
Molecular dynamics simulations of simple aromatic compounds adsorption on single-walled carbon nanotubes
Han Ding ,
Xin Shen,
Chao Chen* and
Xiaojian Zhang
,
Xin Shen,
Chao Chen* and
Xiaojian Zhang
School of Environment, Tsinghua University, Beijing, 100084, P. R. China. E-mail: chen_water@tsinghua.edu.cn
First published on 9th August 2016
Abstract
The adsorption of three simple aromatic compounds (benzene, phenol and 4-chlorophenol) on single-walled carbon nanotubes (SWCNTs) has been investigated at a molecular level using molecular dynamics (MD) simulation. The adsorption energies of adsorbates on SWCNTs were calculated. The effects of adsorbate substituents and SWCNT size on the adsorption energy were analysed. The adsorption capacities of SWCNTs for adsorbates are positively correlated with the corresponding adsorption energy. The configurations of adsorbed molecules show that a second layer was formed on (5, 5) SWCNT and diminished with the increase in SWCNT diameter. The orientations of adsorbed molecules inside were limited by the pore size of the SWCNT. The angular distribution of adsorbed molecules indicates that small angles are more likely to be formed on a less curved surface. The non-bond interaction energy is negatively correlated with the binding strength of adsorbates to the SWCNT. The distances between adsorbed molecules and tube surface were determined by radial distribution function (RDF). The distances for benzene, phenol and 4-chlorophenol were 5.00 Å, 4.85 Å and 4.75 Å respectively, longer than those in vacuum conditions. The visualisation of non-bond interactions indicates that the π–π interaction is rather complicated, consisting of attractive and repulsive interactions.
1. Introduction
Since their discovery by Iijima1 in 1991, carbon nanotubes (CNTs) have attracted great interest due to their excellent physical properties and potential applications in various fields.2 CNTs have a large specific surface area and high hydrophobicity, indicating they can be utilized as a promising adsorbent in environmental remediation.3,4 In recent years, many studies have found that CNTs are very effective in removing organic contaminants from aqueous environment.5–9 The π–π interactions between CNTs surface and aromatic compounds can significantly enhance the adsorption affinity.10 As a result, CNTs may be specifically suitable for the adsorption of aromatic compounds. The adsorption capacity of single-walled carbon nanotubes (SWCNTs) is higher than that of multi-walled carbon nanotubes (MWCNTs) due to the larger specific surface area and smaller diameter of SWCNTs.11 According to the results of previous reports, the driving forces for CNTs adsorbing organic compounds include hydrophobic interactions, π–π interactions, hydrogen bonds and electrostatic interactions.4 However, the limitation of experimental research makes it difficult to elucidate adsorption mechanisms at molecular level, which means the characterization of adsorbate molecules orientations and dynamics on CNTs' surface is hard to achieve.12With the improvement of computational technology, molecular dynamics (MD) simulation has developed into a powerful tool for analysing molecular-level energy, structure and dynamics information that are hard to be obtained in normal experiments.13 MD simulation can provide valuable information about interactions between adsorbate molecules and adsorbent surface during adsorption process. It can reflect the most favourable adsorption sites, measure the binding energy, interpret adsorption data and facilitate system optimization.14 Recently, MD simulation has been used to study the adsorption mechanisms between organic compounds and SWCNTs. These organic compounds include DNA,15,16 amino acids,17 peptides,18 dyes19,20 and surfactants.21 The purposes of these studies are to improve the solubility of SWCNTs in water or to investigate whether SWCNTs can be used as delivery agents or sensors. To the best of our knowledge, only three published reports employed MD simulation to investigate the adsorption mechanisms of organic contaminants removal from water by SWCNTs. The adsorption of simple benzene derivatives,22 nitromethane23 and perfluorooctanesulfonate12 were investigated, with the size effects of SWCNT, adsorption energy and adsorbed molecule configuration discussed. Comer et al.24 used MD simulation to predict adsorption equilibrium constants of aromatic compounds on MWCNTs, which turned out to be highly correlated with experimental results. These pioneering works indicate that the utility of MD simulation in environmental remediation field is worth investigation.
Simple aromatic compounds are typical pollutants in the aqueous environment.25,26 They are ideally suitable for studying the mechanism of interaction on CNTs surface and characterizing the properties of CNTs.26 In the present work, we used MD simulations to explore the adsorption of three simple aromatic compounds (benzene, phenol and 4-chlorophenol) on SWCNTs with different diameters and functional groups in water. The adsorption amount, non-bond interaction energy, orientation of adsorbate molecules on the SWCNT in aqueous condition were measured and analysed. The impacts of temperature and functional groups on the adsorption capacity of SWCNT was discussed. The radial distribution function (RDF) was applied to describe the density of adsorbate molecules around SWCNT. The adsorption energies of water and three adsorbate molecules were calculated. In the end, the interactions between SWCNT surface and adsorbate molecules were visualised for further analysis. Our research provide detailed information about the adsorption of simple aromatic contaminants on SWCNTs at molecular level and can be used to judge the applicability of MD simulation in environmental remediation field.
2. Methods
2.1 Model construction
As shown in Fig. 1, the initial model was composed of a water box, an SWCNT and adsorbate molecules. The water box, which contained 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 010 water molecules, had the dimension of 7 × 7 × 7 nm3. The water molecules were characterized by the TIP3P27 model. The SWCNT was fixed in the centre of the water box. Three kinds of armchair SWCNTs, namely (5, 5), (8, 8) and (10, 10), were chosen as the adsorbent. They were all 3 nm in length. The carbon atoms of SWCNT was represented as neutral sp2-like aromatic carbons in the CHARMM27 general force field.23,28 Benzene, phenol and 4-chlorophenol were selected as the adsorbates due to their simple and typical structures. There were 32 adsorbate molecules uniformly distributed around the SWCNT. The parameters for these adsorbate molecules were extracted from the CHARMM36 general force field.29 The initial coordinates of the molecules' centres were exactly the same for each of the three adsorbates. To investigate the effect of oxidation on the adsorption process, the hydroxyl groups were placed on the top of the SWCNT and the tube surface was kept intact. The whole system was neutral. All the components in the model were constructed by VMD1.9.2.30
010 water molecules, had the dimension of 7 × 7 × 7 nm3. The water molecules were characterized by the TIP3P27 model. The SWCNT was fixed in the centre of the water box. Three kinds of armchair SWCNTs, namely (5, 5), (8, 8) and (10, 10), were chosen as the adsorbent. They were all 3 nm in length. The carbon atoms of SWCNT was represented as neutral sp2-like aromatic carbons in the CHARMM27 general force field.23,28 Benzene, phenol and 4-chlorophenol were selected as the adsorbates due to their simple and typical structures. There were 32 adsorbate molecules uniformly distributed around the SWCNT. The parameters for these adsorbate molecules were extracted from the CHARMM36 general force field.29 The initial coordinates of the molecules' centres were exactly the same for each of the three adsorbates. To investigate the effect of oxidation on the adsorption process, the hydroxyl groups were placed on the top of the SWCNT and the tube surface was kept intact. The whole system was neutral. All the components in the model were constructed by VMD1.9.2.30
 | ||
| Fig. 1 Initial status of the model system for molecular dynamics simulation of benzene. Water molecules were described as red sticks. The models for the other two adsorbates resemble this one. | ||
2.2 Molecular dynamics simulation
All the simulations were carried out with NAMD2.8.31 The periodic boundary conditions were applied to the model system in three dimensions. The cutoff distance, beyond which the non-bond interactions were neglected, was set as 1.2 nm. The switching distance, at which special functions were used to smoothly adjust the non-bond interactions to zero at cutoff distance, was 1.0 nm. The distance within which NAMD would search for atoms likely to interact with each other was 1.4 nm. The particle mesh Ewald method was applied to calculate the long-range electrostatic interactions.32 The time step was 2 fs and the non-bond interactions were calculated once a time step. The output trajectories were stored every 500 fs.The temperature and pressure of the system were maintained at 298 K and 1 atm during the simulations using Langevin dynamics33 and Langevin piston method.34 The bonds involving hydrogen were regarded as rigid. Potential minimization was performed on the system for 1000 time steps before the simulation started. Then the system was equilibrated for 15 ns. Adsorbate molecules within 0.5 nm of the SWCNT surface were defined as adsorbed molecules. The number of adsorbed molecules was extracted from the trajectories. The non-bond interaction energy was analysed by VMD package.
2.3 Adsorption energy and radial distribution function
The adsorption energy between adsorbate molecules and SWCNT was calculated using the following equation:35| Eadsorption = Eadsorbate + ESWCNT − Eadsorbate+SWCNT | (1) |
The radial distribution function (RDF) describes the probability of finding adsorbate molecules at distance r from the carbon atoms of SWCNT.37 It was calculated as follows:38
 | (2) |
2.4 Non-bond interactions visualisation
The reduced density gradient (RDG) method39 was used to visualise the non-bond interactions between adsorbate molecules and SWCNT. First, an isosurface which distinguished non-bond interactions from other interactions was drawn using RDG function. The RDG function has the following formula:
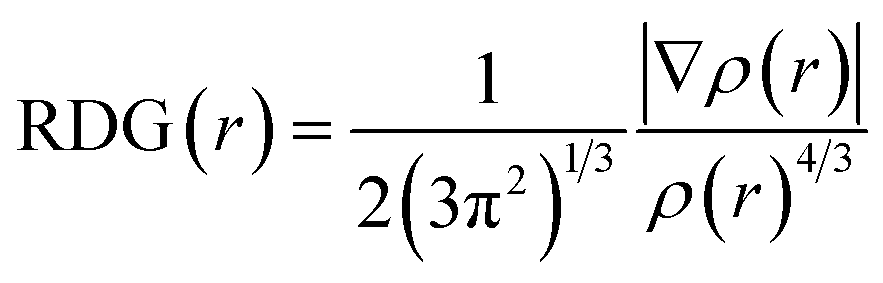 | (3) |
3. Results and discussion
3.1 Adsorption energy
The adsorption energies between adsorbate molecules and SWCNTs are listed in Table 1. The adsorption energies of internal adsorption were much higher than that of external adsorption. This indicates that adsorbate molecules are more stable when confined in the inner pore of SWCNT. The internal adsorption energy declined drastically with the diameter increasing. For instance, the adsorption energy of benzene on (10, 10) was 9.4039 kcal mol−1 lower than on (8, 8). This indicates that the attractive force in the inner pore will decrease sharply with the diameter increasing. On the contrary, the external adsorption energy varied slightly with the change less than 1.7798 kcal mol−1. Thus the size effect on the external adsorption is small.| Adsorbates | Adsorption energy (kcal mol−1) | |||||
|---|---|---|---|---|---|---|
| (5, 5) | (8, 8) | (10, 10) | ||||
| Inner surface | External surface | Inner surface | External surface | Inner surface | External surface | |
| Benzene | — | 6.9766 | 23.9503 | 7.0881 | 14.5464 | 8.1649 |
| Phenol | — | 13.5168 | 32.2670 | 13.0213 | 25.4004 | 14.8616 |
| 4-Chlorophenol | — | 14.4043 | 34.3320 | 14.5673 | 27.0764 | 16.1841 |
| Water | 38.8614 | 17.4136 | 33.2325 | 14.8479 | 25.0267 | 18.0776 |
The internal adsorption energies of (8, 8) were higher than that of (10, 10). This is due to the smaller diameter of (8, 8) which shortened the distances between carbon atoms and confined adsorbate molecules. Thus the attraction between adsorbate molecules and (8, 8) inner surface was increased, which was reflected by the higher adsorption energy. In contrast, the external adsorption energy has a tendency to increase when the diameter of SWCNT gets larger. This is consistent with previous reports.17,41 We suggest that it is related to the curvature of SWCNT. The increase in diameter will decrease the curvature of SWCNT. Thus the contact area between adsorbate molecules and SWCNT surface will increase, which in turn makes the interactions stronger. However, the degree of sp2-hybridisation of SWCNT depends highly on the curvature.42 The increase of hybridisation would promote the reactivity of SWCNT surface to interact with surrounding molecules.43 As a result, the adsorption energy would increase with the curvature. According to our results, the effect of contact area overwhelm the effect of hybridisation. Liu et al.26 reported the adsorption energy of benzene, phenol and 3-chlorophenol on (3, 3) SWCNT were 6.886 kcal mol−1, 8.50 kcal mol−1 and 10.13 kcal mol−1, respectively. These smaller results may be attributed to the size effect of SWCNT on the adsorption energy.
The adsorption energies of three adsorbates, whether internal or external, decreased in the following order: 4-chlorophenol > phenol > benzene. This is consistent with the π electron density of adsorbate molecules' benzene ring. The chlorine and hydroxyl groups both have strong tendencies to gain electrons. As the SWCNT surface is rich in π electrons, the π–π electron-donor–acceptor interaction10 between adsorbate molecules and SWCNT will get stronger with these electron-withdrawing substituents adding to the benzene ring. The adsorption energy gap between phenol and benzene is much larger than that between 4-chlorophenol and phenol. This indicates that the hydroxyl group has stronger ability to gain electron than the chlorine group.
As shown in Table 1, the adsorption energy of water is higher than those of benzene and phenol and close to that of 4-chlorophenol. In the aqueous condition, the hydrophobic effect44 excludes water from the SWCNT surface and enables the adsorption of adsorbate molecules on SWCNT. We propose that the hydrophobic effect is significant so that the energy gaps between water and adsorbates could be overcome.
The most stable geometric configurations of water and three adsorbates on (5, 5) SWCNT after energy minimization are shown in Fig. 2. The hydrogen atom which lost its electron to the oxide atom is closest to the SWCNT, as shown in Fig. 2(a), (c) and (d). This suggests that the π electrons of SWCNT interact with the electron-lacking hydrogen atom. The benzene molecule is parallel to the SWCNT surface, which is consistent with the most favourable configuration of benzene dimer.45 The angles and distances between SWCNT and adsorbate plane are listed in Table 2. The phenol configuration has a larger angle and longer distance than 4-chlorophenol. The chlorine group of 4-chlorophenol increased the benzene ring's affinity with SWCNT, thus pulling the centre of benzene ring closer to SWCNT. While the nearest distance of 4-chlorophenol changed slightly from that of phenol. As a result, the angle was decreased.
 | ||
| Fig. 2 The configurations of adsorbed molecules on (5, 5) SWCNT: (a) water; (b) benzene; (c) phenol; (d) 4-chlorophenol. The hydrogen, carbon, oxide, and chlorine atoms are coloured as white, grey, red and green, respectively. | ||
3.2 Adsorption capacities of different SWCNTs
Fig. 3 compares the dynamic adsorption process of three adsorbates on the same SWCNT. The equilibrium was reached after about 12 ns simulation. As shown in Fig. 3(a), the adsorption capacities of (5, 5) SWCNT for three adsorbates decrease in the following order: 4-chlorophenol > phenol > benzene. This is consistent with their adsorption energies on (5, 5) SWCNT, which implies that the adsorption energies obtained in vacuum condition can be used to compare the adsorption amount of different adsorbates in aqueous condition qualitatively. However, the adsorption capacity of SWCNT for benzene obtained in experiment was much higher than that of phenol under the same equilibrium concentration.46 We propose that the difference may be attributed to the phenol dissociation, SWCNTs aggregation and strong interaction between water and phenol molecules in actual aqueous conditions. Considering the small number of adsorbate molecules, the dissociation of phenol was neglected in this study. However, the dissociation would increase the solubility of phenol in water and depress the adsorption of phenol. On the other hand, the high hydrophobicity of SWCNTs makes them easy to aggregate in water. The interstitial adsorption sites are more available to benzene which has smaller molecular volume. Thus, benzene would be more easily adsorbed by SWCNT bundles than phenol. Further research may be needed to quantitatively investigate the impact of adsorbate dissociation and SWCNTs aggregation. Finally, the hydroxyl group enhances the interaction between phenol and SWCNT as discussed previously, which we suggest overwhelms the attraction from water molecules to hydroxyl group in the simulation. However, in real condition, phenol is the most hydrophilic in water among the three adsorbates.9 Thus the attraction from water molecules to phenol molecules is the strongest, which is consistent with its lowest adsorption amount on SWCNT in experiment. The disparity between simulation and experiment results indicates that ideally simplified simulation may not be suitable for the prediction of SWCNTs' adsorption capacity in real conditions. | ||
| Fig. 3 Comparison of three adsorbates adsorption on (a) (5, 5) SWCNT; (b) (8, 8) SWCNT; (c) (10, 10) SWCNT. | ||
The size effect of SWCNT on its adsorption capacity is shown in Fig. 4. The time-averaged number of adsorbed molecules in the last 2 ns on three SWCNTs decreases in the following order: (10, 10) ≈ (8, 8) > (5, 5). The inner cavity of (5, 5) SWCNT was not accessible to the adsorbate molecules. As a result, it has the lowest adsorption capacity. When the diameter of SWCNT is large enough to hold the adsorbate molecules, the increase in diameter will not improve its adsorption capacity significantly.
 | ||
| Fig. 4 The adsorption of three adsorbates on SWCNTs with different sizes: (a) benzene; (b) phenol; (c) 4-chlorophenol. | ||
As shown in Fig. 5(a), the increase in temperature enhances the adsorption capacity of (5, 5) SWCNT for benzene. When temperature is increased, the molecules move faster. Thus their probability of interacting with SWCNT and being adsorbed gets higher. However, the adsorbed molecules are easier to be desorbed at higher temperatures. Whether the rise in temperature will increase the adsorption capacity of SWCNT depends on the relative strength of the two effects mentioned above. In our simulation the extra adsorbed molecules outnumbered the extra desorbed molecules when the temperature was increased. In actual experiments, the thermodynamic effects on adsorption are rather complicated. The physisorption process is generally considered as exothermic.11 Zhou et al.47 also reported the adsorption of triclosan on CNTs was more favourable at lower temperatures. However, the thermodynamic calculation results of other reports48,49 indicated that the overall adsorption process on CNTS could be endothermic. The contradictory results reflect the complexity of adsorption on CNTs and further research may be needed to reveal the mechanisms.
 | ||
| Fig. 5 The adsorption of benzene on (a) (5, 5) SWCNT at different temperatures; (b) (5, 5) SWCNTs with different oxide concentrations. | ||
The effects of oxide concentrations on the adsorption of benzene is shown in Fig. 5(b). The addition of two hydroxyl groups on top of (5, 5) SWCNT enhanced the adsorption capacity. However, the adsorption capacity was affected slightly with further increase in oxide concentration. Only the carbon atoms on top of the tube were oxidized so that the tube surface was kept unbroken. The results suggest that oxidation could improve the adsorption capacity of SWCNT on condition that the specific surface area is not reduced. We propose that the hydroxyl groups can attract water molecules to the top of tube from the tube surface, thus leaving more adsorption sites for adsorbate molecules. Terzyk et al.22 also observed aggregation of water molecules around the carbonyl groups of oxidised SWCNT in simulation. However, in experiments, the oxidation which caused huge loss of specific surface area of SWCNT would reduce the adsorption capacity greatly.9
3.3 Configurations of adsorbed molecules
The orientations of adsorbed molecules on the surface of SWCNTs when equilibrium was reached are shown in Fig. 6. A two-layer configuration of adsorbate molecules was formed on (5, 5) SWCNT, as shown in Fig. 6(a). As the diameter increased, more adsorption sites were available and the two-layer configuration diminished. The benzene molecules were not as perfectly parallel to the SWCNT surface as they were in the vacuum condition. This was due to the attraction from water molecules and other benzene molecules around. Many of the adsorbed phenol and 4-chlorophenol molecules in the monolayer had their hydroxyl group heading towards the tube surface. It indicates that the interaction between SWCNT and hydroxyl group could overcome the strong attraction from water. It is consistent with our previous suggestion that π electrons of SWCNT interact with the electron-lacking hydrogen atom of hydroxyl group. | ||
| Fig. 6 Snapshots for configurations of equilibrated systems for adsorbates on (a) (5, 5) (b) (8, 8) (c) (10, 10). Hydrogen, oxide and chlorine atoms are coloured as white, red and green, respectively. Water molecules are neglected for clarity. | ||
The orientations of molecules adsorbed internally changed dramatically with the increase in SWCNT diameter. The molecules confined in the pore of (8, 8) SWCNT were perpendicular to the internal surface (Fig. 6(b)), while those adsorbed inside (10, 10) were parallel to the internal surface (Fig. 6(c)). This drastic change in configuration was contrary to the results of Terzyk et al.22 This disparity is due to that the tube used in Terzyk's study had much greater diameter than those adsorbate molecules. Our results indicate that the configurations of molecules inside the tube are limited by the tube diameter. From Table 1, we can see that the confinement of molecules in tube which can barely hold them leads to highest adsorption energy.
The angular distribution of adsorbed adsorbate molecules were investigated to quantitatively reveal the orientations. The angle formed between the benzene ring plane and the axis of SWCNT is measured. We divide 90° to nine equal intervals. Each interval is 10-degrees in length. Fig. 7(a) and (b) show the probability of adsorbed molecules' angles falling into each interval. Fig. 7(c) and (d) show the cumulative probability curve of angles for adsorbed molecules. As shown in Fig. 7(a), the benzene molecules have the highest probability to form a small angle (<20°) with (10, 10) SWCNT. Meanwhile, the probability of forming sharp angles on (10, 10) SWCNT is the lowest. The probability of forming angles smaller 40° on (10, 10) is above 0.8 (Fig. 7(c)). It indicates that small angles are more likely to be formed on less curved surface. Large angles are most likely to occur on (8, 8) SWCNT, which can be seen from Fig. 7(a) and (c). This is due to the benzene molecules adsorbed inside the tube, which are nearly perpendicular to the internal surface. As shown in Fig. 7(b) and (d), the probabilities of angles for different adsorbates were quite close to each other. This indicates that the substituents on benzene ring only affect the orientations of adsorbed molecules slightly.
 | ||
| Fig. 7 (a) The probabilities of angles falling into different intervals for adsorbed benzene molecules on three SWCNTs. (b) The probabilities of angles falling into different intervals for three adsorbates on (5, 5) SWCNT. (c) Cumulative probability curves for angles of adsorbed benzene molecules on three SWCNTs. (d) Cumulative probability curves for angles of three adsorbates on (5, 5) SWCNT. | ||
3.4 Non-bond interaction energy analysis
The non-bond interaction energies between adsorbate molecules and SWCNT are shown in Fig. 8. The non-bond interaction energies for all adsorbates decreased gradually and fluctuated around a stable value at the end of the simulation. The non-bond interaction energies for three adsorbates decreased in the following order: benzene > phenol > 4-chlorophenol. The non-bond interaction energy is negatively correlated with the binding strength of adsorbates to the SWCNT. The sharp decrease in the energy for 4-chlorophenol in Fig. 8(b) is consistent with the sudden rise in number of its adsorbed molecules in Fig. 4(c). By analysing the trajectories of adsorption process, we found that the molecules aggregated with each other before being adsorbed to SWCNT together. As shown in Fig. 8(b), the non-bond interaction energy of 4-chlorophenol decreased slower than benzene and phenol before the sudden drop. This suggests that the interactions among 4-chlorophenol molecules are weaker than that between 4-chlorophenol molecules and SWCNT. | ||
| Fig. 8 The non-bond interaction energies for three adsorbates on (a) (5, 5); (b) (8, 8); (c) (10, 10). | ||
3.5 Radial distribution function analysis
The RDF is not applicable when the inner pore of SWCNT is accessible to the adsorbates because the molecules inside will be counted twice. Thus we only analyse the RDF of (5, 5) SWCNT within a limited distance of 8 Å to avoid double counting of adsorbed molecules. As shown in Fig. 9, g(r) is zero at short distances, which is due to the strong repulsive forces that cause particle separations. The peak of RDF curve represents that adsorbate molecules aggregate in shells around SWCNT.50 Thus the peak position represents the distance between adsorbed molecules and SWCNT. As shown in Fig. 9(a), the distances for benzene, phenol and 4-chlorophenol are about 5.00 Å, 4.85 Å and 4.75 Å, respectively. They are longer than those obtained in vacuum condition. The disparity is attributed to the attraction from water molecules around. The peak positions for three adsorbates are close to each other, which means similar favourite distances for them. The peak heights for three adsorbates decreased in the following order: 4-chlorophenol > phenol > benzene. This is in accord with the adsorption capacities of (5, 5) SWCNT for these adsorbates. The peaks are rather broad, indicating the adsorbed molecules are not strongly confined in their position. | ||
| Fig. 9 Radial distribution function of (a) three adsorbates to (5, 5) SWCNT; (b) water to (5, 5) SWCNT in three systems. | ||
The RDF curves of water to (5, 5) SWCNT in three systems are shown in Fig. 9(b). The peaks are relatively sharp, indicating that water molecules are firmly adsorbed to the tube surface. Their g(r) values are in reverse order when compared with those of adsorbates. This proves that adsorbate molecules could replace adsorbed water molecules and occupy the adsorption site. With more adsorbate molecules adsorbed to the tube surface, the number of water molecules decreases, reflected by its g(r) value. The peaks of water in three systems all appear at the same distance of 3.55 nm, much smaller than those of adsorbates. Since water molecules adsorbed inside contributed a lot to the peaks' g(r) values, we propose that 3.55 nm reflects the distance between adsorbed water molecule and SWCNT inner surface.
3.6 Visualisation of non-bond interactions
The visualization of non-bond interactions of adsorbate molecules and SWCNTs are shown in Fig. 10. The configurations were extracted from the trajectories of MD simulations when equilibrium was reached. The coloured isosurface can be used to identify the type and location of non-bond interactions. The regions with red colour in the centre of benzene rings manifest strong steric effect. The big pieces of isosurfaces filling the interlayer spaces between adsorbates and SWCNTs represent the π–π interactions,39 as shown in Fig. 10(a) and (c). The isosurfaces consist of green and brown regions, which represent weak attractive and repulsive forces respectively. Thus the π–π interaction is a hybrid of attractive and repulsive interactions. When adsorbate molecules were confined in the inner pore of (8, 8) SWCNT, no big piece of isosurface existed (Fig. 10(b)). The π–π interaction disappeared due to the limitation of pore size. | ||
| Fig. 10 The visualisation of non-bond interactions between adsorbate molecules and different SWCNTs: (a) (5, 5); (b) (8, 8); (c) (10, 10). | ||
4. Conclusions
MD simulations and RDG calculations have been conducted to reveal the adsorption mechanisms of simple aromatic compounds adsorption on SWCNTs. Adsorption energies were calculated to evaluate the binding strength of different adsorbates on SWCNTs. The external adsorption energy tended to rise with the increase in SWCNT diameter. The electron-withdrawing substituents strengthened the π–π electron-donor–acceptor interaction, leading to the highest adsorption energy of 4-chlorophenol. Water molecules bound most strongly to the tube surface in vacuum condition, indicating the hydrophobic effect plays a very important role in the selective adsorption of adsorbates in aqueous environment. The equilibrium was reached after 12 ns simulation. The adsorption capacities of (5, 5) SWCNT for three adsorbates were consistent with their adsorption energies. The inaccessibility of the inner pore of (5, 5) SWCNT limited its adsorption capacity. Increase in temperature and adding hydroxyl groups to the top of tube both facilitated adsorbates adsorption. A second layer of adsorbate molecules was formed on (5, 5) SWCNT, while it diminished with the increase in tube diameter. The interaction between SWCNT and hydroxyl group could overcome the strong attraction from water, reflected by the hydroxyl groups of adsorbed molecules heading towards the tube surface. The angular distribution of adsorbed adsorbate molecules was calculated, indicating that small angles are more likely to be formed on less curved surface. The non-bond interaction energy was negatively correlated with the binding strength of adsorbates to the SWCNT. The RDF curves showed that the distances between adsorbates (benzene, phenol and 4-chlorophenol) and (5, 5) SWCNT were 5.00 Å, 4.85 Å and 4.75 Å, respectively. They were much larger than those obtained in vacuum condition, due to the attraction from water molecules. The visualisation of non-bond interactions showed that π–π interaction is rather complicated, consisting of attractive and repulsive interactions.Acknowledgements
This study was supported by Tsinghua National Laboratory for Information Science and Technology, National Water Major Project (No. 2015ZX07402-002), and the National Natural Science Foundation of China (No. 51290284).References
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS
.
- Q. Zhang, J. Q. Huang, W. Z. Qian, Y. Y. Zhang and F. Wei, Small, 2013, 9, 1237–1265 CrossRef CAS PubMed
- L. Ji, W. Chen, L. Duan and D. Zhu, Environ. Sci. Technol., 2009, 43, 2322–2327 CrossRef CAS PubMed
- B. Pan and B. Xing, Environ. Sci. Technol., 2008, 42, 9005–9013 CrossRef CAS PubMed
- K. Yang and B. Xing, Chem. Rev., 2010, 110, 5989–6008 CrossRef CAS PubMed
- K. Yang, L. Zhu and B. Xing, Environ. Sci. Technol., 2006, 40, 1855–1861 CrossRef CAS PubMed
- C. Lu, Y.-L. Chung and K.-F. Chang, Water Res., 2005, 39, 1183–1189 CrossRef CAS PubMed
- F. M. Machado, C. P. Bergmann, E. C. Lima, M. A. Adebayo and S. B. Fagan, Mater. Res., 2014, 17, 153–160 CrossRef CAS
- H. Ding, X. Li, J. Wang, X. Zhang and C. Chen, J. Environ. Sci., 2016, 43, 187–198 CrossRef PubMed
- W. Chen, L. Duan and D. Zhu, Environ. Sci. Technol., 2007, 41, 8295–8300 CrossRef CAS PubMed
- O. G. Apul and T. Karanfil, Water Res., 2015, 68, 34–55 CrossRef CAS PubMed
- Y. Y. Li, J. F. Niu, Z. Y. Shen and C. H. Feng, Chemosphere, 2013, 91, 784–790 CrossRef CAS PubMed
- P. Shan, J.-W. Shen, D.-H. Xu, L.-Y. Shi, J. Gao, Y.-W. Lan, Q. Wang and X.-H. Wei, RSC Adv., 2014, 4, 23730–23739 RSC
- M. Calvaresi and F. Zerbetto, J. Mater. Chem. A, 2014, 2, 12123–12135 CAS
- M. V. Karachevtsev and V. A. Karachevtsev, J. Phys. Chem. B, 2011, 115, 9271–9279 CrossRef CAS PubMed
- W. P. Lv, Chem. Phys. Lett., 2011, 514, 311–316 CrossRef CAS
- S. Az'hari and Y. Ghayeb, Mol. Simul., 2014, 40, 392–398 CrossRef
- A. Kyani and B. Goliaei, J. Mol. Struct.: THEOCHEM, 2009, 913, 63–69 CrossRef CAS
- T. Panczyk, P. Wolski, A. Jagusiak and M. Drach, RSC Adv., 2014, 4, 47304–47312 RSC
- V. V. Chagovets, M. V. Kosevich, S. G. Stepanian, O. A. Boryak, V. S. Shelkovsky, V. V. Orlov, V. S. Leontiev, V. A. Pokrovskiy, L. Adamowicz and V. A. Karachevtsev, J. Phys. Chem. C, 2012, 116, 20579–20590 CAS
- N. Poorgholami-Bejarpasi and B. Sohrabi, J. Mol. Model., 2013, 19, 4319–4335 CrossRef CAS PubMed
- A. P. Terzyk, P. A. Gauden, S. Furmaniak, R. P. Wesolowski, P. J. F. Harris and P. Kowalczyk, Phys. Chem. Chem. Phys., 2009, 11, 9341–9345 RSC
- Y. Z. Liu, W. P. Lai, T. Yu, Y. Kang and Z. X. Ge, Phys. Chem. Chem. Phys., 2015, 17, 6995–7001 RSC
- J. Comer, R. Chen, H. Poblete, A. Vergara-Jaque and J. E. Riviere, ACS Nano, 2015, 9, 11761–11774 CrossRef CAS PubMed
- H. Yan, H. Wu, K. Li, Y. Wang, X. Tao, H. Yang, A. Li and R. Cheng, ACS Appl. Mater. Interfaces, 2015, 7, 6690–6697 CAS
- Y. Liu, J. Zhang, X. Chen, J. Zheng, G. Wang and G. Liang, RSC Adv., 2014, 4, 58036–58046 RSC
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS
- Z. J. He and J. Zhou, Carbon, 2014, 78, 500–509 CrossRef CAS
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. MacKerell, J. Comput. Chem., 2010, 31, 671–690 CAS
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS
- R. W. Pastor, B. R. Brooks and A. Szabo, Mol. Phys., 1988, 65, 1409–1419 CrossRef
- S. E. Feller, Y. Zhang, R. W. Pastor and B. R. Brooks, J. Chem. Phys., 1995, 103, 4613–4621 CrossRef CAS
- Y. V. Shtogun, L. M. Woods and G. I. Dovbeshko, J. Phys. Chem. C, 2007, 111, 18174–18181 CAS
- N. L. Allinger, L. Yan and K. Chen, J. Comput. Chem., 1994, 15, 1321–1330 CrossRef CAS
- A. T. Nasrabadi and M. Foroutan, Comput. Mater. Sci., 2012, 61, 134–139 CrossRef CAS
- F. Cuadros and A. Mulero, Chem. Phys., 1992, 159, 89–97 CrossRef CAS
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
- E. Zaminpayma and K. Mirabbaszadeh, Comput. Mater. Sci., 2012, 58, 7–11 CrossRef CAS
- S. Park, D. Srivastava and K. Cho, Nano Lett., 2003, 3, 1273–1277 CrossRef CAS
- M. Wisniewski, K. Werengowska-Ciecwierz and A. P. Terzyk, Chem. Phys. Lett., 2015, 619, 219–222 CrossRef CAS
- D. Chandler, Nature, 2005, 437, 640–647 CrossRef CAS PubMed
- M. O. Sinnokrot, E. F. Valeev and C. D. Sherrill, J. Am. Chem. Soc., 2002, 124, 10887–10893 CrossRef CAS PubMed
- H. Ding, C. Chen and X. Zhang, SAR QSAR Environ. Res., 2016, 27, 31–45 CrossRef CAS PubMed
- S. Zhou, Y. Shao, N. Gao, J. Deng and C. Tan, Clean: Soil, Air, Water, 2013, 41, 539–547 CrossRef CAS
- X. D. Xin, M. Q. Wang, X. Ge, Q. H. Zhao, S. H. Sun and R. B. Jia, Monatsh. Chem., 2014, 145, 747–754 CrossRef CAS
- F. M. Machado, C. P. Bergmann, T. H. M. Fernandes, E. C. Lima, B. Royer, T. Calvete and S. B. Fagan, J. Hazard. Mater., 2011, 192, 1122–1131 CrossRef CAS PubMed
- J.-P. Hansen and I. R. McDonald, Theory of Simple Liquids, Academic Press, London, GB, 2006 Search PubMed
| This journal is © The Royal Society of Chemistry 2016 |