
Pyrite nanoparticles: an Earth-abundant mineral catalyst for activation of molecular hydrogen and hydrogenation of nitroaromatics†
Ben Maab,
Xili Tong*a,
Congxiu Guoab,
Xiaoning Guoa,
Xiangyun Guo*a and
F. J. Keilc
aState Key Laboratory of Coal Conversion Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, China. E-mail: tongxili@sxicc.ac.cn
bUniversity of the Chinese Academy of Sciences, Beijing 100039, China
cInstitute of Chemical Reaction Engineering, Hamburg University of Technology, D-21047, Germany
First published on 2nd June 2016
Abstract
Pyrite (FeS2) nanoparticles, a kind of Earth-abundant mineral, can efficiently activate molecular hydrogen under mild conditions. At ambient pressure, H2 can be dissociatively adsorbed by FeS2 nanoparticles, react with nitrobenzene, and selectively produce aniline. The catalytic activity for hydrogenation of nitrobenzene is comparable to most non-precious metal catalysts.
Pyrite (FeS2), also known as fool's gold, is a kind of Earth-abundant mineral, and it might have participated in some prebiotic chemical processes involved in the origin of early life.1,2 For a long time, FeS2 has been employed to enhance the liquefaction of coal and lignin under elevated H2 pressure and high temperatures.3 Researchers have paid attention to the material and studied its potential applications in lithium ion batteries,4 electrochemical sensors5 and hydrogen evolution.6 Only recently, FeS2 was noticed to have a catalytic function for photodegradation of organic dyes7 and hydrodeoxygenation of dibenzyl ether into toluene.8 Due to the ubiquity of FeS2 in the early Earth,9 it is necessary to investigate its catalytic property, which may provide us important clues to understand the origin of life.
Hydrogenation processes are frequently employed to produce various fine chemicals, for example, the synthesis of aniline and its derivatives from nitroaromatics.10 Although aniline and other aromatic amines can be obtained by reductive hydrogen atom transfer reactions,11 the direct hydrogenation of nitroaromatics using molecular H2 is widely used in industrial processes. The cleavage of the bond of molecular H2 can be achieved using a proper metal.12 Up to now, the metals employed for activating molecular H2 and reducing nitroaromatics include Ni,13 Au14 and others. Among them, noble metals show high catalytic activity but are limited by scarce resource and high cost; non-noble metal catalysts require supplementary conditions to reach satisfactory yields, such as high H2 pressure, elevated temperature and long reaction time. Recently, non-metal catalyst that can split H2 has attracted considerable interests, such as potassium tert-butanolate,15 organic metal complexes,16 aminoboran,17 and fullerenes.12 For example, Beller's group developed nanoscale iron oxide and cobalt oxide catalysts by pyrolysis of Fe or Co complexes, and found that the oxide catalysts showed excellent catalytic activity for hydrogenation of nitroaromatics.18,19 However, these catalysts were prepared by complicated routes, and were thus expensive.
Herein, we report that the nanoparticles of FeS2 can activate molecular H2 and efficiently catalyze the hydrogenation of nitroaromatics under mild conditions. Compared with traditional metal catalysts, FeS2 nanoparticles are Earth-abundant and have low environmental impact and affordable cost.20 Therefore, the present work will demonstrate a green hydrogenation route for the synthesis of aromatic amines.
FeS2 nanoparticles were prepared by a solvothermal process of ferrous chloride (FeCl2·4H2O) and sulfur powders in a stainless steel autoclave. The preparation is similar to previously reported method.21 From the X-ray diffraction (XRD) patterns shown in Fig. 1A, all of the strong diffraction peaks are identified as a pure phase of FeS2 with a cubic structure indexed to Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) [205] space group.22 Transmission electron microscopy (TEM) image (Fig. 1B) shows that the FeS2 nanoparticles are uniform and have a mean size of about 8 nm. The lattice spacing of the nanoparticles is around 0.24 nm, which is in well agreement with the (200) plane of cubic FeS2 (inset of Fig. 1B). From the patterns of X-ray photoelectron spectroscopy (XPS) shown in Fig. 1C, the binding energy of Fe 2p3/2 at 707.2 eV and 2p1/2 at 720.1 eV can be attributed to Fe2+ in FeS2, and the binding energy at 708.4 and 721.2 eV, 710.2 and 723.4 eV, 711.2 and 724.3 eV, and 713 and 725.5 eV indicate the presence of a little ferrous iron compounds as FeO, Fe2O3, FeOOH and FeS, respectively.23,24 In Fig. 1D, the binding energy at 162.4 and 163.6 eV are the characteristic of S22− species in pyrite or marcasite, and the binding energies at 164.2 and 168.2 eV, and 161.6 and 168.7 eV are assigned to S8 and SO42−, respectively.8,23,25 Therefore, there are a small amount of FeO, FeOOH, Fe2O3, FeS and S8 in the FeS2 sample.
[205] space group.22 Transmission electron microscopy (TEM) image (Fig. 1B) shows that the FeS2 nanoparticles are uniform and have a mean size of about 8 nm. The lattice spacing of the nanoparticles is around 0.24 nm, which is in well agreement with the (200) plane of cubic FeS2 (inset of Fig. 1B). From the patterns of X-ray photoelectron spectroscopy (XPS) shown in Fig. 1C, the binding energy of Fe 2p3/2 at 707.2 eV and 2p1/2 at 720.1 eV can be attributed to Fe2+ in FeS2, and the binding energy at 708.4 and 721.2 eV, 710.2 and 723.4 eV, 711.2 and 724.3 eV, and 713 and 725.5 eV indicate the presence of a little ferrous iron compounds as FeO, Fe2O3, FeOOH and FeS, respectively.23,24 In Fig. 1D, the binding energy at 162.4 and 163.6 eV are the characteristic of S22− species in pyrite or marcasite, and the binding energies at 164.2 and 168.2 eV, and 161.6 and 168.7 eV are assigned to S8 and SO42−, respectively.8,23,25 Therefore, there are a small amount of FeO, FeOOH, Fe2O3, FeS and S8 in the FeS2 sample.
 | ||
| Fig. 1 XRD patterns (A), TEM images (B), XPS profiles (C and D) of the FeS2 nanoparticles, and the size distribution of FeS2 nanoparticles (inset of (B)). | ||
The catalytic performance of FeS2 nanoparticles for the hydrogenation of nitrobenzene under different reaction conditions (T/temperature, P/H2 pressure and t/time) in an autoclave (see ESI†) is shown in Table 1. Elevating the reaction temperature from 25 to 40, 50 and 60 °C (entries 1 to 4), the conversion of nitrobenzene increases from 5.4% to 38%, 66% and 99%, correspondingly. Similarly, higher H2 pressure benefits the hydrogenation reaction. When H2 pressure is increased from 0.25 to 0.4 and 0.5 MPa (entries 4–6), the conversion increases from 39% to 71% and 99%. Shortening the reaction time to 6 and 5 h (entries 7 and 8), the conversion decreases to 94% and 66%. Employing ethanol as the solvent, the reaction can achieve a conversion of 88% under the same conditions (entry 9). However, the reaction in water only achieves a conversion 1% (entry 10), possibly due to the low solubility of nitrobenzene in water, or H2O molecules occupying the catalytic active sites of FeS2.26 The catalytic activity of the FeS2 nanoparticles for nitrobenzene hydrogenation in isopropanol is 1.75 mmol h−1 gFeS2−1.
|
|||||
|---|---|---|---|---|---|
| Entry | T/°C | P/MPa | t/h | Conv. (%) | Select. (%) |
| a Reaction conditions: 1.0 mmol nitrobenzene and 80 mg FeS2 (the mole ratio is 1.49) in 10 ml isopropanol was heated at a specific temperature under a certain pressure of H2 for a specific time.b Ethanol as solvent.c Water as solvent. | |||||
| 1 | 25 | 0.5 | 7 | 5.4 | 99 |
| 2 | 40 | 0.5 | 7 | 38 | 99 |
| 3 | 50 | 0.5 | 7 | 66 | 99 |
| 4 | 60 | 0.5 | 7 | 99 | 99 |
| 5 | 60 | 0.25 | 7 | 39 | 99 |
| 6 | 60 | 0.4 | 7 | 71 | 99 |
| 7 | 60 | 0.5 | 5 | 66 | 99 |
| 8 | 60 | 0.5 | 6 | 94 | 99 |
| 9b | 60 | 0.5 | 7 | 88 | 99 |
| 10c | 60 | 0.5 | 7 | 1 | 99 |

The above results indicate that the FeS2 nanoparticles can efficiently activate molecular hydrogen under mild conditions. To confirm this, we performed hydrogen temperature-programmed desorption (H2-TPD) using a commercial chemisorption analyser. The measured adsorption capacity of the FeS2 nanoparticles in a flow of argon and H2 (∼10% in volume) is ∼6.46 μmol m−2, and the spectra of H2-TPD spectra show that the adsorbed hydrogen can be desorbed as H2 at the temperature above 500 K (Fig. 2A), indicating that the adsorption of H2 on the FeS2 nanoparticles is chemical and dissociative. Based on the H2-TPD spectra, the desorption energy was calculated.27 The desorption energy calculated from the H2-TPD spectra is 93.9 kJ mol−1 (Fig. 2B), which corresponds to an adsorption energy of −0.97 eV. We also performed density function theory (DFT) calculations of H2 adsorption on perfect FeS2 (100) surface. There are two modes for hydrogen adsorption on FeS2 surface, chemical and physical adsorption. In the reaction, the chemisorption plays a key role due to the H2 dissociation is prerequisite for the hydrogenation nitrobenzene. Therefore, only the chemisorption is studied. The calculation results show that the adsorption of H2 can occur on two Fe sites (Fig. S1A†), two S sites (Fig. S1B†), or one Fe site and one S site (Fig. S1C†), with adsorption energies of −0.91, −0.29 and −1.11 eV, respectively. These results mean that H2 can be easily chemisorbed on perfect FeS2 (100) surface. In the H2-TPD experiment, there are defective sites on the surface of FeS2 nanoparticles. However, the calculated adsorption energies from DFT are performed on perfect FeS2 surface without defectives. Therefore, there is deviation between the experimental results and calculated values. It is reasonable that experimental data stands in the range of theoretical values, since the H2 adsorptions on two Fe sites, two S sites, and one Fe site and one S site occur together in the H2-TPD process.
 | ||
| Fig. 2 H2-TPD (A) spectra at different heating rates and the desorption energy calculation (B) based on the spectra. | ||
We prepared FeS2 nanoparticles with different sizes by varying the contents of ferrous chloride and sulphur powders (Table S1†). The particle size was controlled in ranges of 30–50 nm, 50–100 nm and 200–300 nm (Fig. S2†). These size-different nanoparticles show different catalytic activities for hydrogenation of nitrobenzene (Table 2). The smaller the particle size is, the higher the catalytic activity is. To understand the different catalytic activities of size-different nanoparticles, we measured the BET surface area and adsorption capacity of H2 on FeS2 (Table 2). With the decrease of particle size, the amount of adsorbed hydrogen increases, leading to a gradually increase in the catalytic activity.
| Entry | Size/nm | SBET, m2 g−1 | H2/μmol m−2 | Conv. (%) |
|---|---|---|---|---|
| a Reaction conditions: 1.0 mmol nitrobenzene and 80 mg catalyst (the mole ratio is 1.49) in 10 ml isopropanol at 60 °C and 0.5 MPa of H2, reaction time 7 h. | ||||
| 1 | 8 | 37.47 | 6.46 | 99 |
| 2 | 30–50 | 32.69 | 6.45 | 56 |
| 3 | 50–100 | 28.00 | 6.41 | 36 |
| 4 | 200–300 | 24.49 | 6.30 | 21 |
The reduction of nitrobenzene to aniline on metallic catalysts usually follows direct pathway and/or indirect pathway.28–30 The former is featured as hydroxyl amine intermediate, and the latter produces azoxybenzene intermediate.30 The reactant solutions after reactions were analysed by gas chromatography-mass spectroscopy (GC-MS), and the results showed that the reaction product was only aniline and no other by-products were detected. To further understand the reaction path, the adsorption of nitrobenzene on FeS2 (100) surface was also calculated by DFT method. According to the calculation results, nitrobenzene molecule is vertically adsorbed on FeS2 (100) surface via forming two N–O–Fe bonds (Fig. S2D†), with the adsorption energy of −2.29 eV. Under the action of atomic H from dissociated H2 on FeS2 surfaces, nitrobenzene is transformed to aniline (Fig. S3†). However, the detailed mechanism still needs further investigation.31
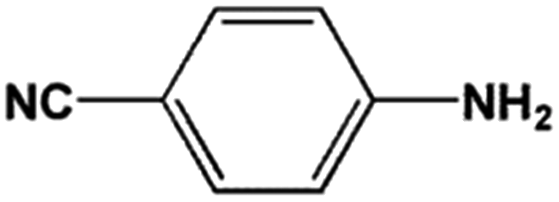
The FeS2 nanoparticles also exhibit general activity for catalytic reduction of other aromatic nitro compounds. From Table 3, electron-withdrawing (entries 1–4) or electron-donating (entries 8–9) substitutes in benzene ring do not significantly affect their reactivity with hydrogen. The complete hydrogenation of p-dinitrobenzene requires longer reaction time because each molecule has two nitro groups (entry 1). It is worth noting that I- or HO-substituted nitrobenzene has slightly lower reactivity (entries 5 and 7).
|
||||
|---|---|---|---|---|
| Entry | Reactant | Main product | Conv. (%) | Select. (%) |
| a Reaction conditions: 1.0 mmol reactant and 80 mg FeS2 catalyst (the mole ratio is 1.49) in 10 ml isopropanol at 60 °C and 0.5 MPa of H2, reaction time 7 h.b The reaction time is 12 h. | ||||
| 1b | 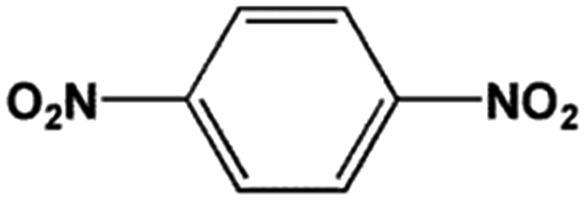 |
 |
99 | 99 |
| 2 | 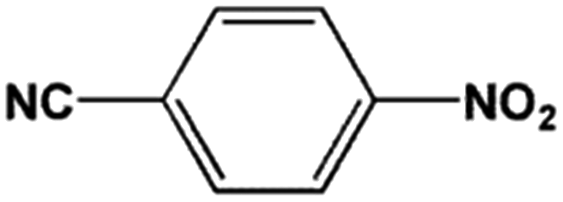 |
 |
99 | 99 |
| 3 |  |
 |
99 | 99 |
| 4 |  |
 |
99 | 99 |
| 5 |  |
 |
75 | 99 |
| 6 |  |
 |
99 | 99 |
| 7 |  |
 |
74 | 99 |
| 8 |  |
 |
99 | 99 |
| 9 |  |
 |
99 | 99 |

The stability is a highly desirable property for an effective heterogeneous catalyst. A decrease in the catalyst activity was observed after five reaction cycles, with the conversion decreasing from 99% in the first cycle to 75% in the fifth cycle (Fig. 3A). It can be seen the intensity of the peak of Fe2O3 and FeOOH significantly increased after five cycles (Fig. 3B).23,24 Due to the oxidation of nitrobenzene,32 part of Fe(II) have been oxidized to Fe(III) in the reaction process, which led to the decrease in the catalyst activity.
 | ||
| Fig. 3 Recyclability (A) of the FeS2 nanoparticles for the hydrogenation of nitrobenzene and XPS profiles (B) of the used FeS2 nanoparticles. | ||
We compared the catalytic performance of the FeS2 nanoparticles with other metal or non-metal catalysts, and found that it was comparable to most of non-precious metal catalysts (Table S2†). The FeS2 nanoparticles were also employed to reduce KNO3 aqueous solution (0.05 M, 10 ml) under the same H2 pressure and temperature. It was found that KNO3 was reduced and NH3 was produced. The pH value of reactant solution increased from 7.0 to 8.8. The reduction of inorganic nitrates and oxidation-disappearance of pyrite are considered as important events involved in the origin of life in the Earth,33,34 therefore it is necessary to further investigate the catalytic properties of FeS2 nanoparticles.
Pyrite has been suggested to be involved in some important prebiotic chemical reactions. The present work further demonstrates that the FeS2 nanoparticles can activate and dissociate molecular hydrogen, one of the gases in primordial atmosphere around the Earth. Dissociated hydrogen atoms by pyrite nanoparticles can participate in many hydrogenation reactions, such as the hydrogenation of nitroaromatics and reduction of nitrates. Besides, the present work suggests a new class of heterogeneous catalysts, FeS2 nanoparticles, which can efficiently catalyze the hydrogenation of nitroaromatics to aromatic amines under mild conditions. The catalytic activity of the FeS2 nanoparticles with a size smaller than 10 nm is comparable to most of non-previous metal catalysts. Because pyrite is an Earth-abundant natural mineral, this provides an alternative route for environmentally friendly production of aromatic amines.
Acknowledgements
The work was financially supported by National Natural Science Foundation of China (21403275, 21473232 and 21403270), and by SKLCC (2014BWZ006 and J16-17-909).Notes and references
- E. Drobner, H. Huber, G. Wachtershäusert, D. Rose and K. O. Stetter, Nature, 1990, 346, 742–744 CrossRef CAS.
- A. C. Tessis, A. Penteado-Fava, M. Pontes-Buarque, H. S. de Amorim, J. A. P. Bonapace, F. de Souza-Barros and A. Vieyra, Origins Life Evol. Biospheres, 1999, 29, 361–374 CrossRef CAS.
- A. Montano, Y. C. Lee, A. Yeyeodu and C. H. Chien, ACS Symp. Ser., 1986, 301, 416–429 CrossRef.
- L. Li, M. Caban-Acevedo, S. N. Girard and S. Jin, Nanoscale, 2014, 6, 2112–2118 RSC.
- B. Chakraborty, B. Show, S. Jana, B. C. Mitra, S. K. Maji, B. Adhikary, N. Mukherjee and A. Mondal, Electrochim. Acta, 2013, 94, 7–15 CrossRef CAS.
- D. Jasion, J. M. Barforoush, Q. Qiao, Y. Zhu, S. Ren and K. C. Leonard, ACS Catal., 2015, 5, 6653–6657 CrossRef CAS.
- S. L. Liu, M. M. Li, S. Li, H. L. Lin and L. Yan, Appl. Surf. Sci., 2013, 268, 213–217 CrossRef CAS.
- N. Ji, X. Y. Wang, C. Werdenthaler, B. Spliethoff and R. Rinaldi, ChemCatChem, 2015, 7, 960–966 CrossRef CAS.
- C. Huber and G. Wachtershäusert, Science, 1997, 276, 245–247 CrossRef CAS PubMed.
- T. Kahl, K.-W. Schröder, F. R. Lawrence, W. J. Marshall, H. Höke and R. JÄckh, Ullmann's encyclopedia of industraial chemistry, Wiley-VCH Verlag GmbH, Co. KGaA, Weinheim, 2012, vol. 3, pp. 465–477 Search PubMed.
- H. M. Yang, X. J. Cui, X. C. Dai, Y. Q. Deng and F. Shi, Nat. Commun., 2015, 6, 6478 CrossRef PubMed.
- B. J. Li and Z. Xu, J. Am. Chem. Soc., 2009, 131, 16380–16382 CrossRef CAS PubMed.
- N. Mahata, A. F. Cunha, J. J. M. Orfa and J. L. Figueiredo, Appl. Catal., A, 2008, 351, 204–209 CrossRef CAS.
- M. Makosch, J. Sa, C. Kartusch, G. Richner, J. A. von Bokhoven and K. Hungerbühler, ChemCatChem, 2012, 4, 59–63 CrossRef CAS.
- A. Berkessel, T. J. S. Schubert and T. N. Müller, J. Am. Chem. Soc., 2002, 124, 8693–8698 CrossRef CAS PubMed.
- M. K. Karunananda and N. P. Mankad, J. Am. Chem. Soc., 2015, 137, 14589–14601 CrossRef PubMed.
- M. Lindqvist, K. Borre, K. Axenov, B. Kótai, M. Nieger, M. Leskelä, I. Pápai and T. Repo, J. Am. Chem. Soc., 2015, 137, 4038–4041 CrossRef CAS PubMed.
- F. A. Wesyerhaus, R. V. Jagadeesh, G. Wienhöfer, M. M. Pohl, J. Radnik, A. E. Surkus, J. Rabeah, K. Junge, H. Junge, M. Nielsen, A. Brückner and M. Beller, Nat. Chem., 2013, 5, 537–543 CrossRef PubMed.
- R. V. Jagadeesh, A. E. Surkus, H. Junge, M. M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schünemann, A. Brückner and M. Beller, Science, 2013, 342, 1073–1076 CrossRef CAS PubMed.
- Y. C. Wang, D. Y. Wang, Y. T. Jiang, H. A. Chen, C. C. Chen, K. C. Ho, H. L. Chou and C. W. Chen, Angew. Chem., Int. Ed., 2013, 52, 6694–6698 CrossRef CAS PubMed.
- C. X. Guo, X. L. Tong and X. Y. Guo, Mater. Lett., 2015, 161, 220–223 CrossRef CAS.
- X. Wen, X. L. Wei, L. W. Yang and P. K. Shen, J. Mater. Chem. A, 2015, 3, 2090–2096 CAS.
- Y. F. Cai, Y. G. Pan, J. Y. Xue, Q. F. Sun, G. Z. Su and X. Li, Appl. Surf. Sci., 2009, 255, 8750–8760 CrossRef CAS.
- M. C. Biesingera, B. P. Payne, A. P. Grosvenord, L. W. M. Laua, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef.
- L. Samad, M. Cabán-Acevedo, M. J. Shearer, K. Park, R. J. Hamers and S. Jin, Chem. Mater., 2015, 27, 3108–3114 CrossRef.
- J. H. Chen, X. H. Long and Y. Chen, J. Phys. Chem. C, 2014, 118, 11657–11665 CAS.
- J. M. Guevremont, D. R. Strongin and M. A. A. Schoonen, Am. Mineral., 1998, 83, 1246–1255 CrossRef CAS.
- F. Haber, Z. Elektrochem., 1898, 22, 506–514 Search PubMed.
- A. Mahata, R. K. Rai, I. Choudhuri, S. K. Singh and B. Pathak, Phys. Chem. Chem. Phys., 2014, 16, 26365–26374 RSC.
- L. Y. Zhang, J. H. Jiang, W. Shi, S. J. Xia, Z. M. Ni and X. C. Xiao, RSC Adv., 2015, 5, 34319–34326 RSC.
- Q. Xiao, S. Sarina, E. R. Waclawik, J. F. Jia, J. Chang, J. D. Riches, H. S. Wu, Z. F. Zheng and H. Y. Zhu, ACS Catal., 2016, 6, 1744–1753 CrossRef CAS.
- D. Y. Li, H. J. Chen and P. N. Liu, Org. Lett., 2014, 16, 6176–6179 CrossRef CAS PubMed.
- T. W. Lyons, C. T. Reinhard and N. J. Planavsky, Nature, 2014, 506, 307–315 CrossRef CAS PubMed.
- S. Singireddy, A. D. Gorden, A. Smirnov, M. A. Vance, M. A. A. Schoonen, R. K. Szilagyi and D. R. Strongin, Origins Life Evol. Biospheres, 2012, 42, 275–294 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra10785k |
| This journal is © The Royal Society of Chemistry 2016 |