used as received. A stock solution of free chlorine (HClO) was prepared from 5% liquid sodium hypochlorite (Sinopharm Chemical Reagent Co., Ltd., China) and standardized by the DPD (N,N-diethyl-p-phenylenediamine, Sigma, >99%) colorimetric method.16 All reagents, which were purchased through commercial companies and used without further purification, were prepared in solutions using Milli-Q water (from a Millipore Milli-Q Ultrapure Gradient A10 purification system).
2.2. Experimental procedures
Chlorination experiments were conducted under pseudo first-order conditions where HOCl was in excess (≥10 folds of CAP mole) with a reaction time of 30 min. In a typical experiment, a 200 mL volume of FF solution (5 mg L−1) was prepared in a batch reactor. FAC solution was subsequently added to initiate reactions at oxidant: substrate molar ratios ranging from 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 5:1. The chlorination was quenched at fixed time intervals with Na2S2O3, which molar is 120% of chlorine. A matrix of experiments was conducted with aqueous solutions of FF to examine the influence on the removal efficiency. Matrix variables included initial chlorine concentration (0.625–10 mg L−1, as Cl2), Cl− (15, 25 and 50 mmol L−1), Br−/HOCl (0.1–1) and humic acid (0–1 mg L−1). NaCl (100 mM) and KBr (100 mM) were added to some reactors to examine the effects of Cl− and Br− respectively. All experiments were conducted in duplicates or triplicates.
:1 to 5:1. The chlorination was quenched at fixed time intervals with Na2S2O3, which molar is 120% of chlorine. A matrix of experiments was conducted with aqueous solutions of FF to examine the influence on the removal efficiency. Matrix variables included initial chlorine concentration (0.625–10 mg L−1, as Cl2), Cl− (15, 25 and 50 mmol L−1), Br−/HOCl (0.1–1) and humic acid (0–1 mg L−1). NaCl (100 mM) and KBr (100 mM) were added to some reactors to examine the effects of Cl− and Br− respectively. All experiments were conducted in duplicates or triplicates.
2.3. Chemical analysis
The samples (1000 mL) of water from different DWTPs were extracted and preconcentrated to 5 mL using Supelclean ENVI-18 SPE tubes. Prior to solid phase extraction, samples were passed through 0.45 mm cellulose nitrate membrane filters (Schleicher and Schuell, Germany). Analyses for the FF substrates were performed by an LC-MS/MS system, equipped with a chromatographic column MGIII C18 column (150 mm × 2.1 mm, 3 μm), thermostat (40 °C) at a flow rate (0.4 mL min−1). The composition of the mobile phase A and D was 0.1% formic acid and acetonitrile, while gradient elution programs were 0–2.0 min 90%–50% mobile phase A, 2.0–2.5 min 50–0% mobile phase A, 2.5–4.5 min 0% mobile phase A, 4.5–5 min 0–90% mobile phase A. MS analyses were conducted using negative mode electro spray ionization (ESI−), monitoring the transitions 357 → 153.3 m/z for FF. The mass collision energy voltage was typically set to 20 eV. The capillary temperature was set at 350 °C. The products of FF were measured using high-performance liquid chromatography tandem mass spectrometry (HPLC-MSMS). First, a full scan mass spectrum was obtained from a low possible m/z value to a large m/z, and then a selected ion monitor (SIM) scan with appropriate mass range was acquired. The injection volume is 10 μL. Chromatographic column: MGIII C18 column (150 mm × 2.1 mm, 3 μm) had mobile phase flow rate: 0.25 mL min−1, column temperature: 40 °C, injection volume: 10 μL, mobile phase A pure water (including 0.1% formic acid), mobile phase D acetonitrile, and isocratic elution program of 0–20.0 min 90% mobile phase A and 10% mobile phase D. Electrospray ionization (ESI) was performed in the positive mode with sheath gas pressure and aux gas pressure with the optimum values set at 20 and 5 psi respectively. The capillary temperature was set at 320 °C. The ion spray voltage was set at 3500 V. DBPs were analyzed by a Thermo TSQ Quantum XLS Triple Quadruple GC-MSMS (Thermo Fisher, USA), with medium polarity column (TG-5MS, 30 m × 0.25 mm × 0.50 μm), based on the method ever used.17,18
3. Results and discussion
3.1. Removal of FF at two different DWTPs
Chlorine is usually added at the beginning (e.g., prechlorination) or at the end (e.g., disinfection) of the water treatment process, depending on the treatment objectives. This study investigated the fate of FF in two different drinking water treatment plants. One DWTP uses sediment, filtration, ozone–BAC and disinfection as treatment process, while the other DWTP uses prechlorination for add. The chlorine doses used in prechlorination for raw water were 1–2 mg L−1. According to Table 1, prechlorination was effective for the removal of FF, which was completely degraded in this process with a range concentration of 11.7, 30.6, 1.62 ng L−1 respectively. However, the FF was limited degradation with sediment, filtration and ozone–BAC with a removal rate of 2.94%, 21.21% and 34.62% respectively. The results were in accordance with previous studies showing chlorine was more effective for the elimination of antibiotics.17,19,20
Table 1 Removal rate of FF during drinking water treatment
| Process |
FF (ng L−1) |
Removal rate |
Process |
FF (ng L−1) |
Removal rate |
| Raw water |
6.12 |
|
Raw water |
11.7, 30.6, 1.62 |
|
| Sediment |
5.94 |
2.94% |
Prechlorination |
ND |
100% |
| Filtration |
4.68 |
21.21% |
Sediment |
ND |
|
| Ozone–BAC |
3.06 |
34.62% |
Filtration |
ND |
|
| Finished water |
1.08 |
64.71% |
Ozone–BAC |
ND |
|
| |
|
|
Finished water |
ND |
|
3.2. Removal of FF by free chlorine oxidation
Kinetic analysis of FF with FAC is modeled using the assumption of second-order kinetics, that is, first order with respect to each reactant, and the reaction can be described by the following equation:| |
 | (1) |
where kapp represents the apparent second-order rate constants for the overall reaction. [Cl2]t is in excess with respect to [FF]t, the total concentration of chlorine remains almost constant during the reaction and pseudo-first-order kinetics takes place, being kobs the corresponding pseudo first-order rate constant. First-order kinetics with respect to FF were verified by conducting experiments under excess FAC conditions (5, 10, 20 and 50 folds) where FF loss on a log scale was well correlated with time (r2 > 0.98) for all replicates. This may suggest that the presence of reaction products in solution hasn't significantly affected the effective collisions between FF and FAC, which is similar with other organic pollutions.21,22 The pseudo-first-order rate constants (kobs) observed for reactions of FF with free chlorine were obtained from the slopes of regression lines fitted to plots of ln([FF]) vs. time in the presence of excess FAC, which was clearly dependent on chlorine dose, as shown in Fig. 1. For FAC concentrations of 7 × 10−5, 1.4 × 10−4, 2.8 × 10−4 and 7 × 10−4 mol, the kobs were 0.043, 0.062, 0.120 and 0.312 min−1. FF elimination was also fast, being almost completely removed after half an hour, when the FAC dose was 7.0 × 10−4 mol. The half time (t1/2) of FF was calculated in this study, which can be used to predict a given residual FAC concentration in the water distribution system. While FAC concentrations increase from 7 × 10−5 mol to 70 × 10−5 mol, the t1/2 decreases from 15.75 to 2.28 min. It can be observed that the concentration increases of the chlorine will accelerate the reaction, as it occurs with other micropollutants.23
 |
| | Fig. 1 The evolution of kobs and t1/2 with chlorine dose (temperature 15 ± 2 °C, pH 5.7 and chlorination dose 2.8 × 10−4 mol). | |
Investigation of FF removal by FAC was then carried out at various initial FF concentrations of 0.37–10 mg L−1. As can be seen from Table 1, increasing initial FF concentration resulted in the decrease of the removal efficiency. The reaction rate kobs were 0.24, 0.22, 0.14 and 0.08 min−1 for the initial toxin concentrations of 1.37, 2.5, 5.01 and 10 mg L−1. It is obvious that higher concentrations need longer reaction time. As the initial pharmaceuticals concentration increased (0.37–10 mg L−1), the amount of time increased from 10 to 60 min for FF removal with >90% elimination. The correlation between kobs and initial FF concentration was then analyzed with linear fitting of the kinetics results shown in Table 2. Poor linear correlation coefficients r2 with 0.90 suggested that kobs had poor dependence on the initial FF concentration, because of the shortage of FAC in high FF concentration. A similar trend was observed for other pharmaceuticals, including antipyrine.24
Table 2 kobs values for initial concentrations of FF at temperature of 15 ± 2 °C, pH of 5.7, reaction time of 30 min, initial 0.37–10 mg L−1, and chlorine dosage of 2.8 × 10−4 M
| [FF]0 (mg L−1) |
kobs (min−1) |
r2 |
[FF]0 (mg L−1) |
kobs (min−1) |
r2 |
| 0.37 |
0.34 |
0.97 |
3.64 |
0.21 |
0.98 |
| 0.46 |
0.38 |
0.99 |
4.48 |
0.18 |
0.99 |
| 0.66 |
0.27 |
0.96 |
5.01 |
0.14 |
0.99 |
| 1.37 |
0.24 |
0.99 |
7.50 |
0.11 |
0.97 |
| 2.50 |
0.22 |
0.99 |
10.00 |
0.08 |
0.95 |
3.3. Influence of chloride and bromide
The influence of chloride (Cl−) and bromide (Br−) have been studied in this experiments performed with an initial chlorine dose of 2.8 × 10−4 mol. As seen in Fig. 2, the chlorination is sensitive to chloride, while kobs increased from 0.14 to 0.20 min−1 when 15 mM chloride was added. In addition, the reaction rate constants increased to 0.33 and 0.45 min−1, which was observed under 25 and 50 mM chloride respectively. This behavior, which is similar to that reported with other anthropogenic chemicals, suggests the presence of Cl− in solution could obviously improve the degradation rates.25,26 The plausible explanation can be derived from the observation that under these conditions, Cl2 can be formed from HOCl and Cl−.27 The reversible formation of Cl2 is shifted to the left in the presence of chloride, as seen in eqn (2). Since Cl2 is more electrophilic than HOCl, its presence could accelerate FF degradation.| |
 | (2) |
 |
| | Fig. 2 Effect of different chloride concentration on FF degradation by chlorination (temperature 15 ± 2 °C, pH 5.7 and chlorination dose 2.8 × 10−4 M). | |
It is well known that naturally occurring bromide in raw waters is readily incorporated into the degradation during water chlorination.28 To investigate the role of bromide during chlorination of FF, the [Br−]/[HOCl] ratio was investigated ranging from 0 to 1. As seen in Fig. 3, FF oxidation was significantly enhanced in the presence of bromide. The observed pseudo-first-order rate (kobs) increased linearly (r2 = 0.96) with an increasing bromide concentration. Similar results were also observed in other drugs chlorination in the presence of bromide.29
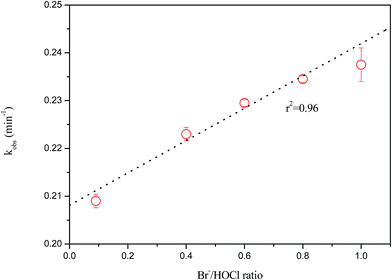 |
| | Fig. 3 Effect of bromide concentration on FF degradation by chlorination (temperature 15 ± 2 °C, pH 5.7 and chlorination dose 2.8 × 10−4 M). | |
This positive effect of bromine is due to the higher values of bromination rate constants compared to chlorination, while bromide is oxidized by chlorination, as seen in eqn (3).
| | |
HOCl + Br− → HOBr + Cl−, k1 = 1550 M−1 s−1
| (3) |
HOBr (E0red = +1.630 V) has a higher redox potential than HOCl (E0red = +1.331 V), and its reactions with some groups of unsaturated compounds have rate constants several orders of magnitude higher than those of HOCl.30,31 Moreover, the activities of electrophilic substitution are more favorable for the Br atom due to its higher electron density and smaller bond strength relative to the Cl atom.32 Under our experimental conditions, the reaction rate increased with the Br−/HOCl ratio increase, which implies the oxidation power of HOBr was likely to exceed that of HOCl. It should be mentioned that the bromination reaction of antibiotics was faster and more efficient than chlorination, and this difference may also affect the environmental risk of antibiotics.
3.4. Chlorination in presence of humic acid (HA)
Humic acid (HA) containing a significant amount of the organic carbon is ubiquitous in surface waters, and it is an important precursor of DBPs in drinking water resulting from the reaction with chlorine.33,34 The concentrations of HA vary depending on the vegetation near the water source, the concentration of algae in water and the time of the year. In this study, the influence of the HA dose was investigated from 0 to 1 mg L−1, as seen in Fig. 4. The pseudo-first-order degradation rate of FF decreased with an increasing concentration of HA, while the reaction rate decrease since the oxidation rate of FF was decreased from 0.15 to 0.10 min−1 with an HA increase from 0 to 1 mg L−1. In addition, the increase of HA concentration in samples to 100 μg L−1 led to the decrease of FF removal during chlorination from 99% to 97%. Similar results for antibiotics were also reported in other chlorination processes.35
 |
| | Fig. 4 Effect of HA concentration on FF degradation by chlorination (temperature 15 ± 2 °C, pH 5.7 and chlorination dose 2.8 × 10−4 M). | |
In general, unsaturated hybridized carbon is more susceptible to chlorine attack.36 The reaction mechanisms between chlorine and HA have been reported to react via oxidation (i.e., cleaving carbon–carbon double bonds) and/or substitution (i.e., replacement of functional groups by a chlorine molecule), producing organic halides, ketone and aldehyde.37 As a result, these micro-environmental changes would hinder the elimination of FF in the chlorination process. Higher chlorine doses must be used in waters with high humic acid content in order to reach the chlorine required to degrade FF completely.
3.5. Products identification in FF chlorination
Although the degradation of the target compound seems to be completed in the time frame of the reaction, a series of degradation products are produced. There is growing concern about the development of transformation mechanisms of organic pollutants and their inactive byproducts.38,39 In this study, various transformation products may be generated during the chlorination. Hypochlorous acid is more likely to induce small modifications in the parent compound's structures than when oxidized. Based on the information detected by LC-MS/MS, reaction pathways for FF with chlorine are proposed as shown in Fig. 5. In total we identified 21 byproducts for FF, and their chromatograms are provided in ESI Fig. S1–S21.† In addition, the main pathways of FF chlorination undergo the following processes:
 |
| | Fig. 5 Proposed destruction pathways of FF during chlorination. | |
(1) Chlorine substitution. The chlorine electrophilic attack on aromatic ring and the amino group of FF leading to the formation of mono-, di-, tri-, and tetra-FF (products 1–6) by successive chlorination, corresponding to m/z 391.5, 426, 460.5 and 495. The full scan chromatogram of m/z at 391.5 (product 1 and product 2) and 460.5 (product 4 and product 5) has two different peaks (compounds), which is the isomer. A penta-FF was not detected in this study, due to a penta-chloro intermediate being involved in the ring-opening step.40 In addition, the modification of the benzene ring was an important step, while aromatic compounds were the primary contributors to trihalomethanes (THM) formation.41 The N-chlorinated is consistent with results obtained by other researchers for the reaction of other antibiotics such as trimethoprim and sulfamethoxazole with free chlorine.42,43 On the other hand, replacing the methylsulfonyl group by Cl, led to product m/z 314.5 (product 7). The replace of methylsulfonyl was also found in the photodegradation experiments by the photo induced chlorination reaction.44 Subsequently, chlorination convert product 7 to products 8, 9 and 10, which may be by the same mechanism of chlorine substitution on the aromatic ring. As ESI is a soft ionization technique, there was no notable fragmentation of ions in the mass spectrum to confirm the exact position of the chlorine on the aromatic ring.45 Moreover, there is a degradation product corresponding to florfenicol amine resulting in amide bond hydrolysis corresponding to m/z 248 (product 11), which then transform to products 12, 13, 14 and 15 (m/z 282.5, 317, 351.5 and 387) through electrophilic attack of HOCl. The hydrolysis product (product 11) of FF has also been observed in other previous studies.44
(2) Fragmentation cleavage. A number of studies suggest that chlorine could have reacted with the higher organic objects, breaking down the larger molecules to smaller molecules.18 The oxidation of FF leads to the cleavage of the original drug molecule, resulting in the formation of products 16, 17, 18 and 19, with the m/z 279, 96, 106 and 188 respectively. This degradation type was reported in the previous studies for the aqueous chlorination of cimetidine and triazines.46,47 Chlorination then converts intermediate 18 to intermediate 20, which may cleave by a mechanism similar to that forming product 16. Product 20 with aldehydic groups is unsaturated, which can be oxidized to an acid group (product 21, m/z 122) during aqueous chlorination without incorporating chlorine into the molecule. The formation of product 20 should be taken into account as aldehydes in particular have been involved in some odor troubles.48In addition, the formation mechanism of C-DBPs and N-DBPs during chlorination for FF (5 mg L−1) has also been measured at 24 h of contact time with oxidant to substrate molar ratios 20:1. By GC/MSMS analysis, chloroform (TCM), carbon tetrachloride (CTC), bromodichloromethane (BDCM), tetrachloroethylene (PCE), bromochloroacetonitrile (BCAN), trichloroacetone (TCAce), N-nitrosodi-n-propylamine (NDPA) were detected with concentrations of 12.30, 0.01, 8.25, 0.01, 0.01, 0.03 and 0.01 μg L−1. This finding was consistent with previous studies that among the most commonly formed DBPs are the trihalomethanes (THMs), while chloroform (CHCl3) is often predominant.49
4. Conclusions
In this study, FAC was suggested to play an important role in the removal of FF in DWTPs. Several chlorination experiments were conducted in order to evaluate the effects on FF degradation. FF can be removed effectively by chlorination, which is dependent on the initial chlorine dose. The presence of Cl− and Br− are favorable for FF oxidation, while HA hinders elimination. In addition, 21 different byproducts were found during chlorination. This is an important issue which needs more exploration as some of these degradation products may be more toxic than the parent compounds.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 51178321), the National Major Project of Science & Technology Ministry of China (No. 2012ZX07403-001; 2012ZX07403-002; No. 2008ZX07421-002), and the Specialized Research Fund for the Doctoral Program of Higher Education (No. 20120072110050). We are also thankful to the reviewers for their valuable advice to improve this manuscript.
References
- W. Tao, M. H. Lee, J. Wu, N. H. Kim, J.-C. Kim, E. Chung, E. C. Hwang and S.-W. Lee, Inactivation of Chloramphenicol and Florfenicol by a Novel Chloramphenicol Hydrolase, Appl. Environ. Microbiol., 2012, 78, 6295–6301 CrossRef CAS PubMed.
- Y. Belkaid and T. W. Hand, Role of the microbiota in immunity and inflammation, Cell, 2014, 157, 121–141 CrossRef CAS PubMed.
- K. Holmes, D. Bedenice and M. G. Papich, Florfenicol pharmacokinetics in healthy adult alpacas after subcutaneous and intramuscular injection, J. Vet. Pharmacol. Ther., 2012, 35, 382–388 CrossRef CAS PubMed.
- X. Tao, H. Jiang, X. Yu, J. Zhu, X. Wang, Z. Wang, L. Niu, X. Wu, X. Xia, W. Shi and J. Shen, Development and validation of a chemiluminescent ELISA for simultaneous determination of florfenicol and its metabolite florfenicol amine in chicken muscle, Anal. Methods, 2012, 4, 4083–4090 RSC.
- V. P. Syriopoulou, A. L. Harding, D. A. Goldmann and A. L. Smith, In vitro antibacterial activity of fluorinated analogs of chloramphenicol and thiamphenicol, Antimicrob. Agents Chemother., 1981, 19, 294–297 CrossRef CAS PubMed.
- J. D. Bowker, D. Carty and M. P. Bowman, The Safety of Aquaflor (50% Florfenicol) Administered in Feed to Fingerling Yellow Perch, North American Journal of Aquaculture, 2013, 75, 517–523 CrossRef.
- J. A. Settepani, The hazard of using chloramphenicol in food animals, J. Am. Vet. Med. Assoc., 1984, 184, 930–931 CAS.
- S. P. a. S. Schwarz, In Vitro Activities of Florfenicol against Bovine and Porcine Respiratory Tract Pathogens, Antimicrob. Agents Chemother., 2003, 47, 2703–2705 CrossRef.
- R. Del Pozo Sacristán, J. Thiry, K. Vranckx, A. López Rodríguez, K. Chiers, F. Haesebrouck, E. Thomas and D. Maes, Efficacy of florfenicol injection in the treatment of Mycoplasma hyopneumoniae induced respiratory disease in pigs, Vet. J., 2012, 194, 420–422 CrossRef PubMed.
- C. Kehrenberg, J. Mumme, J. Wallmann, J. Verspohl, R. Tegeler, T. Kühn and S. Schwarz, Monitoring of florfenicol susceptibility among bovine and porcine respiratory tract pathogens collected in Germany during the years 2002 and 2003 (ref. 2), J. Antimicrob. Chemother., 2004, 54, 572–574 CrossRef CAS PubMed.
- P. Gaunt, R. Endris, L. Khoo, A. T. Leard, S. Jack, T. Santucci, T. Katz, S. V. Radecki and R. Simmons, Preliminary Assessment of the Tolerance and Efficacy of Florfenicol against Edwardsiella ictaluri Administered in Feed to Channel Catfish, J. Aquat. Anim. Health, 2003, 15, 239–247 CrossRef.
- H. Wang, N. Wang, B. Wang, Q. Zhao, H. Fang, C. Fu, C. Tang, F. Jiang, Y. Zhou, Y. Chen and Q. Jiang, Antibiotics in Drinking Water in Shanghai and Their Contribution to Antibiotic Exposure of School Children, Environ. Sci. Technol., 2016, 50, 2692–2699 CrossRef CAS PubMed.
- R. Wei, F. Ge, M. Chen and R. Wang, Occurrence of ciprofloxacin, enrofloxacin, and florfenicol in animal wastewater and water resources, J. Environ. Qual., 2012, 41, 1481–1486 CrossRef CAS PubMed.
- B. Bektemuroglu and M. Sireli, The effect of chloramphenicol and florfenicol on electrocardiogram in guinea pigs, Ankara Univ. Vet. Fak. Derg., 2011, 58, 155–160 Search PubMed.
- J. Gibs, P. E. Stackelberg, E. T. Furlong, M. Meyer, S. D. Zaugg and R. L. Lippincott, Persistence of pharmaceuticals and other organic compounds in chlorinated drinking water as a function of time, Sci. Total Environ., 2007, 373, 240–249 CrossRef CAS PubMed.
- APHA, Standard Methods for the Examination of Water and Wastewater, APHA, AWWA, WPCF, Washington, DC, 1998 Search PubMed.
- W. Chu, D. Li, N. Gao, D. Yin, Y. Zhang and Y. Zhu, Comparison of free amino acids and short oligopeptides for the formation of trihalomethanes and haloacetonitriles during chlorination: effect of peptide bond and pre-oxidation, Chem. Eng. J., 2015, 281, 623–631 CrossRef CAS.
- Y. Zhang, Y. Shao, N. Gao, W. Chu and Z. Sun, Removal of microcystin-LR by free chlorine: identify of transformation products and disinfection by-products formation, Chem. Eng. J., 2016, 287, 189–195 CrossRef CAS.
- J. L. Acero, F. J. Benitez, F. J. Real, G. Roldan and E. Rodriguez, Chlorination and bromination kinetics of emerging contaminants in aqueous systems, Chem. Eng. J., 2013, 219, 43–50 CrossRef CAS.
- M. Q. Cai, L. Q. Zhang and L. Feng, Influencing factors and degradation behavior of propyphenazone and aminopyrine by free chlorine oxidation, Chem. Eng. J., 2014, 244, 188–194 CrossRef CAS.
- W. Li, J. M. Duan and D. Mulcahy, Kinetic characteristics of oxidation of microcystin-LR at low concentration by chlorine and permanganate, J. Water Supply: Res. Technol.--AQUA, 2012, 61, 82–93 CrossRef CAS.
- H. Cheng, D. Song, Y. Chang, H. Liu and J. Qu, Chlorination of tramadol: reaction kinetics, mechanism and genotoxicity evaluation, Chemosphere, 2015, 141, 282–289 CrossRef CAS PubMed.
- R. Rodil, J. B. Quintana and R. Cela, Transformation of phenazone-type drugs during chlorination, Water Res., 2012, 46, 2457–2468 CrossRef CAS PubMed.
- M. Q. Cai, L. Q. Zhang, F. Qi and L. Feng, Influencing factors and degradation products of antipyrine chlorination in water with free chlorine, J. Environ. Sci., 2013, 25, 77–84 CrossRef CAS.
- J. D. Sivey, C. E. McCullough and A. L. Roberts, Chlorine monoxide (Cl2O) and molecular chlorine (Cl2) as active chlorinating agents in reaction of dimethenamid with aqueous free chlorine, Environ. Sci. Technol., 2010, 44, 3357–3362 CrossRef CAS PubMed.
- D. P. Cherney, S. E. Duirk, J. C. Tarr and T. W. Collette, Monitoring the speciation of aqueous free chlorine from pH 1 to 12 with Raman spectroscopy to determine the identity of the potent low-pH oxidant, Appl. Spectrosc., 2006, 60, 764–772 CrossRef CAS PubMed.
- L. M. Rebenne, A. C. Gonzalez and T. M. Olson, Aqueous chlorination kinetics and mechanism of substituted dihydroxybenzenes, Environ. Sci. Technol., 1996, 30, 2235–2242 CrossRef CAS.
- C. Xue, Q. Wang, W. Chu and M. R. Templeton, The impact of changes in source water quality on trihalomethane and haloacetonitrile formation in chlorinated drinking water, Chemosphere, 2014, 117, 251–255 CrossRef CAS PubMed.
- S. E. Duirk, J. C. Tarr and T. W. Collette, Chlorpyrifos transformation by aqueous chlorine in the presence of bromide and natural organic matter, J. Agric. Food Chem., 2008, 56, 1328–1335 CrossRef CAS PubMed.
- H. Gallard and U. Von Gunten, Chlorination of phenols: kinetics and formation of chloroform, Environ. Sci. Technol., 2002, 36, 884–890 CrossRef CAS PubMed.
- F. Javier Benitez, J. L. Acero, F. J. Real, G. Roldan and F. Casas, Bromination of selected pharmaceuticals in water matrices, Chemosphere, 2011, 85, 1430–1437 CrossRef PubMed.
- K. Inaba, T. Doi, N. Isobe and T. Yamamoto, Formation of bromo-substituted triclosan during chlorination by chlorine in the presence of trace levels of bromide, Water Res., 2006, 40, 2931–2937 CrossRef CAS PubMed.
- A. D. Nikolaou, S. K. Golfinopoulos, T. D. Lekkas and M. N. Kostopoulou, DBP levels in chlorinated drinking water: effect of humic substances, Environ. Monit. Assess., 2004, 93, 301–319 CrossRef CAS PubMed.
- F. Tian, W. J. Liu, G. Guo, Z. M. Qiang and C. Zhang, Kinetics and mechanism of dimethoate chlorination during drinking water treatment, Chemosphere, 2014, 103, 181–187 CrossRef CAS PubMed.
- U. Iriarte-Velasco, J. I. Alvarez-Uriarte and J. R. Gonzalez-Velasco, Kinetics of chloroform
formation from humic and fulvic acid chlorination, J. Environ. Sci. Health, Part A: Environ. Sci. Eng. Toxic Hazard. Subst. Control, 2006, 41, 1495–1508 CrossRef CAS PubMed.
- P. Westerhoff, J. Debroux, G. Aiken and G. Amy, Ozone-induced changes in natural organic matter (NOM) structure, Ozone: Sci. Eng., 1999, 21, 551–570 CrossRef CAS.
- P. Westerhoff, P. Chao and H. Mash, Reactivity of natural organic matter with aqueous chlorine and bromine, Water Res., 2004, 38, 1502–1513 CrossRef CAS PubMed.
- M. Bourgin, E. Bichon, J.-P. Antignac, F. Monteau, G. Leroy, L. Barritaud, M. Chachignon, V. Ingrand, P. Roche and B. Le Bizec, Chlorination of bisphenol A: non-targeted screening for the identification of transformation products and assessment of estrogenicity in generated water, Chemosphere, 2013, 93, 2814–2822 CrossRef CAS PubMed.
- K. Moriyama, H. Matsufuji, M. Chino and M. Takeda, Identification and behavior of reaction products formed by chlorination of ethynylestradiol, Chemosphere, 2004, 55, 839–847 CrossRef CAS PubMed.
- V. L. Heasley, M. E. Anderson, D. S. Combes, D. S. Elias, J. T. Gardner, M. L. Hernandez, R. J. Moreland and D. F. Shellhamer, Investigations of the structure and reactions of the intermediate in the chlorination of resorcinol, Environ. Toxicol. Chem., 1993, 12, 1653–1659 CrossRef CAS.
- D. M. White, D. S. Garland, J. Narr and C. R. Woolard, Natural organic matter and DBP formation potential in Alaskan water supplies, Water Res., 2003, 37, 939–947 CrossRef CAS PubMed.
- M. C. Dodd and C. H. Huang, Transformation of the antibacterial agent sulfamethoxazole in reactions with chlorine: kinetics mechanisms, and pathways, Environ. Sci. Technol., 2004, 38, 5607–5615 CrossRef CAS PubMed.
- W. G. Lai, N. Zahid and J. P. Uetrecht, Metabolism of trimethoprim to a reactive iminoquinone methide by activated human neutrophils and hepatic microsomes, J. Pharmacol. Exp. Ther., 1999, 291, 292–299 CAS.
- L. Ge, J. Chen, X. Qiao, J. Lin and X. Cai, Light-source-dependent effects of main water constituents on photodegradation of phenicol antibiotics: mechanism and kinetics, Environ. Sci. Technol., 2009, 43, 3101–3107 CrossRef CAS PubMed.
- M. Bedner and W. A. Maccrehan, Transformation of acetaminophen by chlorination produces the toxicants 1,4-benzoquinone and N-acetyl-p-benzoquinone imine, Environ. Sci. Technol., 2006, 40, 516–522 CrossRef CAS PubMed.
- J. M. Buth, W. A. Arnold and K. McNeill, Unexpected products and reaction mechanisms of the aqueous chlorination of cimetidine, Environ. Sci. Technol., 2007, 41, 6228–6233 CrossRef CAS PubMed.
- R. Brix, N. Bahi, M. J. L. de Alda, M. Farre, J. M. Fernandez and D. Barcelo, Identification of disinfection by-products of selected triazines in drinking water by LC-Q-ToF-MS/MS and evaluation of their toxicity, J. Mass Spectrom., 2009, 44, 330–337 CrossRef CAS PubMed.
- S. H. A. G. S. Daignault, Potent odor-causing chemicals arising from drinking water disinfection, Water Sci. Technol., 1988, 20, 55–61 Search PubMed.
- E. R. Blatchley, D. Margetas and R. Duggirala, Copper catalysis in chloroform formation during water chlorination, Water Res., 2003, 37, 4385–4394 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra23342b |
| ‡ Present address: 1239 Siping Road, Shanghai, P. R. China. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 






