
Flower-like Bi4O5I2/Bi5O7I nanocomposite: facile hydrothermal synthesis and efficient photocatalytic degradation of propylparaben under visible-light irradiation†
Shunheng Tu,
Mingli Lu,
Xin Xiao*,
Chunxia Zheng,
Huan Zhong,
Xiaoxi Zuo and
Junmin Nan*
School of Chemistry and Environment, South China Normal University, Guangzhou Key Laboratory of Materials for Energy Conversion and Storage, Guangzhou 510006, PR China. E-mail: xiaox@scnu.edu.cn; jmnan@scnu.edu.cn; Fax: +86-20-39310187; Tel: +86-20-39310255
First published on 28th April 2016
Abstract
Nonstoichiometric bismuth oxyiodide materials have exhibited high potential for applications in visible-light photocatalytic environmental cleaning and solar energy conversion. Herein, novel Bi4O5I2/Bi5O7I nanocomposites, BiOI nanosheets, Bi4O5I2 nanoflowers, and Bi5O7I microfibers are synthesized by controlling the alkalinity of reaction solutions in a facile one-pot hydrothermal route. The as-prepared Bi4O5I2/Bi5O7I nanocomposite exhibits excellent visible-light photocatalytic performance for the degradation of propylparaben (PPB, a potential environmental contaminant structure that contains a benzene ring, hydroxyl, and carboxyl), which is approximately 32, 33, and 4 times higher than that of pure BiOI, Bi5O7I, and Bi4O5I2, respectively. The enhanced photocatalytic activity of the Bi4O5I2/Bi5O7I composite can be attributed to enhancement of charge separation by the formation of Bi4O5I2/Bi5O7I interfaces, more positive valence band edge potential at +2.18 V, good absorption from UV to visible light, three-dimensional flower-like morphology composed of number nanoflakes, and large specific surface area with mesoporous features. The band structures of Bi4O5I2 and Bi5O7I, the electrochemical oxidation behaviors of PPB, and the roles of the primary photogenerated oxidative species are analyzed, then a reasonable photocatalytic mechanism is proposed based on the experimental results. In addition, the as-synthesized Bi4O5I2/Bi5O7I heterojunction remains stable throughout photocatalytic process and can be used repeatedly, indicating its potential for practical applications.
1. Introduction
Semiconductor-based heterogeneous photocatalysis for solar energy conversion and environmental remediation has gained extensive interest in recent decades.1 In particular, the exploration of novel visible-light-active photocatalysts has attracted increasing attention, as visible light accounts for more than 40% of the solar irradiation.2 Recently, as a new family of advantageous photocatalysts, bismuth oxyhalides (BiOX, X = Cl,3 Br,4 and I5), have demonstrated remarkable photocatalytic performance due to their special layered structural features. The internal static electric field perpendicular to each layer facilitates the separation of photogenerated charge carriers.6 In addition to BiOX with a stoichiometric ratio Bi![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O:X = 1, approximately ten other oxygen-rich bismuth oxyhalides with different atomic Bi:O:X ratios have also been discovered.7 Since the chemical composition and crystal structure of a semiconductor can greatly affect its energy band structure, optical adsorption, and charge transfer, bismuth oxyhalides with various Bi:O:X ratios will result in diverse photocatalytic properties and environmental applications.
:O:X = 1, approximately ten other oxygen-rich bismuth oxyhalides with different atomic Bi:O:X ratios have also been discovered.7 Since the chemical composition and crystal structure of a semiconductor can greatly affect its energy band structure, optical adsorption, and charge transfer, bismuth oxyhalides with various Bi:O:X ratios will result in diverse photocatalytic properties and environmental applications.
In our previous paper,8 we reported that Bi4O5I2 exhibited superior photocatalytic and mineralization efficiencies compared with BiOI under visible-light irradiation, although BiOI has the smallest band gap among all bismuth oxyhalides and can be excited by most solar light (from UV to ∼700 nm). Similar findings were also described by Liu et al.,9 He et al.,10 and Wang's group.11 The superior photocatalytic activity of Bi4O5I2 over that of BiOI is ascribed to its higher valence band edge potential, which results in a stronger oxidizing ability to react with organic pollutants.8 However, although Bi4O5I2 has presented excellent visible-light photocatalytic activity and photochemical stability, until now, research into controlling its synthesis, evaluating its photocatalytic activity, and exploring its photocatalytic mechanism and environmental applications are very limited. In addition, a reasonable strategy for the construction of Bi4O5I2-based composites is also required. It is well known that forming heterojunction photocatalysts by two or more types of semiconductors with different band structures can effectively improve their photocatalytic activity. Very recently, Xia et al. found that ultrathin C3N4/Bi4O5I2 layered nanojunctions exhibited much higher photocatalytic activity than pure Bi4O5I2,12 and Yin et al. described an one-dimensional BiOI/Bi4O5I2/Bi2O2CO3 p–n–p heterojunction photocatalyst showed superior photodegradation efficiency of dyes under solar light irradiation.13 More studies should be conducted to exploit Bi4O5I2-based composites catalysts and to better understand their activity-enhanced mechanism.
Parabens (alkyl esters of p-hydroxybenzoic acid) have recently been recognized as emerging environmental contaminants,14 thus it is essential to develop effective methods to eliminate these compounds in aqueous environments.15 Recent studies have indicated that the residues of parabens and their metabolites are frequently detected in aqueous environment and even human serum samples.16 In particular, they have potential endocrine-disrupting activity and acute/chronic toxicity,17 and may increase the risk of human breast cancer.18,19 Photocatalytic technology is one of advantageous ways for the treatment of organic pollutants in wastewaters since it can utilize sunlight directly, operate under ambient conditions, and achieve high degradation efficiency.20 However, reports in degrading parabens using photocatalytic process, especially under visible-light irradiation, are very limited. To the best of our knowledge, the photocatalytic degradation of propylparaben (PPB) under visible-light has rarely been investigated so far.21 The degradation of parabens using photocatalytic technology is not an easy job, which may be due to they contain carboxyl groups that have electron donor effects thus may deactivate the aromatic ring and prevent the oxidation decomposition of the parabens.22 Thanks for their potential high photocatalytic activity, high oxidation ability, and good optical absorption property, Bi4O5I2 and Bi4O5I2-based composites may be promising visible-light photocatalysts for the degradation of parabens in water.
In this current work, a novel flower-like inter-bismuth oxyiodides heterostructure photocatalyst, i.e. Bi4O5I2/Bi5O7I, was synthesized through a pH-controlled hydrothermal route. The morphology, structure, composition, photoabsorption, specific surface area, and formation process of Bi4O5I2/Bi5O7I nanocomposite were systemically analyzed. Then, the visible-light photocatalytic elimination of PPB, one of the most applied paraben that used as either in single or in combination,23 was evaluated over the as-synthesized Bi4O5I2/Bi5O7I photocatalyst and compared with pure Bi4O5I2, Bi5O7I, and BiOI. Moreover, the oxidation potential of PPB, energy band structures of Bi4O5I2 and Bi5O7I, roles of the primary oxidative species in the system were explored, and a possible photocatalytic mechanism was proposed based on the experimental results.
2. Experimental
2.1. Materials and methods
Propylparaben was purchased from J&K Chemical Ltd. L-Asparagine was supplied by Shanghai Bo'ao Biological Technology Co., Ltd. Bismuth nitrate pentahydrate (Bi(NO3)3·5H2O) and potassium iodide (KI) were obtained from Tianjin Kermel Chemical Reagent Co., Ltd. All other chemicals were of analytical grade and used without further purification.In a typical procedure, 9 mmol L-asparagine was completely dissolved in 33 mL distilled water at room temperature. Then, 2 mmol Bi(NO3)3·5H2O was added with continuous magnetic stirring until a homogeneous solution was formed. After that, 2 mmol KI was added into the solution. Subsequently, a certain amount of NaOH (10 mol L−1) was added dropwise to the system. After stirring for another 15 minutes, the mixture was then poured into a 45 mL Teflon-lined stainless-steel autoclave and incubated in an oven at 140 °C for 12 h. At the end of the reaction, the precipitates were collected by centrifugation, thoroughly washed with distilled water and absolute ethanol, and finally dried in an oven at 60 °C. The samples synthesized with 0, 1.0, 1.5, and 2.0 mL NaOH were labeled as S1, S2, S3, and S4, respectively.
2.2. Catalyst characterization
The structures and phase compositions of the samples were determined by powder X-ray diffraction (XRD) on a Bruker D8 Advance X-ray diffractometer (Bruker AXS, Germany) using Cu Kα radiation. The morphologies were observed by a field-emission scanning electron microscope (FE-SEM, Zeiss Ultra 55, Germany). Chemical mapping was performed by combining the scanning module of the microscope with the energy dispersive X-ray spectroscopy (EDX) detector. The microstructures and phase interfaces were detected using a field emission transmission electron microscopy (TEM, Tecnai G2 F20, FEI). The states of elements in the sample were analyzed using a X-ray photoelectron spectroscopy (XPS, Thermo Scientific ESCALAB 250Xi, USA) equipped with an Al Kα source. UV-Vis diffuse reflection spectra (DRS) were estimated on a UV-Vis spectrophotometer (UV-3010, Hitachi, Japan) using an integrating sphere with BaSO4 as a reference, and these spectra were converted to absorption spectra by the Kubelka–Munk method. Nitrogen adsorption–desorption isotherms were recorded at 77 K and then estimated by Brunauer–Emmett–Teller analysis (BET, NOVA, Quantachrome). A desorption isotherm was applied to calculate the pore size distribution using the Barrett–Joyner–Halenda (BJH) method.Cyclic voltammetry (CV) experiments were operated on a CHI 910B electrochemistry workstation (Shanghai Chenhua Apparatus Corporation, China) with a scan rate of 50 mV s−1 in 0.1 mol L−1 LiClO4/CH3CN electrolyte solution. A glassy carbon electrode (GCE), a platinum plate, and a saturated calomel electrode (SCE) served as the working, counter, and reference electrodes, respectively. Mott–Schottky experiments were conducted to evaluate the band positions of the catalysts with an electrochemistry workstation (Autolab PGSTAT302, Netherlands). An appropriate amount of each as-prepared sample suspension was deposited on conductive indium tin oxide glass to serve as the working electrode, and a platinum plate and a SCE served as the counter and reference electrodes, respectively. A 0.5 mol L−1 Na2SO4 aqueous solution was used as the electrolyte. The potential range was from −0.6 to +0.8 V at a constant frequency of 1000 Hz with steps of 10 mV. Photocurrent measurements were carried out on a CHI 660C electrochemical station (Shanghai Chenhua Apparatus Corporation, China) in a standard three-electrode configuration containing the as-prepared samples as the working electrodes, a platinum plate as the counter electrode, and a commercial Ag/AgCl electrode as the reference electrode, with a 0.5 mol L−1 Na2SO4 aqueous solution as the electrolyte. The working electrodes were fabricated by dispersing a certain amount of each sample in an ethylcellulose–ethanol solution. This suspension was deposited onto the surface of fluorine-doped transparent conductive oxide (FTO) glass with a defined area controlled by plastic insulation, and the electrode was then dried in an oven at 60 °C to yield a film electrode. A 300 W xenon lamp (PLS-SXE300/300UV, Beijing Trusttech Co. Ltd., China) assembled with a UV light cut-off filter (λ > 420 nm) was used as the visible-light source.
2.3. Photocatalytic activity
The photocatalytic activities of the as-prepared catalysts were evaluated by the degradation of PPB in water under visible-light irradiation using a XPA-VII photochemical reactor (Nanjing Xujiang Machine-electronic Plant, China) equipped with a 1000 W xenon lamp and 420 nm cut-off filters. Each experiment was performed with 50 mg of as-synthesized photocatalyst and 50 mL of PPB aqueous solution (10 mg L−1). Prior to irradiation, the suspension was kept in the dark under magnetic stirring for 90 min to ensure the adsorption–desorption equilibration of the system. At given time intervals, approximately 3 mL of the suspension was collected, the solids were separated from the solution using a 0.45 μm nitrocellulose filter, and the filtrate was measured by recording the absorbance at the characteristic band of 255 nm using UV-Vis spectroscopy (UV-1800, Shimadzu, Japan) to determine the PPB concentrations.2.4. Analysis of the photogeneration intermediates
After the photocatalytic reaction for 30 min, sample solutions were collected, separated, and degradation products of PPB were identified based on headspace solid phase microextraction (HS-SPME) and gas chromatography (GC, Thermo DSQ, Thermo Fisher Scientific, CA, USA). The sample was placed in a 30 mL screw-capped headspace vials with SPME fiber to accurate PPB and its intermediates for 50 min at 90 °C, with a magnetic stirring. Then, the compounds were thermally desorbed from the fiber to the GC injector. Separation was carried out in a OV-1 capillary column (30 m × 0.32 mm i.d., 0.32 μm film thickness). The inlet temperature was 150 °C. The column temperature program was 140 °C (3 min hold) then 5 °C min−1 raised to 250 °C (5 min hold). The FID detector temperature was 300 °C. The carrier gases were N2 (50 mL min−1), H2 (50 mL min−1), and air (400 mL min−1). Some standard compounds were sampled under same experimental conditions to identify the possible metabolites. And diethyl phthalate was used as an internal standard to quantify the concentrations of oxidation products during the photodegradation process.3. Results and discussion
3.1. Characterization of catalysts
The morphologies of the as-synthesized samples were examined by SEM. As observed in Fig. 1, the amount of NaOH added greatly affects the shape and size of final products. The sample S1 (with no addition of NaOH, Fig. 1A) displays a large number of monodisperse nanosheets with a thickness of ∼40 nm and a width of ∼250 nm. Sample S4 (with the maximum amount of added NaOH, Fig. 1D) is composed of micro-scale fibers exhibiting a thickness of ∼200 nm, a breadth of ∼500 nm, and a length of 15–20 μm. The samples S2 and S3, however, exhibit three-dimensional micro/nano flower-like structures approximately 300–600 nm in diameter, which are composed of numerous interconnected nanoflakes, which themselves are ∼15 nm thick and ∼200 nm in width and length. Compared with S2, S3 shows more homogenous, smaller and thinner nanoflakes. | ||
| Fig. 1 SEM images of the as-synthesized samples S1 (A), S2 (B), S3 (C), and S4 (D). | ||
The XRD patterns of the as-prepared samples are shown in Fig. 2. All of the diffraction peaks of sample S1 (a Bi:I feed ratio of 1:1, without adding NaOH) can be unambiguously identified as tetragonal BiOI (JCPDS no. 73-2062), and sample S4 is purity orthorhombic Bi5O7I (JCPDS no. 40-0548). However, the samples S2 and S3 could not be indexed to any pure phase from the powder diffraction standards cards. By carefully comparing the patterns with the experimental data from other bismuth oxyiodides, sample S2 was eventually confirmed as Bi4O5I2,8 and the coexistence of Bi4O5I2 and Bi5O7I can be identified in S3.
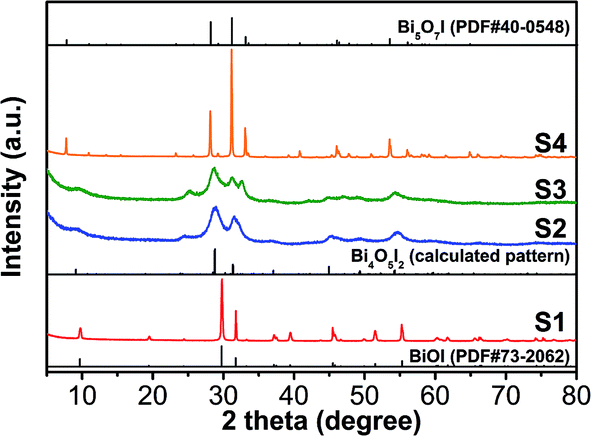 | ||
| Fig. 2 XRD patterns of the as-synthesized samples S1, S2, S3, and S4. | ||
To further investigate the samples' chemical compositions, surface analysis was performed using XPS. The full survey spectra demonstrates that all samples were composed of Bi, O, and I, yet the iodine content declines from S2 to S4 (Fig. 3A). The binding energies (BE) at 159.1 and 164.3 eV in the high-resolution spectra of Bi 4f (Fig. 3B) can be associated with the Bi 4f7/2 and Bi 4f5/2 for Bi3+, respectively.24 Compared with S2 (Bi4O5I2), the peaks of S4 noticeably shift downward, which may be ascribed to that elementary Bi in S4 (Bi5O7I) is surrounded by more O, which is more electronegative than I.25 In addition, the sample S3 (composite) is shown to have similar but slightly lower peaks than those of S2. Similar results can be observed from the O 1s high-resolution spectra (Fig. 3C). In addition, there is little change of BE for I 3d5/2 and I 3d3/2 at 618.9 and 630.3 eV, respectively (Fig. 3D), which indicates the states of I does not vary much different among bismuth oxyiodides. Accordingly, the XPS spectra suggest the coexistence of Bi4O5I2 and Bi5O7I in the sample S3, which validates the XRD results. To determine the percentage of Bi4O5I2 and Bi5O7I in the S3, quantitative analysis of XPS was also performed. The results reveal that atomic ratio of Bi to I in S3 is approximately 2.6:1 on the basis of the peak areas of Bi and I, which implies that Bi4O5I2 accounted for approximately 80% of the composite.
 | ||
| Fig. 3 XPS spectra of the as-synthesized samples S2, S3, and S4. (A) Survey spectra and (B–D) high-resolution XPS spectra of Bi 4f, O 1s, and I 3d, respectively. | ||
To understand the formation mechanism of the as-synthesized materials, a series of samples were fabricated by adding various amounts of NaOH (10 mol L−1, from 0 to 2.5 mL), which were then characterized by XRD. Fig. S1A (ESI†) clearly shows that if less than 0.5 mL NaOH is added, the products are pure phase BiOI, which can be described by reaction (1). Thanks to the coordination effect of asparagine (which can prevent the rapid hydrolysis of Bi(NO3)326) and its tetragonal phase nature, the products (pure BiOI) are well-dispersed nanoscale sheet structures (Fig. 1A). With increasing amounts of NaOH, Bi4O5I2 gradually appears through the reaction of BiOI with OH− under hydrothermal conditions (reaction (2)), and when the amount of added NaOH reaches 1.0 mL, pure Bi4O5I2 can be achieved (Fig. S1A†). With the increasing of alkalinity in the system, Bi3+ and I− are able to react strongly. Then, many freshly precipitated Bi4O5I2 nuclei can develop into thinner nanoflakes due to their intrinsic crystalline nature and rapid growth. These nanoflakes tend to aggregate into larger particles to reduce their surface energy, thus forming flower-like microspheres (Fig. 1B).27 By further increasing the amount of OH−, Bi5O7I can also form (Fig. S1B and S1C†) according to reactions (3) and (4). When the percentage of Bi5O7I is low in the composite, the flower-like structures remain (Fig. 1C), while with further additions of NaOH (>1.7 mL), pure Bi5O7I will be attained, which displays a rod-like or fibrous shape (Fig. 1D). For an attempt to understand the formation of Bi4O5I2/Bi5O7I composites rather than yielding Bi7O9I324 or its mixture, two other samples were also prepared using a different base (KOH and ammonia), as shown in Fig. S2 (ESI†). The results revealed that using different types of base does not affect the structure and composition of products, which suggests that the final product is mainly related to the presence of the particular complexing agent (L-asparagine) and current reaction temperature.
| Bi3+ + I− + H2O → BiOI↓ + 2H+ | (1) |
| 4BiOI + 2OH− → Bi4O5I2↓ + 2I− + H2O | (2) |
| 5Bi4O5I2 + 6OH− → 4Bi5O7I↓ + 6I− + 3H2O | (3) |
| 5BiOI + 4OH− → Bi5O7I↓ + 4I− + 2H2O | (4) |
To understand the relationship between Bi4O5I2 and Bi5O7I in the composite, SEM mapping and high-resolution TEM (HRTEM) analyses of sample S3 were performed. As presented in Fig. 4A, the SEM mapping images clearly reveals that Bi, O, and I are uniformly distributed on the surface of S3. The HRTEM image (inset of Fig. 4B) further confirms that there are two different lattice sets with d spacings of 0.29 and 0.31 nm, which correspond to the (004) plane of Bi5O7I and the (−4−11) plane of Bi4O5I2, respectively. The results suggest that heterojunctions were successfully built between Bi4O5I2 and Bi5O7I in the sample S3 with good interfacial contact.
 | ||
| Fig. 4 (A) SEM image and EDX mapping distribution of Bi, O, and I of sample S3. (B) TEM and high-resolution TEM (HRTEM) images of sample S3. | ||
Fig. 5 shows the UV-Vis diffuse reflection spectra (DRS) of the as-synthesized samples. The maximal absorbance of S1 (BiOI), S2 (Bi4O5I2), S3 (Bi4O5I2/Bi5O7I), and S4 (Bi5O7I) are measured to be 677, 589, 510, and 418 nm, respectively. Correspondingly, the products display very different colors ranging from red through yellow to white (inset of Fig. 5). Their band gap energies (Eg) can be determined from the following equation (eqn (5)):28
| α(hν) = A(hν − Eg)n/2 | (5) |
 | ||
| Fig. 5 (A) UV-Vis diffuse reflection spectra (DRS) of samples S1, S2, S3, and S4; the circular insets show digital photos of the corresponding samples. (B) Plots of (αhν)1/2 vs. photon energy (hν). | ||
The nitrogen adsorption–desorption experiments of the as-prepared samples were performed, and the results are shown in Fig. 6. The type IV isotherm with type H3 hysteresis loop at high relative pressures can be observed in the samples S1–S3 along with the IUPAC classification, suggesting their mesoporous features.30 The BET surface areas of these samples, analyzed by their isotherms, were determined to be 21.99, 41.32, 51.63, and 3.265 m2 g−1 for S1, S2, S3, and S4, respectively. Then, the pore size distributions were evaluated using the BJH method, and the results are presented in the inset of Fig. 6, which discloses the average pore diameters were 24.68, 16.26, 16.15, and 33.99 nm for S1, S2, S3, and S4, respectively, with relatively wide pore size distributions. The mesoporous features with broadly distributed pores are probably due to the inter-nanosheet spacing in these products.31 The results clearly indicate that the specific surface areas of the samples are directly related to their three-dimensional flower-like structures and the thickness of nanosheets. The sample S4 having a poor specific surface area is attributable to the fact that it has larger particle size (micro-scale fibers) without mesoporous structures. The large surface area of samples S2 and S3 may result in their good photocatalytic performance.
 | ||
| Fig. 6 Nitrogen adsorption–desorption isotherms and pore-size distribution curves (inset) of the sample S1 (BiOI), S2 (Bi4O5I2), S3 (Bi4O5I2/Bi5O7I), and S4 (Bi5O7I). | ||
3.2. Photocatalytic degradation of PPB
The photocatalytic performances of the as-synthesized photocatalysts were evaluated for the degradation of PPB under visible-light irradiation. Prior to the photodegradation, PPB aqueous solution and photocatalysts were mixed and kept stirring for 90 min in dark to ensure the adsorption–desorption equilibration and to estimate the adsorption efficiency of the catalysts. As shown in Fig. S3 (ESI†), the adsorption rates of four catalysts are consistent with their specific surface area, and the highest adsorption take place in sample S3 (about 16%). Under equal visible-light irradiation for 120 min, as shown in Fig. 7A, the photodegradation efficiencies of PPB over the synthetic samples S1–S4 reveal a significant difference, and the S3 exhibited the highest photocatalytic activity. Then reaction kinetics were investigated by fitting the data to the pseudo first-order kinetics equation (eqn (6)):32
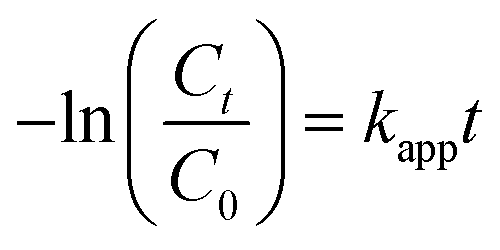 | (6) |
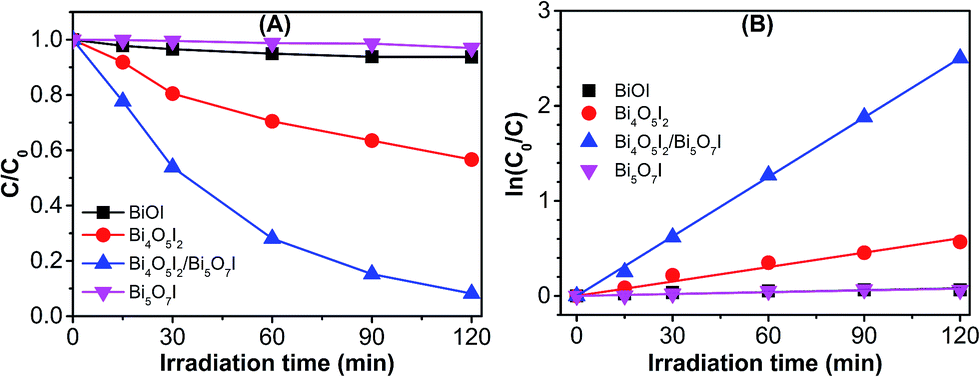 | ||
| Fig. 7 (A) Photocatalytic degradation kinetics of PPB over the sample S1 (BiOI), S2 (Bi4O5I2), S3 (Bi4O5I2/Bi5O7I), and S4 (Bi5O7I) under visible-light irradiation. (B) Linear plots of ln(C0/Ct) versus degradation time. | ||
According to Fig. 7, there was no observable removal of PPB under the photocatalytic process using sample S4 (Bi5O7I), which can be easily explained by its limited absorption in visible light and the smallest specific surface area. Nevertheless, it is surprising that the BiOI nanosheets (sample S1), which has excellent visible-light absorption, was also shown to have very poor photodegradation performance. To understand the underlying intrinsic mechanism, the band structures of the synthesized catalysts and the electrochemical oxidation behaviors of PPB were studied using electrochemical techniques. As shown in Fig. 8A, a Mott–Schottky analysis was carried out to evaluate the positions of the flat band potential (Vfb) of the as-prepared samples.29 The results show that the Vfb of BiOI, Bi4O5I2, and Bi5O7I electrodes are 0, −0.22, and −0.43 V vs. SCE, respectively. It is generally accepted that the conduction band minimum (CBM) of many semiconductors are approximately 0.1 V more negative than their Vfb. Thus, combined with the results of bandgap energy assessed from the DRS analysis (Fig. 5), the valence band (VB) edge potentials of synthetic BiOI, Bi4O5I2, and Bi5O7I were eventually estimated to be 1.99, 2.18, and 2.68 vs. normal hydrogen electrode (NHE), respectively, which suggests their different oxidizing capacities. In addition, the electrochemical behavior of PPB was investigated using cyclic voltammetry (CV). As shown in Fig. 8B, there are clearly two oxidation peaks in the applied potential range, one starting from +1.25 V and another starting from +1.90 V vs. SCE (i.e., 1.49 and 2.14 V vs. NHE). The first peak can be attributed to the hydroxyl group in PPB, which is easily oxidized, while the other peak can be assigned to the carboxyl group, which is more difficult to oxidize than the hydroxyl group.22 According to these results, BiOI (VB potential at 1.99 V) is sufficient to oxidize the phenolic hydroxyl group (>1.49 V) in PPB, yet it is not enough to oxidize carboxyl group (>2.14 V), which may be the key reason behind its poor catalytic activity.
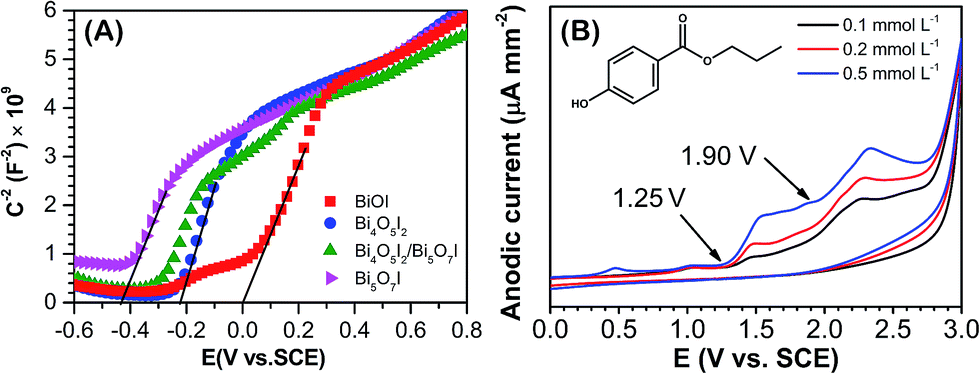 | ||
| Fig. 8 (A) Mott–Schottky plots for the as-synthesized samples S1–S4. (B) The cyclic voltammogram of PPB at various concentrations. | ||
Although pure Bi4O5I2 is thermodynamically suitable (VB potential at +2.18 V) to degrade PPB, its demonstrated photocatalytic activity is not yet advanced enough (Fig. 7), which may be associated with its lower transfer and separation capability for photo-generated carriers. To better understand the electron transfer process, the transient photocurrent responses of the as-synthesized samples S1–S4 were measured with intermittent visible-light irradiation for three on–off cycles. As shown in Fig. 9, the photocurrent quickly increased once the light irradiation was turned on, and it then dropped quickly after light was turned off, which could be directly related to the separation efficiency of the photogenerated carriers.33 Note that the photocurrent of Bi4O5I2/Bi5O7I clearly revealed a much higher photocurrent density than that of pure Bi4O5I and the other two samples, which indicates that the heterojunction is more effective at separating the photogenerated electron–hole pairs and thus that this sample can achieve higher photocatalytic activity (Fig. 7).
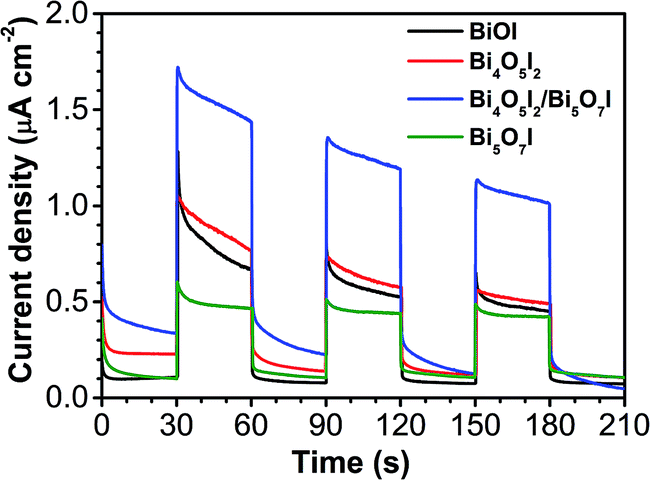 | ||
| Fig. 9 Photocurrent transient responses for the as-synthesized samples S1–S4. | ||
Instead of Bi4O5I2/Bi5O7I, another Bi4O5I2-based heterostructured photocatalyst, i.e. Bi4O5I2/BiOI can also be obtained by controlled NaOH added from 0.6 to 0.9 mL (Fig. S1A, ESI†). Although all of Bi4O5I2 and BiOI has a fine energy-band structure and had shown high visible-light photocatalytic activity in various studies, Bi4O5I2/BiOI heterojunction may not be good photocatalysts due to their mismatched band structure (the VB edge potential of Bi4O5I2 is more positive than BiOI, and its CB potential is more negative than BiOI, as revealed in Fig. S5, ESI†). And the experimental results confirm the speculation. As shown in Fig. S6 (ESI†), the photocatalytic performances of Bi4O5I2/BiOI heterojunctions show lower activities which only slightly better than that of BiOI.
The involvement of photogenerated reactive species, such as holes (h+), electrons (e−), hydroxyl radical (˙OH), and superoxide (˙O2−), were considered to be crucial to the photocatalytic reactions. To investigate the contributions of the various reactive species in the PPB degrading over the as-prepared Bi4O5I2/Bi5O7I photocatalyst, the photocatalytic performance was evaluated in the presence of various scavengers and compared with the blank (no scavenger). As shown in Fig. 10, the addition of isopropanol as a scavenger for ˙OH, the photocatalytic efficiency caused no noteworthy change compared with the efficiency without any scavengers, indicating the minor role of ˙OH in this photodegradation process. This result may ascribed to the standard redox potential of Bi5+/Bi3+ (E0 = 1.59 V at pH 0), which is more negative than that of ˙OH/OH− (E0 = 1.99 V at pH 0) and ˙OH/H2O (2.73 eV at pH 0), thus the photogenerated holes from the bismuth oxyiodides cannot oxidize ambient OH− to form ˙OH.34 However, obvious inhibitory effects were observed when potassium bromate, TEMPOL, and N2 were added to remove photogenerated e−, ˙O2−, and dissolved oxygen, respectively. Similarly, a significant decline in the photocatalytic efficiency was found after adding sodium oxalate as a scavenger for photogenerated h+. These phenomena suggest that both h+ and ˙O2− (which originated from photogenerated e− reacted with O2) may play important roles during the photocatalytic reactions. It is well known that the redox potential of O2/˙O2− is −0.33 V vs. NHE, and thus, the photogenerated electrons in the lower conduction band potential of the as-synthesized Bi4O5I2 (−0.08 V vs. NHE according to the Mott–Schottky analysis), seemingly cannot react with O2 to yield ˙O2−. Actually, under visible-light irradiation (λ ≥ 420 nm and E ≤ 2.95 eV), some electrons can be excited to higher energy states in the conduction bands of Bi4O5I2. As a result, ˙O2− could be produced by reacting these higher energy electrons with dissolved oxygen.
 | ||
| Fig. 10 Photodegradation of PPB over the as-synthesized flower-like Bi4O5I2/Bi5O7I composite in the presence of various scavengers: no scavenger, isopropanol, sodium oxalate, potassium bromate, 4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy (TEMPOL), and N2. | ||
Metabolites and degradation products of PPB were identified by HS-SPME and GC techniques. As shown in Fig. 11, PPB was eluted at a retention time (RT) of 11.3 min. After degradation for 30 min, three potential intermediates eluted at RT of 5.13, 8.28, and 9.03 min were observed. Though compared the GC peaks with retention times of some probable substance under same GC condition, the intermediates eluted at RT 9.03 and 5.13 were identified as 4-hydroxybenzoic acid and hydroquinone, respectively, while the substance at RT 8.28 cannot be recognized in the current analyzed conditions. Moreover, by using diethyl phthalate as an internal standard, the contents of PPB, 4-hydroxybenzoic acid, and hydroquinone were quantified to be about 4.7, 3.1, and 0.4 mg L−1, respectively, at 30 min photodegradation reaction. It was found that the degradation intermediates did not include any hydroxylated products, which illustrates the main photocatalytic route in the system are ring-opening, chain broken, and oxygenation process. In other word, the reaction pathways are mainly dominated by the photogenerated holes and superoxide radicals oxidation rather than reacted by ˙OH.
 | ||
| Fig. 11 GC chromatograms of PPB degradation over an as-synthesized Bi4O5I2/Bi5O7I sample after visible-light irradiation for 30 min. | ||
Based on the above results and analyses, we can then propose realistic reaction mechanisms for the degradation of PPB over the as-synthesized samples, as presented in Scheme 1. Under visible-light irradiation (i.e., at wavelengths λ ≥ 420 nm at which the energy of light is less than 2.95 eV), the light is insufficient to excite Bi5O7I (2.97 eV) to produce photogenerated carriers, thus PPB cannot be degraded by Bi5O7I. Although BiOI is able to absorb most of the visible-light and can be successfully excited to form photogenerated electron–hole pairs, its valence band potential (1.99 V) is still lower than the oxidation potential of the carboxyl group in PPB (2.14 V), and therefore BiOI exhibits poor efficiency in removal of PPB. Compared with BiOI and Bi5O7I, Bi4O5I2 can yield effectively oxidized species (i.e., photogenerated holes and superoxide radicals) under visible-light irradiation and thus is thermodynamically suitable for the photocatalytic removal of PPB. Moreover, when the Bi4O5I2 and Bi5O7I were developed together to form a heterojunction, the partially photogenerated electrons generated by Bi4O5I2 can transfer to the conduction bands of Bi5O7I to inhibit the recombination of photogenerated holes and electrons, thereby prolonging their lifetimes. Meanwhile, the holes formed in the Bi4O5I2 retain their strong oxidizing ability, which can thus enhance the photocatalytic performance.
 | ||
| Scheme 1 Schematic illustration of reaction mechanism of photodegradation of PPB over BiOI, Bi4O5I2, Bi4O5I2/Bi5O7I, and Bi5O7I under visible-light irradiation. | ||
The ability of a photocatalyst to be recycled is very important for its practical application. After the photocatalytic reaction, the as-synthesized Bi4O5I2/Bi5O7I composite was collected by centrifuge and then reused under identical conditions, then their dark adsorption and photoreaction activities under visible-light light were shown in Fig. S7 (ESI†) and Fig. 12, respectively. As shown in Fig. 12, the Bi4O5I2/Bi5O7I photocatalyst kept its high photocatalytic performance after four successive cycles, suggesting a good stability for recycling. In addition, XRD analysis of the Bi4O5I2/Bi5O7I sample before and after the reaction suggests that its composition remained the same (Fig. S8, ESI†), which further confirms the structure stability of the catalyst.
 | ||
| Fig. 12 Recycling stability of the photocatalytic degradation of PPB over as-synthesized Bi4O5I2/Bi5O7I composite. | ||
4. Conclusion
In summary, we constructed a novel flower-like Bi4O5I2/Bi5O7I heterojunction photocatalyst by using a simple hydrothermal route. The as-prepared composite was used as an excellent visible-light photocatalyst for the degradation of PPB, an emerging contaminant with a high oxidation potential. The enhanced photocatalytic performance of the as-synthesized Bi4O5I2/Bi5O7I can be ascribed to its good visible-light absorption, more positive valence band edge potential, strong ability to separate photogenerated carriers between the Bi4O5I2 and Bi5O7I, large specific surface area, and high interface-to-volume ratio thanks to its three-dimensional structures with high anti-aggregation capability. The photogenerated holes and superoxide radicals produced over Bi4O5I2/Bi5O7I were confirmed to be the crucial active species in the photocatalytic reaction. Because of its low cost, easy preparation, high efficiency, and good stability, the flower-like Bi4O5I2/Bi5O7I photocatalyst synthesized by this facile method is a promising photocatalyst for practical applications in environmental purification.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21477040) and the Guangdong Province Natural Science Foundation (No. 2015A030313393, S2012040007074).References
- K. Pirkanniemi and M. Sillanpaa, Chemosphere, 2002, 48, 1047 CrossRef CAS PubMed.
- D. Chatterjee and S. Dasgupta, J. Photochem. Photobiol., C, 2005, 6, 186 CrossRef CAS.
- K. L. Zhang, C. M. Liu, F. Q. Huang, C. Zheng and W. D. Wang, Appl. Catal., B, 2006, 68, 125 CrossRef CAS.
- M. Shang, W. Z. Wang and L. Zhang, J. Hazard. Mater., 2009, 167, 803 CrossRef CAS PubMed.
- X. Xiao and W. Zhang, J. Mater. Chem., 2010, 20, 5866 RSC.
- J. Wang, Y. Yu and L. Zhang, Appl. Catal., B, 2013, 136–137, 112 CrossRef CAS.
- X. Xiao, C. Liu, R. Hu, X. Zuo, J. Nan, L. Li and L. Wang, J. Mater. Chem., 2012, 22, 22840 RSC.
- X. Xiao, C. Xing, G. He, X. Zuo, J. Nan and L. Wang, Appl. Catal., B, 2014, 148–149, 154 CrossRef CAS.
- Q. Liu, D. Ma, Y. Hu, Y. Zeng and S. Huang, ACS Appl. Mater. Interfaces, 2013, 5, 11927 CAS.
- R. He, S. Cao, J. Yu and Y. Yang, Catal. Today, 2016, 264, 221 CrossRef CAS.
- C. Liu and X. Wang, Dalton Trans., 2016 10.1039/c6dt00530f.
- J. Xia, M. Ji, J. Di, B. Wang, S. Yin, Q. Zhang, M. He and H. Li, Appl. Catal., B, 2016, 191, 235 CrossRef CAS.
- Y. Peng, P. Yu, H. Zhou and A. Xu, New J. Chem., 2015, 39, 8321 RSC.
- X. Feng, Y. Chen, Y. Fang, X. Wang, Z. Wang, T. Tao and Y. Zuo, Sci. Total Environ., 2014, 472, 130 CrossRef CAS PubMed.
- B. Albero, R. A. Perez, C. Sanchez-Brunete and J. L. Tadeo, J. Hazard. Mater., 2012, 239–240, 48 CrossRef CAS PubMed.
- X. Ye, X. Zhou, L. Wong and A. M. Calafat, Environ. Sci. Technol., 2012, 46, 12664 CrossRef CAS PubMed.
- H. Yamamoto, I. Tamura, Y. Hirata, J. Kato, K. Kagota, S. Katsuki, A. Yamamoto, Y. Kagami and N. Tatarazako, Sci. Total Environ., 2011, 410–411, 102 CrossRef CAS PubMed.
- P. D. Darbre and A. K. Charles, Anticancer Res., 2010, 30, 815 CAS.
- S. Khanna, P. R. Dash and P. D. Darbre, J. Appl. Toxicol., 2014, 34, 1051 CrossRef CAS PubMed.
- A. Kubacka, M. Fernandez-Garcia and G. Colon, Chem. Rev., 2011, 112, 1555 CrossRef PubMed.
- H. Fang, Y. Gao, G. Li, J. An, P. Wong, H. Fu, S. Yao, X. Nie and T. An, Environ. Sci. Technol., 2013, 47, 2704 CrossRef CAS PubMed.
- J. R. Steter, R. S. Rocha, D. Dionísio, M. R. Lanza and A. J. Motheo, Electrochim. Acta, 2014, 117, 127 CrossRef CAS.
- M. G. Soni, G. A. Burdock, S. L. Taylor and N. A. Greenberg, Food Chem. Toxicol., 2001, 39, 513 CrossRef CAS PubMed.
- X. Xiao and W. Zhang, RSC Adv., 2011, 1, 1099 RSC.
- M. Long, P. Hu, H. Wu, Y. Chen, B. Tan and W. Cai, J. Mater. Chem. A, 2015, 3, 5592 CAS.
- R. Chen, C. Gao, Y. Wu, H. Wang, H. Zhou, Y. Liu, P. Sun, X. Feng and T. Chen, Chem. Commun., 2011, 47, 8136 RSC.
- J. H. Yao, K. R. Elder, H. Guo and M. Grant, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 8173 CrossRef.
- X. Zhang, Z. H. Ai, F. L. Jia and L. Z. Zhang, J. Phys. Chem. C, 2008, 112, 747 CAS.
- J. Cao, X. Li, H. Lin, B. Xu, B. Luo and S. Chen, Mater. Lett., 2012, 76, 181 CrossRef CAS.
- K. Sing, D. H. Everett, R. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- J. Zhang, F. Shi, J. Lin, D. Chen, J. Gao, Z. Huang, X. Ding and C. Tang, Chem. Mater., 2008, 20, 2937 CrossRef CAS.
- J. Xu, W. Meng, Y. Zhang, L. Li and C. Guo, Appl. Catal., B, 2011, 107, 355 CrossRef CAS.
- F. Dong, Z. Zhao, T. Xiong, Z. Ni, W. Zhang, Y. Sun and W. Ho, ACS Appl. Mater. Interfaces, 2013, 5, 11392 CAS.
- Y. Li, J. Wang, H. Yao, L. Dang and Z. Li, J. Mol. Catal. A: Chem., 2011, 334, 116 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra03988j |
| This journal is © The Royal Society of Chemistry 2016 |