
Rapid and selective determination of pitavastatin calcium in presence of its degradation products and co-formulated drug by first-derivative micelle-enhanced and synchronous fluorimetric methods
Ahmed S. Fayed*a,
Maha A. Hegazya,
Enas E. Abbasb and
Nahla N. Salamab
aAnalytical Chemistry Department, Faculty of Pharmacy, Cairo University, Cairo, Egypt. E-mail: ahmed.fayed@pharma.cu.edu.eg; fayedaeg@yahoo.com; Tel: +20 1001494990
bNational Organization for Drug Control and Research (NODCAR), Abu Hazem Street, Pyramids Avenue, Giza, Egypt
First published on 25th October 2016
Abstract
New spectrofluorimetric methods are presented for the rapid and selective determination of pitavastatin calcium (PIT) in the presence of its hydrolytic degradation products and the co-formulated drug ezetimibe (EZE). In the first method (method A), PIT was determined in the presence of its hydrolytic degradation products by measuring the first derivative (1D) of its enhanced native fluorescence using sodium dodecyl sulfate (SDS) as an anionic micelle enhancer at a pH of 4.1, which was adjusted using Britton–Robinson buffer in aqueous media. PIT was subjected to stress conditions of 5 M hydrochloric acid and 5 M sodium hydroxide. The 1D peak amplitude was measured at a λem of 415 nm using a λex of 252 nm. The effect of different surfactants on the fluorescence of PIT was studied; the quantum yield of PIT was in the following order: SDS (0.30) > cetrimide (0.23) > Tween (0.02). A second method (method B) was developed for the simultaneous determination of PIT and the co-formulated drug EZE in the presence of degradation products of PIT. This method utilized synchronous fluorescence spectroscopy (SFS) in an acetonitrile medium. The wavelengths that were used were λex = 260 nm and λem = 330 and 272 nm for PIT and EZE, respectively, using Δλ = 40 nm. Validation of the proposed methods was performed according to the ICH guidelines. The methods were employed for the determination of PIT (methods A and B) and EZE (method B) in bulk powder, pharmaceutical preparations of co-formulated drug substances and laboratory-prepared mixtures containing hydrolytic degradation products of PIT. Statistical analysis proved that the investigated assays were excellent. It was concluded that method A was inexpensive and environmentally friendly.
1. Introduction
Pitavastatin calcium (PIT) is chemically designated as (3R,5S,6E)-7-[2-cyclopropyl-4-(p-fluorophenyl)-3-quinolyl]-3,5-dihydroxy-6-heptenoic acid calcium salt (Fig. 1). It is a novel fully synthetic statin, which has more potent cholesterol-lowering activity than other drugs in its class. PIT is an inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase. It is used in the treatment of hyperlipidemia and can reduce the risk of cardiovascular diseases in everyday medical practice.1–3 On the basis of preclinical findings, PIT has been widely used as a first-line agent in lipid-modifying therapies.4 | ||
| Fig. 1 Chemical structures of the investigated drugs. | ||
Ezetimibe (EZE) has the systematic name 1-(4-fluorophenyl)-3-(3-(4-fluorophenyl)-3-hydroxypropyl)-4-(4-hydroxyphenyl)-azetidin-2-one (Fig. 1). It lowers levels of cholesterol by preventing its absorption from dietary and biliary sources via blocking the transport of cholesterol through the intestinal wall.2,5 A previous study demonstrated that the co-administration of statins such as atorvastatin or PIT with EZE has an additive effect. The concomitant use of PIT and EZE, according to a European patent, has a remarkable blood cholesterol level lowering effect in comparison to that of the concomitant use of another HMG-CoA reductase inhibitor and EZE.6,7
A literature survey revealed that few methods have been used for the simultaneous determination of PIT and EZE. These include thin-layer chromatography (TLC) and high-performance liquid chromatography (HPLC).8,9 Several analytical methods such as spectrophotometry,10,11 spectrofluorometry,12 HPTLC,13,14 HPLC,15–20 stability-indicating HPLC,21 ultra-performance liquid chromatography (UPLC),22 LC-MS/MS23,24 and LC-ESI-MS25,26 have been reported for the determination of PIT in pharmaceutical dosage forms and/or biological samples.
There is no reported spectrofluorometric method for the determination of PIT in the presence of its degradation products or EZE. The aim of this study is to develop selective, sensitive, cheap and eco-friendly spectrofluorometric methods for the determination of the abovementioned drugs.
The first proposed method is a first-derivative micelle-enhanced spectrofluorometric process. This method was developed for the determination of PIT in the presence of its acid and base hydrolytic degradation products. Derivative spectrofluorimetry has been used to resolve spectral interference in emissions to enable the simultaneous determination of components of a mixture.27–29 Derivatization of overlapping curves will enable the appearance of a zero-crossing point, where one of the curves gives a zero reading while the other has a pronounced value, which allows it to be determined.
In the second suggested method, advantage is taken of the ability of synchronous fluorescence spectroscopy (SFS) to increase the selectivity and resolution of interfering spectra. An SFS method is presented for the simultaneous determination of both PIT and EZE in the presence of degradation products of PIT. Upon employing SFS, shifting and narrowing of the obtained emission spectra of components of a mixture occurs, which enables spectral resolution. These effects arise from permitting simultaneous emission and excitation scanning to take place with a difference in the values of λ used for scanning.30,31
2. Experimental
2.1. Instrumentation
A Cary Eclipse fluorescence spectrofluorometer (Agilent Technologies) was used, which was operated with Cary Eclipse Scan Application software version 1.2 (147).2.2. Materials and reagents
2.3. Solutions
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (50:50, v/v), whereas a stock standard solution of EZE was prepared by dissolving 10.00 mg EZE in 100 mL acetonitrile.:water (50:50, v/v) or by using acetonitrile as the diluting solvent for methods A and B, respectively. On the other hand, a working standard solution of EZE was prepared by diluting 5 mL of its stock standard solution to 50 mL with acetonitrile.
:water (50:50, v/v), whereas a stock standard solution of EZE was prepared by dissolving 10.00 mg EZE in 100 mL acetonitrile.:water (50:50, v/v) or by using acetonitrile as the diluting solvent for methods A and B, respectively. On the other hand, a working standard solution of EZE was prepared by diluting 5 mL of its stock standard solution to 50 mL with acetonitrile.2.3.3.1 Stock solutions (1.00 mg mL−1). Stock solutions of hydrolytic degradation products of PIT were separately prepared by heating an accurately weighed amount of 50.00 mg PIT with 50 mL of either 5 M hydrochloric acid or 5 M sodium hydroxide for 6 h at 100 °C. The solutions were neutralized and evaporated on a water bath. The residues were separately dissolved and diluted to 50 mL with acetonitrile
:water (50:50, v/v).
2.3.3.2 Working standard solutions (10.00 and 100.00 μg mL−1). Two working solutions were prepared for both acid and base degradation products. The first solutions were obtained by diluting 1 mL of the stock solutions of the degradation products to 100 mL to achieve a final concentration of 10.00 μg mL−1. The second working solutions were prepared by diluting 5 mL of each of the stock solutions of the degradation products to 50 mL to achieve a final concentration of 100.00 μg mL−1. Dilutions were performed using a solvent mixture of acetonitrile
:water (50:50, v/v).
2.4. Procedures
2.5. Method validation35
2.5.1.1 Method A. Accurately measured aliquots equivalent to 0.10–10.00 μg mL−1 PIT were transferred into a series of 10 mL volumetric flasks and then 1 mL BRB buffer (pH 4.1) and 1 mL 0.04% SDS were added. The volumes were made up with water to achieve a final concentration range of 0.01–1.00 μg mL−1, and the procedure mentioned under method A was followed. The calibration graph was constructed by relating the peak amplitudes at 392 nm to the corresponding concentrations of PIT and the regression equation was computed.
2.5.1.2 Method B. Accurately measured aliquots equivalent to 2.50–30.00 μg mL−1 PIT and 2.00–50.00 μg mL−1 EZE were transferred into a series of 10 mL volumetric flasks. The volumes were made up with acetonitrile to obtain standard solutions in the concentration ranges of 0.25–3.00 μg mL−1 for PIT and 0.20–5.00 μg mL−1 for EZE, and then the procedure mentioned under method B was followed. The fluorescence intensities for each compound were plotted against the corresponding concentrations and the regression equations were computed.
2.5.3.1 Laboratory-prepared mixture with acid degradation products.
Method A. Aliquots equivalent to 0.80–0.99 μg mL−1 PIT were separately transferred from its stock standard solution into a series of 10 mL volumetric flasks followed by aliquots equivalent to 0.20–0.01 μg mL−1 of the acid degradation products. Then, 1 mL BRB buffer (pH 4.1) and 1 mL 0.04% SDS were added and the volumes were made up with water.
Method B. Aliquots equivalent to 2.10–2.97 μg mL−1 of the binary mixture (3 μg mL−1 PIT; 3 μg mL−1 EZE) were transferred from the stock solutions into a series of 10 mL volumetric flasks followed by aliquots equivalent to 0.90–0.03 μg mL−1 of the acid degradation products (1–30%). The volumes were made up with acetonitrile.
The procedure described under Linearity was then followed. The concentrations of the intact drugs were calculated from the corresponding regression equations.
2.5.3.2 Laboratory-prepared mixture with alkaline degradation products.
Method A. Aliquots equivalent to 0.80–0.99 μg mL−1 PIT were separately transferred from its stock solution into a series of 10 mL volumetric flasks followed by aliquots equivalent to 0.20–0.01 μg mL−1 of the base degradation products.
Method B. Aliquots equivalent to 2.10–2.97 μg mL−1 of the binary mixture (3 μg mL−1 PIT; 3 μg mL−1 EZE) were transferred from the stock solutions into a series of 10 mL volumetric flasks followed by aliquots equivalent to 0.90–0.03 μg mL−1 of the acid degradation products (1–30%). The volumes were made up with acetonitrile.
The procedure described under Linearity was then followed. The concentrations of intact drugs were calculated from the corresponding regression equations.
Synthetic mixtures of the binary mixture were prepared with different ratios (1:1, 1:2, and 1:5) of PIT and EZE, respectively. The procedure described under Linearity was followed. The concentration of each drug was calculated from the corresponding regression equations.
2.6. Application to pharmaceutical formulations
:water (50:50, v/v) to reach a final concentration of 10 μg mL−1. Then, the procedure described under method A was followed.3. Results and discussion
Two rapid, sensitive and selective spectrofluorimetric stability-indicating methods were developed, optimized and validated for the determination of PIT in the presence of its hydrolytic degradation products (acid and alkaline) and the co-formulated drug EZE.PIT is characterized by displaying native fluorescence owing to its fused aromatic rings and extended conjugated structure (Fig. 1). The excitation and emission spectra of PIT are shown in Fig. 2, which shows a λem of 418 nm after excitation at a λex of 252 nm in water. Moreover, it is liable to degradation under both acidic and alkaline conditions. The acid and alkaline degradation products that were formed exhibited detectable fluorescence at the emission maximum of the drug.
 | ||
| Fig. 2 Excitation (-----) and emission (——) spectra of native fluorescence of pitavastatin calcium (0.8 μg mL−1) in 1 mL BRB (pH 4.1), 1 mL SDS and water as solvent at a λem of 418 nm and a λex of 252 nm. | ||
The first stability-indicating method was based on measuring the amplitude of the first derivative (1D) at a λem of 293 nm of the micelle-enhanced native fluorescence of PIT. The 1D spectra of the hydrolytic degradation products display zero-crossing at the abovementioned value of λem. Forced degradation studies of PIT were carried out using 5 M hydrochloric acid and 5 M sodium hydroxide. Complete degradation was confirmed by HPLC. To increase the sensitivity of the method, advantage was taken of the enhancement of fluorescence by the addition of a surfactant at a concentration above its critical micellar concentration. This has resulted in an increase in the fluorescence quantum yield of a fluorophore in many cases.36 This phenomenon has long been studied. It is suggested that in an organized micellar assembly enhancement of media occurs owing to the isolation and protection of the analyte from quencher molecules by micelles, as well as changes in the physical properties of the environment around it, e.g., increases in viscosity, polarity, etc. The enhancement depends on the type of micelle, the structure of the substrate and the interaction between them.37,38 The fluorescence properties of PIT in various micellar media were studied. There was a fourfold increase in fluorescence intensity in the presence of SDS in comparison with that in aqueous solution. PIT has a pKa of 4.12 and in an acidic medium it will be protonated; this would allow interaction to take place with the anionic surfactant SDS. Fig. 3 shows the excitation and emission spectra of PIT in water and upon adding 1 mL BRB buffer (pH 4.1) and 1 mL 0.04% SDS. The spectra of its acid and alkaline degradation products displayed considerable interference. The first derivative of the micelle-enhanced fluorescence spectrum was computed to cancel the interference of the acid and alkaline degradation products of PIT. Zero-crossing was observed at a λem of 293 nm and therefore this was selected as the wavelength for analysis (Fig. 4).
 | ||
| Fig. 3 Emission spectra of native fluorescence of pitavastatin calcium in media enhanced by sodium dodecyl sulfate micelles at a pH of 4.1: BRB and water as diluting solvent (——), pitavastatin calcium in water (-----), acid degradation products (……), alkaline degradation products (-.-.-.-.-) and blank reagent (_.. _) at a λem of 418 nm and a λex of 252 nm (1 μg mL−1). | ||
 | ||
Fig. 4 First-derivative spectra of: (A) pitavastatin calcium ( ) and its acid ( ) and its acid ( ) and alkaline ( ) and alkaline ( ) degradation products at a λem of 392 nm and a λex of 252 nm (1.0 μg mL−1). (B) Different concentrations of pitavastatin calcium (0.01–1.00 μg mL−1) at a λem of 392 nm and a λex of 252 nm. Inset: calibration curve correlating the peak amplitudes of the first-derivative fluorescence intensities of pitavastatin calcium determined using method A with the corresponding concentrations (0.01–1.00 μg mL−1). ) degradation products at a λem of 392 nm and a λex of 252 nm (1.0 μg mL−1). (B) Different concentrations of pitavastatin calcium (0.01–1.00 μg mL−1) at a λem of 392 nm and a λex of 252 nm. Inset: calibration curve correlating the peak amplitudes of the first-derivative fluorescence intensities of pitavastatin calcium determined using method A with the corresponding concentrations (0.01–1.00 μg mL−1). | ||
In the second method, a synchronous fluorescence spectroscopy technique (SFS) was employed for the simultaneous determination of both PIT and EZE in co-formulated preparations and in the presence of acid and alkaline degradation products of PIT. SFS plays an important role in the simultaneous determination of compounds in multicomponent mixtures owing to its remarkable advantages of spectral simplification, reduction in light scattering, and improvement in selectivity over that of conventional fluorescence spectroscopy.39–41 Taking advantage of the spectral resolution achieved by the shifting and narrowing of the emission spectra upon adding SFS and increasing the sensitivity by the enhancement of fluorescence using organized micellar media has enabled the analysis of multicomponent mixtures of polycyclic aromatic hydrocarbons.42,43
An emphasis is made in this work on the development of a rapid and simple synchronous fluorescence-based method for the determination of a binary mixture of PIT and EZE in the presence of hydrolytic degradation products of PIT. Fig. 5 shows the normal emission spectrum of a binary mixture of PIT and EZE in acetonitrile. Both PIT and EZE exhibit native fluorescence with λmaximum for PIT and EZE occurring at values of λem of 373 nm and 310 nm, respectively, after excitation of both at 260 nm, where no complete separation was observed (Fig. 5). Therefore, the SFS technique was performed, which resulted in complete spectral resolution and enabled the simultaneous determination of both PIT and EZE in their drug formulations and pharmaceutical preparations, as well as in the presence of degradation products of PIT. Fig. 6 shows the emission spectra of a mixture of PIT and EZE obtained by employing SFS with the optimum chosen parameters, in which complete resolution of the spectra of PIT and EZE was observed.
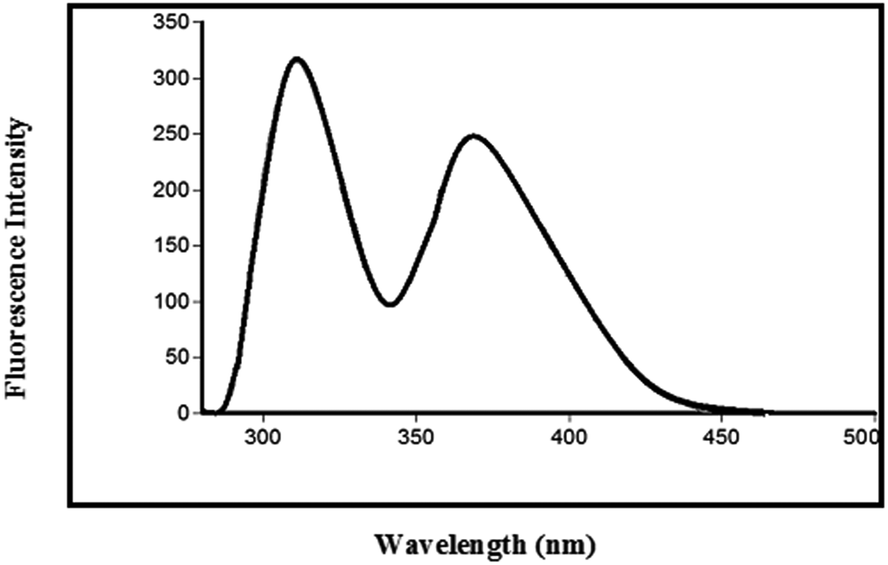 | ||
| Fig. 5 Intensity of the native fluorescence spectrum of a mixture of pitavastatin calcium and ezetimibe at values of λem of 373 nm and 310 nm, respectively, after excitation of both at 260 nm in acetonitrile (3 μg mL−1 each). | ||
 | ||
Fig. 6 Intensity of the synchronous fluorescence spectra of a synthetic mixture of pitavastatin calcium, ezetimibe and 30% of the degradation products of pitavastatin (acid  and alkaline and alkaline  ) at Δλ = 40 nm at values of λem of 330 nm and 272 nm ( ) at Δλ = 40 nm at values of λem of 330 nm and 272 nm ( ) for both drugs, respectively, in acetonitrile (3 μg mL−1 each). ) for both drugs, respectively, in acetonitrile (3 μg mL−1 each). | ||
3.1. Optimization of experimental conditions
Several factors were found to affect the fluorescence. These factors were studied and optimized; for example, the effect of different diluting solvents including water, methanol, ethanol, acetonitrile, isopropanol and acetone was studied. It was observed that by using water and acetonitrile, the strongest fluorescence was obtained for PIT and EZE, respectively. However, upon using acetonitrile:water (50:50, v/v) and employing SFS (method B), a considerable shift in the synchronous spectrum of EZE to longer wavelengths occurred, which caused interference with the spectrum of PIT; therefore, acetonitrile alone was used as the diluting solvent.
Different model surfactant systems were tested, whereby the maximum fluorescence signals were obtained using SDS as an anionic surfactant, whereas little enhancement was observed upon using Tween 80 as an example of a non-ionic surfactant. The cationic surfactant cetrimide caused a considerable enhancement of fluorescence, but this was still less than that for SDS. The quantum yields [QY]44,45 upon adding different surfactants were calculated and the order of the quantum yields for the surfactants was SDS > cetrimide > Tween (Table 1).
| Surfactant | Abs | Integrated emission | QYa |
|---|---|---|---|
| a QY = Ys(Fu/Fs)(As/Au), QY = quantum yield, As = absorbance of standard, Au = absorbance of unknown, Ys = quantum yield of standard, Fu = integrated emission of unknown, Fs = integrated emission of standard. | |||
| Quinine sulphate (standard) | 0.018 | 30480 |
— |
| SDS | 0.012 | 24383 |
0.30 |
| Cetrimide | 0.014 | 16597 |
0.23 |
| Tween | 0.014 | 12490 |
0.02 |
For testing the effect of pH, a universal buffer (Britton–Robinson buffer) with various pH values ranging from 3.1 to 8.1 was used. The optimum pH was found to be 4.1. The influence of pH values above 4.1 caused a gradual decrease in FI. As the fluorescence intensity for protonated species is always higher than that for neutral species, it can be inferred that the protonated form of a species interacts more strongly with anionic micelles of SDS than the neutral form of the drug. The influences of the concentration and volume of SDS on the FI were studied using different volumes of 0.04% SDS (w/v). It was found that an increase in the volume of the SDS solution resulted in a corresponding increase in FI up to 1 mL, after which no further increase was observed. Therefore, 1 mL SDS was chosen as the optimum volume for PIT. The synchronous parameters investigated for method B included the scanning range and the effect of Δλ on SFS. The synchronous wavelength range of 220–600 nm was scanned while changing Δλ. The effect of different values of Δλ on the resolution of the emission spectra of PIT and EZE from Δλ = 10 nm to Δλ = 70 nm is shown in Fig. 7, in which the optimum value of Δλ was found to be 40 nm with λem values of 330 nm and 272 nm for PIT and EZE, respectively, using a λex of 260 nm. Fig. 8 and 9 show the intensity of synchronous fluorescence for different concentrations of PIT in the presence of EZE and vice versa.
 | ||
Fig. 7 Intensity of synchronous fluorescence at different Δλ values of 10–70 nm ( ), with the best spectrum at Δλ = 40 nm ( ), with the best spectrum at Δλ = 40 nm ( ), of a synthetic mixture of pitavastatin calcium and ezetimibe (each at 2 μg mL−1) at λem values of 330 nm and 272 nm, respectively, in acetonitrile. ), of a synthetic mixture of pitavastatin calcium and ezetimibe (each at 2 μg mL−1) at λem values of 330 nm and 272 nm, respectively, in acetonitrile. | ||
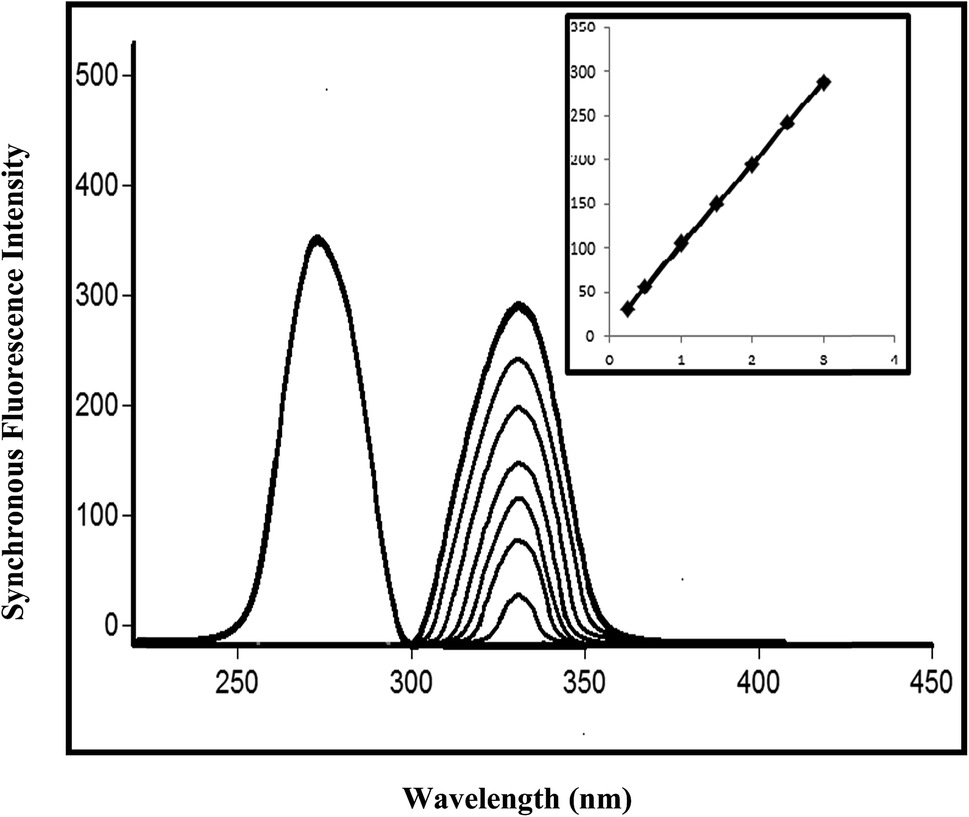 | ||
| Fig. 8 Intensity of synchronous fluorescence for different concentrations of pitavastatin calcium (0.25–3.00 μg mL−1) and ezetimibe (3.00 μg mL−1) at Δλ = 40 nm and λem = 330 nm and 272 nm for both drugs, respectively, in acetonitrile. Inset: calibration curve correlating the intensities of the synchronous fluorescence of pitavastatin calcium with the corresponding concentrations. | ||
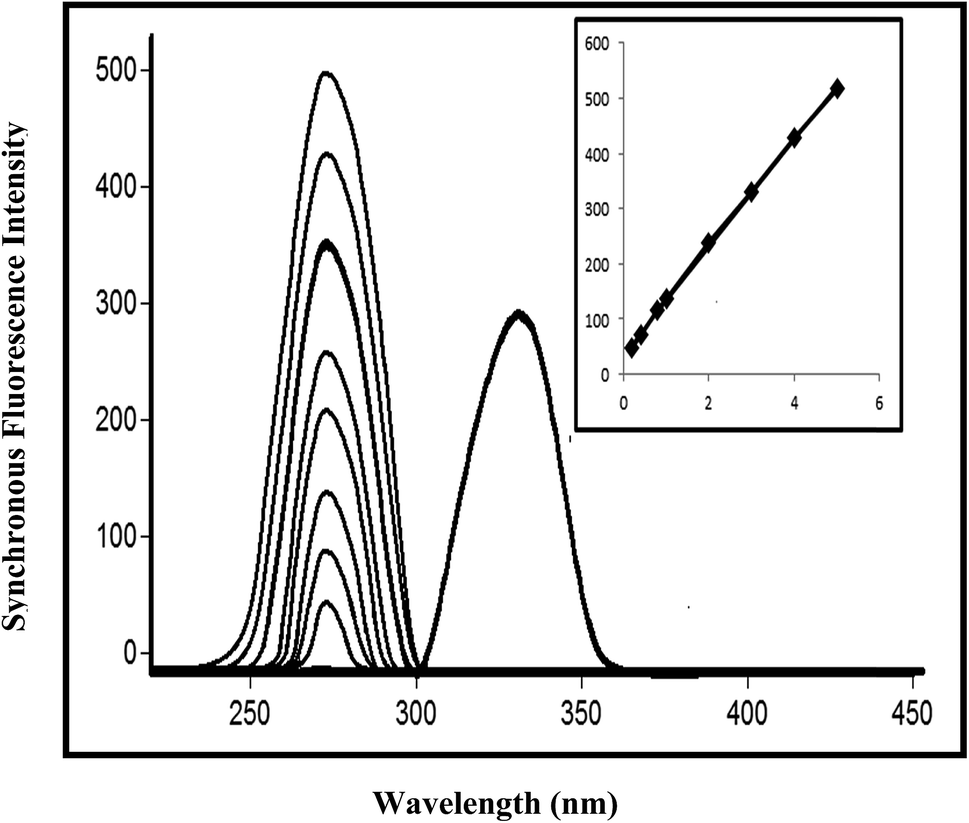 | ||
| Fig. 9 Intensity of synchronous fluorescence for different concentrations of ezetimibe (0.20–5.00 μg mL−1) and pitavastatin calcium (3.00 μg mL−1) at Δλ = 40 nm and λem = 272 nm and 330 nm for both drugs, respectively, in acetonitrile. Inset: calibration curve correlating the intensities of the synchronous fluorescence of ezetimibe to the corresponding concentrations. | ||
3.2. Method validation
Calibration curves were constructed, which represented the relationship between the fluorescence intensities and the corresponding concentrations in the ranges of 0.01–1.00 μg mL−1 and 0.25–3.00 μg mL−1 for PIT using methods A and B, respectively, whereas the range was 0.20–5.00 μg mL−1 for EZE using method B (Fig. 8 and 9). The characteristic parameters for the regression equations were computed. The LOD and LOQ were calculated and the results are presented in Table 2. The developed methods were found to be accurate for the determination of PIT, where values of the recovery percentage ± RSD of 100.25 ± 1.026% and 100.22 ± 1.091% for methods A and B, respectively, were obtained, as shown in Table 2.| Parameter | Method A | Method B | |
|---|---|---|---|
| PIT | PIT | EZE | |
| *Mean ± SD, n = 5.a is the intercept,b is the slope. | |||
| Linearity range (μg mL−1) | 0.01–1.00 | 0.25–3.00 | 0.20–5.00 |
| LOD (μg mL−1) | 0.0022 | 0.047 | 0.049 |
| LOQ (μg mL−1) | 0.0075 | 0.155 | 0.165 |
| Accuracy* (mean ± SD) | 99.68 ± 0.828% | 99.73 ± 1.548% | 100.12 ± 1.366% |
| Slopeb | 254.699 | 93.110 | 97.411 |
| SE of slope | 2.146 | 0.824 | 1.211 |
| Confidence limit of the slope | 249.623 to 259.774 | 90.993 to 95.227 | 94.448 to 100.373 |
| Intercepta | 21.719 | 9.337 | 36.234 |
| SE of intercept | 1.064 | 1.487 | 3.199 |
| Confidence limit of intercept | 19.202 to 24.236 | 5.515 to 13.159 | 28.407 to 44.060 |
| Correlation coefficient (r) | 0.9994 | 0.9996 | 0.9991 |
| SE of estimation | 2.237 | 2.068 | 5.707 |
The repeatability and intermediate precision of the proposed methods were determined for three concentration levels (0.30, 0.50, and 0.90 μg mL−1 and 0.40, 0.70, and 1.40 μg mL−1 for methods A and B, respectively, for PIT and 1.50, 2.00, and 2.50 μg mL−1 for EZE) by assaying three times on the same day and in triplicate on three successive days using the developed methods (n = 9). The results in Table 3 indicate the satisfactory precision of the proposed methods. For testing the selectivity of the methods, different laboratory-prepared mixtures containing different percentages of the studied drugs and their different degradation products were prepared and analyzed by the proposed methods. The methods were found to be selective, as indicated by the results in Table 4. The presence of 1–20% and 1–30% of hydrolytic degradation products of PIT, respectively, did not affect the recovery of the pure drug substances by both methods. Moreover, a set of laboratory-prepared mixtures of the two investigated drugs in variable proportions were analyzed to determine the specificity of the suggested methods (Table 4).
| Method B | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| PIT | EZE | ||||||||
| Taken (μg mL−1) | Intra-day | Inter-day | Taken (μg mL−1) | Intra-day | Inter-day | ||||
| Founda (μg mL−1) ± SD | Precision (RSD%) | Founda (μg mL−1) ± SD | Precision (RSD%) | Founda (μg mL−1) ± SD | Precision (RSD%) | Founda (μg mL−1) ± SD | Precision (RSD%) | ||
| 0.40 | 0.40 ± 0.001 | 0.25 | 0.41 ± 0.004 | 0.86 | 1.50 | 1.51 ± 0.020 | 1.32 | 1.51 ± 0.013 | 0.84 |
| 0.70 | 0.74 ± 0.006 | 0.91 | 0.70 ± 0.007 | 0.96 | 2.00 | 2.03 ± 0.036 | 1.78 | 2.03 ± 0.025 | 1.24 |
| 1.40 | 1.41 ± 0.012 | 0.86 | 1.41 ± 0.022 | 1.56 | 2.50 | 2.50 ± 0.047 | 1.26 | 2.50 ± 0.006 | 0.23 |
| Method B | |||||
|---|---|---|---|---|---|
| Acid degradation products% (w/w) | Recovery%a of intact drugs | Alkaline degradation products% (w/w) | Recovery%a of intact drugs | ||
| PIT | EZE | PIT | EZE | ||
| 1 | 99.47 | 99.50 | 1 | 98.82 | 100.50 |
| 5 | 99.98 | 100.80 | 5 | 100.33 | 98.90 |
| 10 | 99.53 | 98.53 | 10 | 99.17 | 99.62 |
| 20 | 98.87 | 99.74 | 20 | 98.99 | 101.20 |
| 30 | 99.95 | 101.45 | 30 | 99.92 | 100.80 |
| Mean ± RSD% | 99.58 ± 0.52 | 100.04 ± 1.14 | Mean ± RSD% | 99.45 ± 0.65 | 100.20 ± 0.93 |
| Synthetic mixture (μg mL−1) | Recovery%a | ||
|---|---|---|---|
| PIT | EZE | PIT | EZE |
| 1 | 1 | 99.58 | 98.54 |
| 1 | 2 | 99.46 | 99.72 |
| 1 | 5 | 98.89 | 101.22 |
| Mean ± RSD | 99.31 ± 0.37 | 99.83 ± 1.35 | |
The accuracy of the developed spectrofluorimetric methods was also determined by spiking previously analyzed drug products with extra concentrations of PIT and EZE. Satisfactory recoveries were obtained indicating no interference from tablets' excipients. The results are presented in Table 5.
| Method A | ||||
|---|---|---|---|---|
| Drug product | Recoverya of claimed amount ± RSD% | Amount taken (μg mL−1) | Standard added (μg mL−1) | Recovery%b of standard added |
| a Mean ± RSD of five determinations.b Mean of three determinations. | ||||
| Livazo 2 mg PIT per tablet | 92.83 ± 1.402 | 0.20 | 0.05 | 101.05 |
| 0.10 | 99.63 | |||
| 0.20 | 100.26 | |||
| 0.60 | 98.49 | |||
| 0.80 | 101.21 | |||
| Mean ± RSD%a | 100.13 ± 1.11 | |||
| Method B | ||||||
|---|---|---|---|---|---|---|
| Drug product | Recoverya of claimed amount ± RSD% | Amount taken (μg mL−1) | Standard added (PIT) (μg mL−1) | Recovery%b of standard added | Standard added (EZE) (μg mL−1) | Recovery%b of standard added |
| Livazo 2 mg PIT per tablet | 94.33 ± 1.381 | 0.40 PIT and 2.00 EZE | 0.40 | 101.046 | 2.00 | 99.44 |
| Choletimb 10 mg EZE per tablet | 91.43 ± 0.906 | 0.50 | 99.64 | 2.50 | 99.55 | |
| 0.60 | 101.21 | 3.00 | 101.6 | |||
| Mean ± RSD% | 100.63 ± 0.858 | Mean ± RSD% | 100.19 ± 1.214 | |||
The results obtained by employing the proposed methods for the analysis of the studied compounds in bulk powders and pharmaceutical preparations were statistically compared with those obtained using the manufacturer's methods. The calculated values of t and F were less than the theoretical values, which revealed that there was no significant difference with respect to accuracy and precision,46 as indicated in Table 6.
| Parameter | Drug formulations | |||||
|---|---|---|---|---|---|---|
| Method A | Method B | |||||
| Proposed method | Manufacturer'sa method | Proposed method | Manufacturer's method | |||
| PIT | EZE | PITa | EZEb | |||
| a Manufacturer's HPLC method, using Intersil C18 (5 μm, 250 × 4.6 mm) as the column, phosphate buffer (pH 3.5), acetonitrile in the ratio of 50:50 v/v as the mobile phase with a flow rate of 1.5 mL min−1, an injection volume of 20 μL and detection at 245 nm.b Manufacturer's HPLC method, using Intersil C18 (5 μm, 250 × 4.6 mm) as the column, phosphate buffer (pH 4.5), acetonitrile in the ratio of 65:35 v/v as the mobile phase with a flow rate of 1 mL min−1, an injection volume of 20 μL and detection at 240 nm.c The values in parentheses are the theoretical values of t and F. |
||||||
| Mean | 99.68 | 99.27 | 99.73 | 100.12 | 99.27 | 99.48 |
| SD | 0.828 | 1.490 | 1.548 | 1.366 | 1.490 | 0.971 |
| N | 5 | 5 | 5 | 5 | 5 | 5 |
| Variance | 0.686 | 2.220 | 2.396 | 1.866 | 2.220 | 0.943 |
| SE | 0.372 | 0.666 | 0.692 | 0.611 | 0.666 | 0.434 |
| t value (2.306)c | 0.538 | — | 0.479 | 1.121 | — | — |
| F Value (6.400)c | 3.236 | — | 1.079 | 0.96 | — | — |
The proposed spectrofluorimetric methods have various advantages over published chromatographic methods that are employed to determine PIT and EZE in pharmaceutical preparations, being less tedious and less time-consuming. Moreover, they are stability-indicating methods and can determine PIT in the presence of its degradation products either alone or in a binary mixture with EZE.
4. Conclusion
Two proposed spectrofluorimetric methods are presented. The first is a stability-indicating method based on the determination of pitavastatin calcium in the presence of its acid and alkaline hydrolytic degradation products using first-derivative micelle-enhanced spectrofluorimetry. The method is environmentally friendly and cost-saving as a result of using inexpensive and less toxic reagents. The second is also a stability-indicating method for the determination of pitavastatin calcium and is, moreover, used for the simultaneous determination of pitavastatin calcium and ezetimibe, a co-formulated drug, using synchronous spectrofluorimetry. The proposed methods are characterized by being simple, sensitive, specific and having a shorter analysis time in comparison to other methods, so they can be employed for routine analysis in quality control laboratories.References
- M. Soyka, C. Schuetz and U. Preuss, Drugs R&D, 2002, 3, 58–60 Search PubMed.
- S. C. Sweetman, Martindale: The complete dug reference, The Pharmaceutical press, London, 37th edn, 2011 Search PubMed.
- R. Y. Mukhtar, J. Reid and J. P. Reckless, Int. J. Clin. Pract., 2005, 59, 239–252 CrossRef CAS PubMed.
- K. Kajinami, N. Takekoshi and Y. Saito, Cardiovasc. Drug Rev., 2003, 21(3), 199–215 CrossRef CAS PubMed.
- T. Kosoglou, P. Statkevich, A. O. Johnson-Levonas, J. F. Paolini, A. J. Bergman and K. B. Alton, Clin. Pharmacokinet., 2005, 44, 467–494 CrossRef CAS PubMed.
- T. Aoki, H. Yamazaki and T. Maejima, European Patent, 1785137, 2005.
- ClinicalTrials, gov, NCT00653913.
- J. P. Hiral and N. S. Bhanubhai, J. AOAC Int., 2014, 97(1), 99–104 CrossRef.
- J. P. Hiral and N. S. Bhanubhai, Int. J. PharmTech Res., 2011, 3(4), 2155–2161 Search PubMed.
- M. V. Krishna and D. G. Sankar, Eur. J. Chem., 2007, 4, 272–278 CAS.
- Y. Katsuta, M. Ohsuga, H. Komeichi, S. Shimizu, Y. Kato, A. Miyamoto, K. Satomura and H. Tetada, Jpn. Pharmacol. Ther., 2007, 35, 489–501 Search PubMed.
- R. I. El-Bagary, E. F. El-Kady and A. M. Kadry, Spectroscopy, 2012, 27(2), 83–92 CrossRef CAS.
- N. H. Vadia, P. Vandana and H. N. Bhalara, Indian J. Pharm. Sci., 2008, 70, 649–651 CrossRef CAS PubMed.
- N. K. Satheesh and J. Baghyalakshmi, Anal. Lett., 2007, 40, 2625–2632 CrossRef.
- J. P. Hiral, N. S. Bhanubhai, J. P. Natvarlal and H. P. Bhavesh, J. Planar Chromatogr.-Mod. TLC, 2008, 21, 267–270 CrossRef.
- J. Kojima, H. Fujino, M. Yosimura, H. Morikawa and H. Kimata, J. Chromatogr. B: Biomed. Sci. Appl., 1999, 724, 173–180 CrossRef CAS.
- N. Ramakrishna, M. Koteshwara and K. Vishwottam, J. Pharm. Biomed. Anal., 2007, 44, 379–387 CrossRef PubMed.
- S. K. Nanjappan, N. Narayanan, N. Jayabalan, S. Narayanan and J. Bagyalakshmi, Pharm. Anal. Acta, 2011, 2, 119 CAS.
- J. P. Hiraland and N. S. Bhanubhai, J. AOAC Int., 2009, 92, 158–164 Search PubMed.
- A. Ojha, S. Guttikar, C. Vayeda and H. Padh, Sepu, 2007, 25, 715–718 CAS.
- G. Pawel, V. Giampietro, V. Daniela, D. Francesco, G. Anna and M. Jadwiga, J. Pharm. Biomed. Anal., 2009, 50, 597–601 CrossRef PubMed.
- R. G. Antony, R. R. Pannala, S. Nimmakayala and S. Jadi, Am. J. Anal. Chem., 2010, 2, 83–90 Search PubMed.
- J. P. Hiral and N. S. Bhanubhai, Acta Chromatogr., 2011, 3, 81–94 Search PubMed.
- W. D. Jian, B. K. Kwon, H. Z. Hong, H. L. Kwang and G. S. Jae, Biomed. Chromatogr., 2008, 22, 131–135 CrossRef PubMed.
- L. V. Hua, G. S. Jian, J. W. Guang, Y. Z. Xiao, Z. Ying, H. G. Sheng, L. Yan and S. Jie, Clin. Chim. Acta, 2007, 386, 25–30 CrossRef PubMed.
- T. Lei, H. Yiling, J. Youhong, H. Lu and L. Yishi, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 865, 127–132 CrossRef PubMed.
- M. C. Mahedero, F. Salinas and J. J. Aaron, J. Pharm. Biomed. Anal., 1994, 12(9), 1097–1101 CrossRef CAS PubMed.
- M. A. Ragab and E. I. EL-Kimary, Luminescence, 2015, 30(6), 760–767 CrossRef CAS PubMed.
- H. Maher, M. M. Alshehri and Sh. M. Altaweel, Luminescence, 2015, 30(3), 330–336 CrossRef CAS PubMed.
- J. B. Lloyd, Nature, 1971, 231, 64–65 CAS.
- D. Patra and A. K. Mishra, Talanta, 2001, 55, 143–153 CrossRef CAS PubMed.
- Manufacturer HPLC method supplied by Mash Pharmaceuticals, Egypt Search PubMed.
- Manufacturer HPLC method supplied by EVA Pharm Company, Egypt Search PubMed.
- E. Joseph, J. M. Reynolds, B. V. Russell and M. S. Kyril, Spectral and Redox Properties of the GFP Synthetic Chromophores as a Function of pH in buffered media, The Royal Society of Chemistry, 2013 Search PubMed.
- Validation of Analytical Procedure-Methodology (Q2R1), International Conference on Harmonization (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use, Geneva, Switzerland, 2005 Search PubMed.
- W. L. Hinze, H. N. Singh, Y. Baba and N. G. Harvey, Trends Anal. Chem., 1984, 3(8), 193–199 CrossRef CAS.
- Ch. D. Tran and T. A. Van Fleet, Anal. Chem., 1988, 60(22), 2478–2482 CrossRef CAS.
- W. R. Baeyens and B. L. Lings, J. Pharm. Biomed. Anal., 1989, 7(12), 1385–1394 CrossRef CAS PubMed.
- C. Jiag and J. He, J. Pharm. Biomed. Anal., 2002, 29, 737–742 CrossRef.
- J. L. Vilchez, A. Navalon, J. Rohand, R. Avidad and L. F. Capitian-Vallvey, J. Fluoresc., 1995, 5, 225–229 CrossRef CAS PubMed.
- T. Perez-Ruiz, C. Martinez Lozano, V. Tomas and J. Carpena, J. Anal. Chem., 1998, 361, 492–495 CAS.
- D. Patra, Luminescence, 2003, 18(2), 97–102 CrossRef CAS PubMed.
- D. Patra and T. H. Ghaddar, Talanta, 2009, 77(4), 1549–1554 CrossRef CAS PubMed.
- A. T. Williams, S. A. Winfield and J. N. Miller, Analyst, 1983, 108, 1067–1071 RSC.
- L. E. Abdelfattah, E. A. Taha and T. A. Mohamed, Chem. Ind. Chem. Eng. Q., 2010, 16(1), 31–38 CrossRef.
- M. Spiegel and L. Stephens, Schaum's Outline of Theory and Problems of Statistics, McGraw-Hill, New York, NY, 1999 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |