
Amphiphilic functional block copolymers bearing a reactive furfuryl group via RAFT polymerization; reversible core cross-linked micelles via a Diels–Alder “click reaction”†
Nabendu B. Pramanik and
Nikhil K. Singha*
Indian Institute of Technology Kharagpur, Rubber Technology Centre, Kharagpur 721302, West Bengal, India. E-mail: nks@rtc.iitkgp.ernet.in; Fax: +91 3222 255303; Tel: +91 3222 283178
First published on 21st December 2015
Abstract
Amphiphilic diblock copolymers of poly(furfuryl methacrylate) (PFMA) and poly(polyethylene glycol methyl ether methacrylate) (PPEGMA) (PFMA-b-PPEGMA) were prepared via reversible addition fragmentation chain-transfer (RAFT) polymerization by using PFMA as a macro-RAFT agent. In water, the amphiphilic diblock copolymer, poly(FMA-b-PEGMA) (PFP) formed micelles with PFMA in the core and PPEGMA in the corona. Diels–Alder chemistry was carried out to crosslink the hydrophobic PFMA core by using bismaleimide in water at 60 °C. The de-crosslinking of hydrophobic PFMA core was carried out upon heating the samples at 150 °C for 30 min via retro-DA reaction which was confirmed by 1H-NMR analysis.
Introduction
Amphiphilic block copolymers (Am-BCPs) can self-assemble to form nano-scale particles with spherical, cylindrical and vascular morphologies above their critical micelle concentration (CMC) value.1–9 Block copolymer micelles are widely used in different applications, e.g. drug carrier systems,10 membrane filtration and separation systems,11 electronic materials,12 pharmaceutical and diagnostic applications depending upon their ability for phase separation in solutions as well as in the solid phase. In an aqueous solution the Am-BCPs form micelles in which the hydrophobic block forms the core and hydrophilic block forms the corona.13–15 The different structures and morphologies of self-assembled Am-BCPs could be obtained by regulating different environment stimulus; such as temperature, pH, magnetic field etc.16–21There have been several reports on the preparation of amphiphilic BCP having reactive functional group. For example, Wooley et al. reported cross-linked amphiphilic polymer networks based on furan-functionalized hyperbranched fluoropolymers and maleimide-functionalized linear poly(ethylene glycol) via DA reaction which exhibited healable antibiofouling coating capability.22 Barner-Kowollik et al. have reported the preparation of amphiphilic diblock copolymers of poly(isoprene-co-styrene)-b-poly(triethylene glycol methyl ether acrylate) based on reversible addition-fragmentation chain transfer (RAFT) polymerization via hetero-DA (HAD) ligation to create reversible cleavage of block junctions.23
Poly(furfuryl methacrylate) (PFMA) contains reactive furfuryl group which is used as diene for Diels–Alder (DA) “click reaction” to prepare different advanced materials.24–27 In recent years, different click reactions like DA reaction,28 alkyne–azide Huisgen cycloaddition,29 thiol-ene,30,31 thiol-Michael addition32 and [4 + 2] addition based on 1,2,4 triazolinedione (TAD) chemistry33 have been used to prepare different functional polymeric materials having unique material properties. Among the different click reactions, DA cycloaddition reaction has an added advantage, as this is thermoreversible and provides an opportunity of self-healing properties.34–37 DA chemistry is very versatile, as dienes and dienophiles are not restricted to one class of compounds. DA chemistry is used in polymeric systems for the synthesis of dendritic,38,39 linear,40,41 graft,42,43 block,44 star45,46 and cross-linked polymers.47–49 Some more exciting works also have been done under the title of hetero Diels–Alder (HDA) reaction where dithioesters have been coupled with butadiene and cyclopentadienyl moieties.50–52 But the most popular pairing for DA reaction is between the maleimide-type dienophile and furfuryl-based diene.
Schubert et al. reported the formation of PEG-block-poly(furfuryl glycidyl ethers) diblock copolymers via living anionic ring-opening polymerization (ROP) and studied their self-assembly behavior followed by cross-linking in the core via DA reaction.53 To the best of our knowledge there is no report on the preparation of amphiphilic diblock copolymer of PFMA and PPEGMA (PFMA-b-PPEGMA) via RAFT polymerization and to study their self-assembly in dilute aqueous solution and followed by crosslinking in the core via DA reaction.
In this investigation, Am-BCPs, PFMA-b-PPEGMA were prepared via RAFT polymerization. The BCP undergoes self-assembly in dilute aqueous solution into micelles with hydrophobic PFMA core and hydrophilic PPEGMA corona. The PFMA core domains with reactive furfuryl group were cross-linked by using bismaleimide via DA reaction upon heating the micellar solution at 60 °C. The cross-linking was thermoreversible in nature at higher temperature via retro-DA reaction.
Experimental section
Chemicals
Furfuryl methacrylate (FMA) and poly(ethylene glycol) methyl ether methacrylate (PEGMA) (Mn = 300 g mol−1) monomers were purchased from Sigma-Aldrich, USA, and were purified by passing through basic alumina column to remove the inhibitor. 4-Cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl] pentanoic acid (CDTSPA), 4,4′-azobis(4-cyanovaleric acid) (ABCVA) and 1,1-(methylene di-4,1-phenylene)bismaleimide (BM) were also purchased from Sigma-Aldrich and were used as received. Dimethyl formamide (DMF) (Merck, India) was purified by vacuum distillation over CaH2. Dichloromethane (Merck, India) was dried over CaCl2.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 g mol−1; Mn,GPC = 24200 g mol−1; Đ = 1.45.
600 g mol−1; Mn,GPC = 24200 g mol−1; Đ = 1.45.Characterization
Gel permeation chromatography (GPC) analysis was carried out to determine the molecular weight (Mn) and dispersity (Đ) of the polymer samples. GPC analysis was carried out in a Viscotek GPC instrument equipped with RI detector (model VE 3580) and two Viscotek viscogel columns (model GMHHR-M) having pore size 30–650 Å. Data analysis was carried out in OmniSEC 4.2 software. Tetrahydrofuran (THF) was used as eluent with a flow rate of 1 mL min−1 at ambient temperature and low dispersity poly(methyl methacrylate) (PMMA) was used as calibration standard. The retention time was fixed for 25 min of the polymer solution within the instrument.1H-NMR spectra of the polymer samples were recorded in d6-DMSO on a Bruker, 600 MHz instrument at ambient temperature. Tetramethylsilane (TMS) was used as an internal standard.
The particle size of the micelles of the BCP was determined by using dynamic light scattering (DLS) instrument. DLS analysis was performed at a scattering angle of 90° on a Malvern Nano ZS instrument using a 4 MW He–Ne laser (λ = 632.8 nm) at 25 °C.
The morphology of the micelles was determined by a transmission electron microscope (TEM). TEM analysis was carried out by using High Resolution (HR) TEM (JEOL 2000) operated at accelerated voltage of 200 kV. The sample was prepared using drop casting method. A dilute aqueous solution of polymer sample was drop cast over 300 mesh carbon coated copper grid. The grids were dried over in a desiccator before analysis.
Atomic force microscopy (AFM) analysis was carried out on an Agilent 5500 (USA) instrument operated at room temperature in air. Very dilute aqueous solution of BCP was casted on glass slide by spin coating followed by drying to remove trace amount of solvent to prepare thin film of block copolymer PFP. AFM analysis was carried out in tapping mode.
Differential scanning calorimetric (DSC) analysis was performed on a DSC 200 F3 instrument (Netzsch, Germany). The BCP was heated from −100 °C to +100 °C under nitrogen atmosphere at a heating rate 10 °C min−1. In case of cross-linked DA polymer the sample was heated from −25 °C to +250 °C at a heating rate of 10 °C min−1 under nitrogen atmosphere. Nitrogen was used as an inert atmosphere with a flow rate 50 mL min−1. The temperature against heat flow was recorded. The enthalpy was calibrated with the indium standard supplied by Netzsch.
The surface tension values of self-assembled Am-BCP, PFMA-b-PPEGMA were determined by liquid pendant method using Rame-Hart instrument co. USA, Model no. 260F4, Goniometer/Tensiometer at 25 °C, taking water as a liquid phase. In this case, water pendant drop was suspended in air as gaseous external phase. The surface tension measurement was carried out for the determination of critical micelles concentration (CMC) of BCP in water. Each water droplet volume is employed in this study. With increasing the BCP concentration in water the surface tension value of the solution is deceased. The surface tension vs. the concentration of BCP in solution is plotted for the measurement of CMC of the amphiphilic BCP in water. All the measurements were repeated 10 times and their average value taken for consideration. The polymer concentration was varied from 0.001 to 0.1 mg mL−1 for determining the CMC.
Results and discussion
Am-BCPs of PFMA and PEGMA were prepared via RAFT polymerization using PFMA as macro-RAFT agent (Scheme 1). In this case, first PFMA was prepared via RAFT polymerization of FMA using CDTSPA as RAFT reagent and ABCVA as initiator. The presence of RAFT end group in the PFMA polymer has been shown by 1H-NMR spectra (Fig. S1; ESI†). This PFMA was used as a macro-RAFT agent to prepare different BCPs of PFMA and PPEGMA. Table 1 shows the summary of the different BCPs prepared at different solvents and having varying content of PPEGMA. It was observed that when the PPEGMA content was higher, the reaction was less controlled resulting broad dispersity (Đ). The block copolymerization was carried out in toluene and in DMF. The block copolymerization was better controlled in DMF, as it was observed from the narrow dispersity value (Table 1). This may be due to better solubility of PPEGMA as well as of the RAFT reagent in DMF (Table 1). The Am-BCP was gel free and soluble in various solvents like THF, chloroform and DMSO. | ||
| Scheme 1 Synthesis of amphiphilic block copolymers of PFMA-b-PPEGMA via RAFT polymerization and its micelle formation in aqueous solution. | ||
| Expt. no. | Polymer name | Sample compositionb | [M]:[macro-RAFT]:[I] |
Conv. (%) | Mn,Theo. (g mol−1) | Mn,GPC (g mol−1) | Đ | PFMAc (mol%) | CMCd (mg mL−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Block copolymerization was carried out using PFMA (Mn = 8500 g mol−1, Đ = 1.35) as macro-RAFT.b Subscript denotes the degree of polymerization determined by GPC analysis.c Mol% of PFMA was determined by 1H-NMR.d Determined via surface tension measurement.e In this case toluene was used as solvent. | |||||||||
| 1 | PFP1 | PFMA51-b-PPEGMA72 | 400:4:1 |
90 | 34000 |
30100 |
1.58 | 31 | 0.0202 |
| 2 | PFP2 | PFMA51-b-PPEGMA53 | 200:4:1 |
94 | 22600 |
24200 |
1.45 | 45 | 0.0172 |
| 3 | PFP3 | PFMA51-b-PPEGMA48 | 160:4:1 |
95 | 19900 |
22900 |
1.41 | 49 | 0.0161 |
| 4 | PFP4 | PFMA51-b-PPEGMA33 | 120:4:1 |
96 | 17200 |
18400 |
1.39 | 59 | 0.0143 |
| 5 | PFP5e | PFMA51-b-PPEGMA47 | 200:4:1 |
65 | 18300 |
22700 |
2.01 | 46 | |
Fig. 1 shows the 1H-NMR spectrum of the PFP2 BCP. It shows the resonances at δ = 7.6 ppm, δ = 6.4 ppm for furfuryl group of PFMA and δ = 4.9 ppm for the methylene protons (>CH2) of PFMA unit. The resonances at δ = 3.9 ppm are due to methylene protons (>CH2) of PPEGMA unit. The integral area of these two types of methylene protons in the PFMA and PPEGMA units was used to calculate the mole ratio of PFMA and PPEGMA present in the BCP.
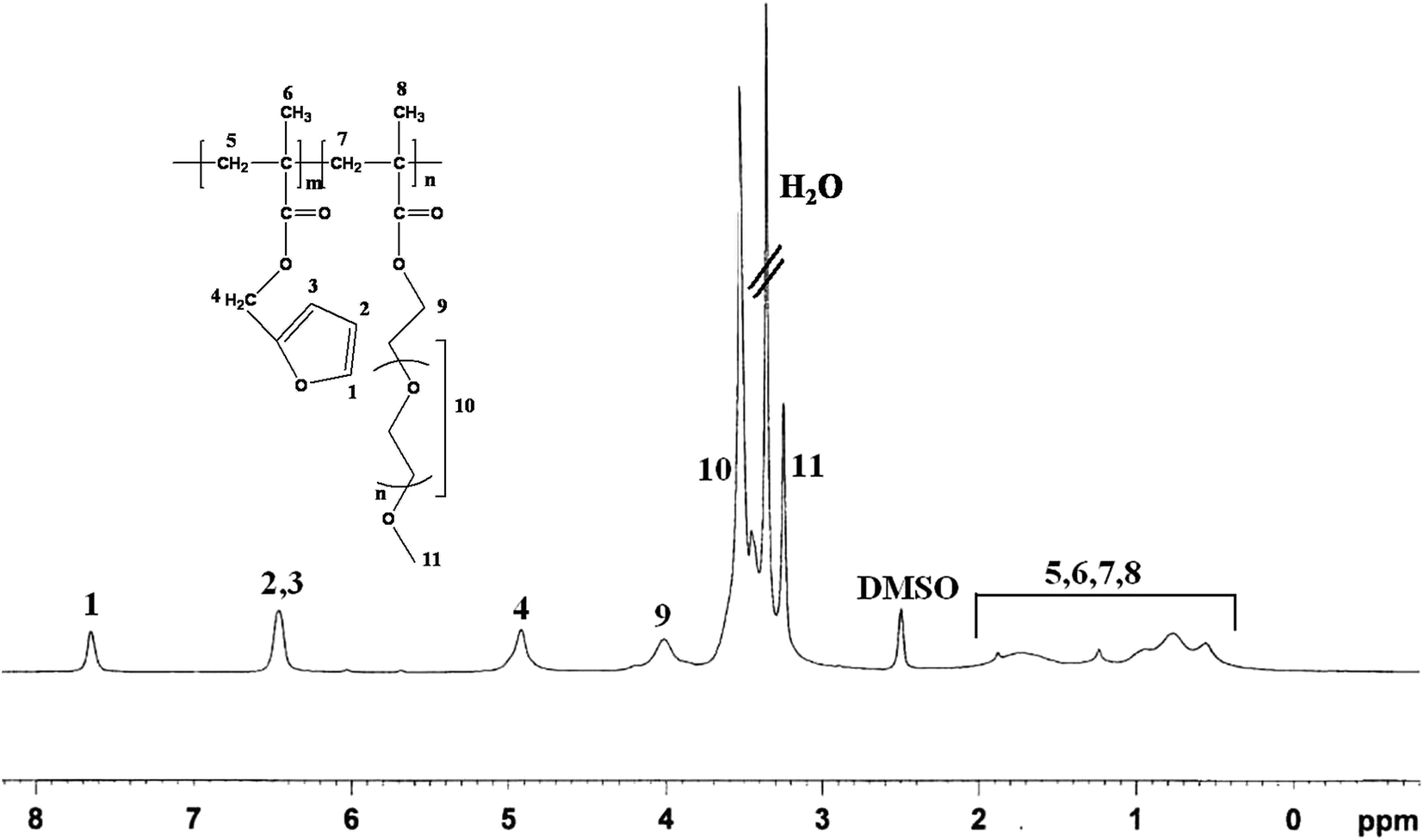 | ||
| Fig. 1 1H-NMR spectrum of the PFP2 BCP. | ||
Molecular weight and the dispersity (Đ) of the BCPs were determined by GPC analysis (Table 1). A typical GPC trace of PFMA macro-RAFT and its BCP PFP2 are shown in ESI (Fig. S2†). Complete shifting of the GPC traces towards the higher molecular weight in case of BCP indicates the successful formation of the BCP. Table 1 also shows the percent of PFMA content calculated via 1H-NMR analysis. The formation of diblock copolymers was also confirmed by DSC analysis. In DSC thermogram the BCP, PFP2 shows two Tsg; one for the hard segment of PFMA at 41 °C and another for the soft segment of PPEGMA at −53 °C (Fig. S3 in ESI†).
Self-assembly of PFMA-b-PPEGMA in water
The BCP, PFMA-b-PPEGMA forms micelles in dilute aqueous solution due to its amphiphilic nature. This micelle formation was demonstrated using DLS, TEM and AFM analyses. The core–shell structures consist of a hydrophobic PFMA in the core and hydrophilic PPEGMA in the corona. DLS analysis of the BCP, PFP2 in aqueous solution (1 mg mL−1) showed that the micelles have broad size distribution of maximum micelles having size around 80 nm (Fig. 2). At higher concentration of 5 mg mL−1, the size distribution of the micelles became very broad. It had the maximum micelles having size of around 190 nm due to aggregation of micellar particles. This is evident from the bimodal distribution of the micelle particles in the DLS analysis (Fig. 2). | ||
| Fig. 2 DLS profiles of Am-BCP, PFMA51-b-PPEGMA53 (PFP2) in water using concentration of 1 mg mL−1 (a) and 5 mg mL−1 (b). | ||
In DLS analysis at low concentration of 1 mg mL−1, no bimodal distribution was observed in case of PFP2 BCP. This may be due to less size variation of the micelles in aqueous solution. But at high concentration (5 mg mL−1) above the CMC value, the bimodal size distribution was observed in DLS analysis. This is due to the formation of micelles of two different sizes in aqueous solution and aggregation of the smaller size micelles at high concentration.
TEM analysis was used to characterize the shape and size of the micelles formed at different concentrations of the BCP (Fig. 3). TEM analysis revealed that the micelles are spherical in size and they have core–shell structure. The dark portion of the core consists of hydrophobic PFMA block and the lighter dark portion in the shell is due to the hydrophilic PPEGMA block in the corona (Fig. 3). When the BCP concentration is 1 mg mL−1, the size of the micelles is 90 nm. But at higher concentration of BCP (5 mg mL−1), the size of the micelles is much bigger and the micellar size distribution is also broad (100–300 nm). This may be due to coalescence of the micelles at higher concentration. TEM analysis of all the BCPs having different block length of PPEGMA was carried out for better understanding of the morphology of the micelles, as the morphology of the BCPs depends on the hydrophilic and hydrophobic block length. In case of the BCP, PFP1, the micelles are bigger in size (Fig. 3c). As the hydrophilic block length increased, the CMC of BCP also increased. Because more amphiphilic macromolecules were needed to form the micelles, leading to the formation of bigger size micelles. When the hydrophilic block length decreased, the CMC of the BCP decreased and the micelle size also decreased. This is the case for PFP4, where the size of the micelles is smaller, as shown in TEM analysis (Fig. 3d).
 | ||
| Fig. 3 TEM images of PFP2 diblock copolymer in aqueous solution at a concentration of 1 mg mL−1 (a), 5 mg mL−1 (b), PFP1 (c) and PFP4 (d) BCP at a concentration of 1 mg mL−1. | ||
The micelle formation of the PFP2 diblock copolymer in aqueous solution was successfully demonstrated by AFM analysis (Fig. 4). AFM analysis showed that the micelles were spherical in shape and had the average micellar size of ∼90 nm in case of concentration of 1 mg mL−1 of BCP in water. The micellar size obtained from the AFM analysis was in good agreement with the micelles size shown by TEM analysis.
 | ||
| Fig. 4 AFM phase image of PFP2 diblock copolymers in aqueous solution (concentration of 1 mg mL−1). | ||
The critical micelle concentration (CMC) of Am-BCP in aqueous solution was determined by surface tension measurement (Fig. 5). With increase in the concentration of Am-BCP, the surface tension gradually decreased. After achieving the concentration, on further increase in the BCP concentration, the surface tension almost remains constant. The CMC value was calculated from the intersection of two lines as shown in Fig. 5. The CMC of PFP2 was calculated to be 17.2 × 10−3 mg mL−1.
 | ||
| Fig. 5 Plot of surface tension vs. the concentration of self-assembled Am-BCP, PFP2. | ||
Diels–Alder reaction in the micellar core
Diels–Alder (DA), the [4 + 2] cycloaddition addition reaction has been used in case of furfuryl system to prepare different types of functional polymeric materials which have potential applications in the formation of self-healing and thermally amendable materials. In the present system the micellar core having reactive furfuryl group was cross-linked via DA reaction using a bismaleimide (Scheme 2). For this purpose the BCP, PFP2 and a bifunctional cross-linker 1,1-(methylene di-4,1-phenylene)bismaleimide (BM) (1:1 mole ratio) were dissolved in DMF. To encapsulate the BM cross-linker into the PFMA hydrophobic core domain, water was added slowly to obtain turbid solution (DMF:water = 1:4). Subsequently, the mixture was heated at 60 °C for 48 h to induce cross-linking inside the core domains. After the reaction, excess DMF was removed from the reaction mixture via dialysis against water. The aqueous solution of the cross-linked micelles (PFP2-BM) was analyzed by DLS analysis. The PFP2 micelles containing BM in the core i.e. the core cross-linked micelles showed the size of approximately 115 nm in water (Fig. 6a). The bimodal distribution in DLS analysis may be due to crosslinking of the micelles of various size. To prove the successful cross-linking via DA reaction in the PFMA core domain, the micelles were transferred to a nonselective solvent, THF. In this case, the cross-linked (PFP2-BM) micellar aqueous solution was transferred into an excess THF solvent, so that the volume ratio of THF:H2O was 6:1. DLS analysis against THF showed the micellar size of approximately 125 nm (Fig. 6b). The micellar size in THF was increased due to the swelling of the cross-linked PFMA core in THF. Again, after several days the swollen cross-linked PFMA core showed that the micellar size was unchanged during DLS analysis. These results clearly indicate the successful cross-linking of PFMA core with BM via [4 + 2] DA cycloaddition reaction. TEM images of core cross-linked micelles were shown in Fig. 7. This TEM image is comparable with the TEM images of the BCP, PFP2 before DA crosslinking.53,54
 | ||
| Scheme 2 Schematic representation of cross-linking of the micellar core via DA reaction. | ||
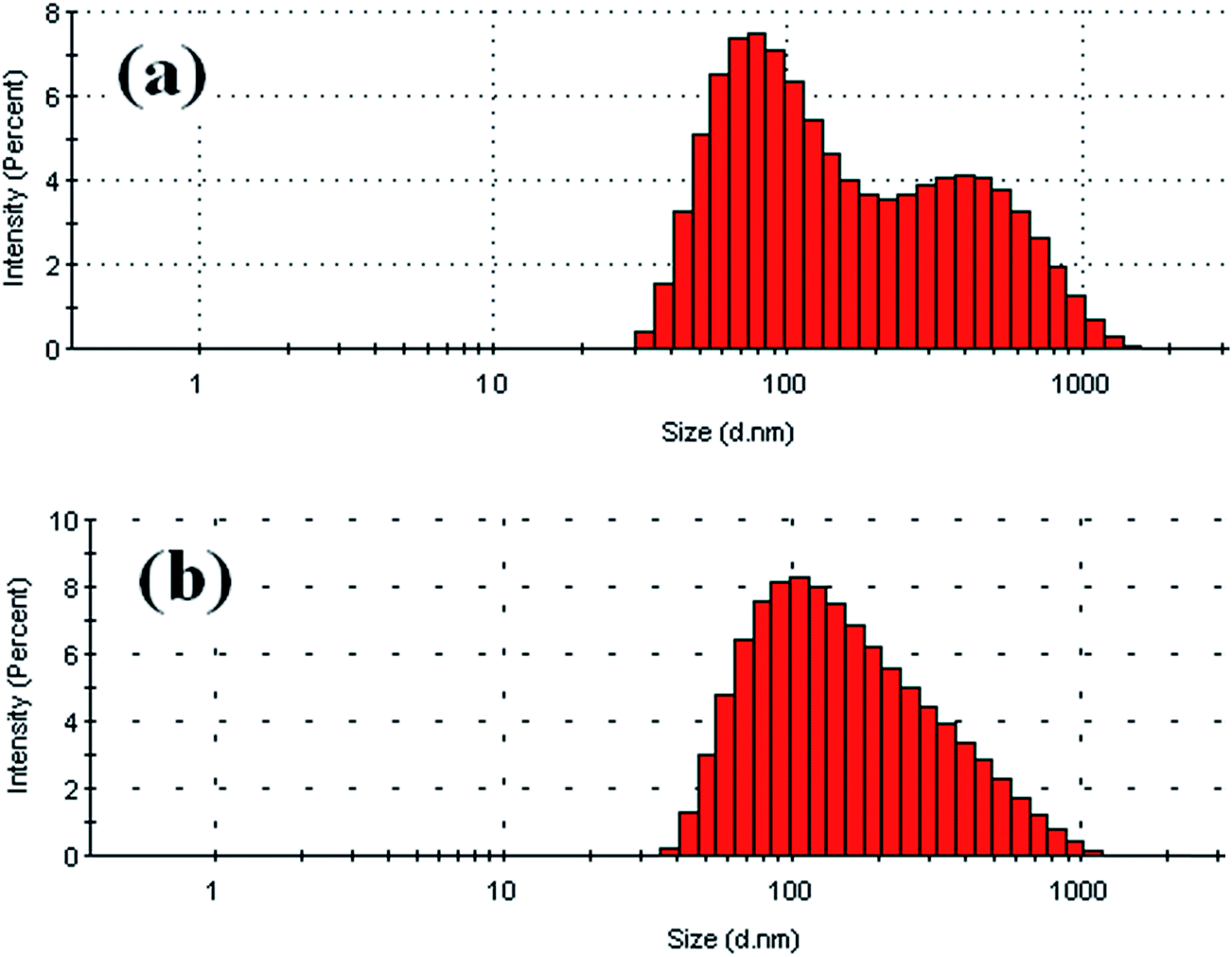 | ||
| Fig. 6 DLS profiles of cross-linked DA BCP (PFP2-BM) micelle in water (a) and in THF (b). | ||
 | ||
| Fig. 7 TEM images of PFP2-BM core cross-linked micelles. | ||
The cross-linking in PFMA core domain with bismaleimide (BM) via DA reaction was also proved by 1H-NMR analysis. Fig. 8 shows the 1H NMR spectra of cross-linked DA polymer (PFP2-BM) and its retro-DA polymer. The cross-linking was proved by the disappearance of the resonance of the protons of furfuryl group at δ = 7.6 ppm and 6.4 ppm in 1H NMR analysis after prolonged heating of PFP2-BM at 60 °C. Complete disappearance of furan protons indicates the complete consumption of furan moieties in reaction with BM by DA reaction. To check its thermoreversibility, the cross-linked micellar solution was heated at 150 °C for 30 min in DMSO for rDA reaction. In 1H NMR spectra the resonances for the protons of furan ring reappear at δ = 7.6 ppm and 6.4 ppm for the rDA product, as shown in Fig. 8B. This indicates that the cross-linked DA micelles are thermoreversible in nature.
 | ||
| Fig. 8 1H NMR of spectrum of PFP2-BM (A) core cross-linked micelles and (B) its retro-DA product. | ||
The DSC analysis of PFP2-BM cross-linked micelles was carried out to study the rDA reaction temperature (Fig. 9). The DSC traces of PFP2-BM showed a broad endothermic peak at 142 °C due to the cleavage of bonds between the furfuryl groups of PFMA and bismaleimide via the retro-DA reaction.
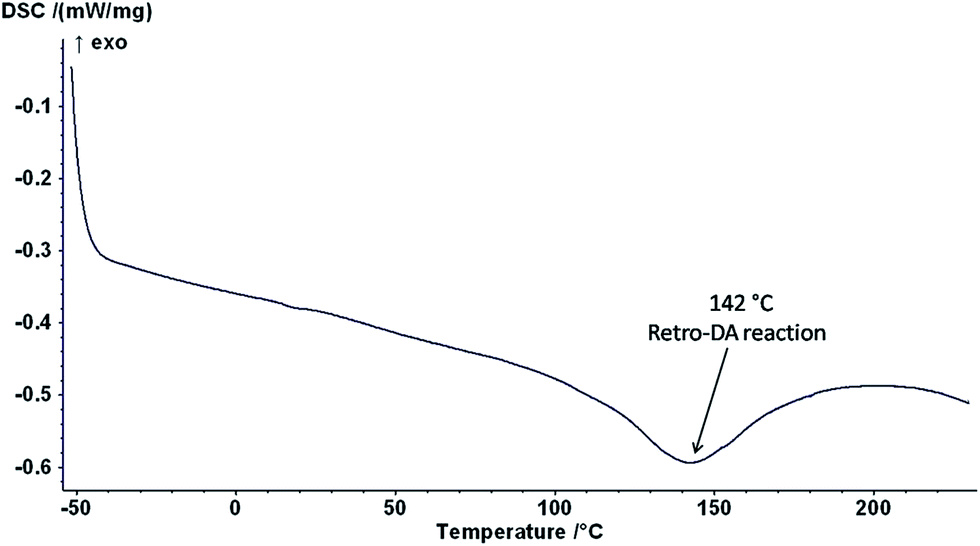 | ||
| Fig. 9 DSC traces of PFP2-BM core cross-linked micelles. | ||
Conclusion
Diblock copolymers containing hydrophilic block of PPEGMA and hydrophobic PFMA block were synthesized via RAFT polymerization using PFMA macro-RAFT. In dilute aqueous solution the diblock copolymers, PFMA-b-PPEGMA formed spherical micelles having core–shell morphology in which hydrophobic PFMA was in the core and hydrophilic PPEGMA was in the corona. The size and shape of the formed micelles were characterized by DLS, TEM and AFM analyses. The PFMA core was cross-linked using BM as a cross-linker via DA reaction. The core-crosslinked micelles retain their structure in non-solvent THF. The cross-linked micelles undergo de-crosslinking via retro-DA reaction when it was heated at 150 °C for 30 min in DMSO solvent. The reversible crosslinking of the core in the spherical core–corona system will be very effective for controlled release of encapsulated materials or surface patterning from non-selective solvents.Acknowledgements
UGC, New Delhi is acknowledged for their financial support.References
- Z. L. Tyrrell, Y. Shen and M. Radosz, Prog. Polym. Sci., 2010, 35, 1128–1143 CrossRef CAS.
- A. Rösler, G. W. M. Vandermeulen and H. A. Klok, Adv. Drug Delivery Rev., 2012, 64, 270–279 CrossRef.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis, S. Pispas and A. Avgeropoulos, Prog. Polym. Sci., 2005, 30, 725–782 CrossRef CAS.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- J. Zhu and W. Jiang, Macromolecules, 2005, 38, 9315–9323 CrossRef CAS.
- J. Rodríguez-Hernández and S. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026–2027 CrossRef PubMed.
- J. Du and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 12800–12801 CrossRef CAS PubMed.
- G. Gaucher, M. H. Dufresne, V. P. Sant, N. Kang, D. Maysinger and J. C. Leroux, J. Controlled Release, 2005, 109, 169–188 CrossRef CAS PubMed.
- F. H. Schachar, P. A. Rupar and I. Manners, Angew. Chem., Int. Ed., 2012, 51, 7898–7921 CrossRef PubMed.
- H. C. Kim, S. M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146–177 CrossRef CAS PubMed.
- M. H. Stenzel, Chem. Commun., 2008, 30, 3486–3503 RSC.
- K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS PubMed.
- J. Liu, Y. Xiao and C. Allen, J. Pharm. Sci., 2004, 93, 132–143 CrossRef CAS PubMed.
- C. Pietsch, U. Mansfeld, C. Guerrero-Sanchez, S. Hoeppener, A. Vollrath, M. Wagner, R. Hoogenboom, S. Saubern, S. H. Thang, C. R. Becer, J. Chiefari and U. S. Schubert, Macromolecules, 2012, 45, 9292–9302 CrossRef CAS.
- F. Liu and A. Eisenberg, J. Am. Chem. Soc., 2003, 125, 15059–15064 CrossRef CAS PubMed.
- E. He, P. Ravi and K. C. Tam, Langmuir, 2007, 23, 2382–2388 CrossRef CAS PubMed.
- A. E. Smith, X. Xu, S. E. Kirkland-York, D. A. Savin and C. L. McCormick, Macromolecules, 2010, 43, 1210–1217 CrossRef CAS.
- M. T. Savoji, S. Strandman and X. X. Zhu, Langmuir, 2013, 29, 6823–6832 CrossRef CAS PubMed.
- Q. Yan and Y. Zhao, J. Am. Chem. Soc., 2013, 135, 16300–16303 CrossRef CAS PubMed.
- P. M. Imbesi, C. Fidge, J. E. Raymond, S. I. Cauët and K. L. Wooley, ACS Macro Lett., 2012, 1, 473–477 CrossRef CAS.
- M. Langer, J. Brandt, A. Lederer, A. S. Goldmann, F. H. Schacher and C. Barner-Kowollik, Polym. Chem., 2014, 5, 5330–5338 RSC.
- N. B. Pramanik, G. B. Nando and N. K. Singha, Polymer, 2015, 69, 349–356 CrossRef CAS.
- N. B. Pramanik, D. S. Bag, S. Alam, G. B. Nando and N. K. Singha, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3365–3374 CrossRef CAS.
- A. A. Kavitha and N. K. Singha, ACS Appl. Mater. Interfaces, 2009, 1, 1427–1436 CAS.
- A. A. Kavitha and N. K. Singha, Macromolecules, 2010, 43, 3193–3205 CrossRef CAS.
- X. X. Chen, M. A. Dam, K. Ono, A. Mal, H. B. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS PubMed.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- S. Billiet, K. D. Bruycker, F. Driessen, H. Goossens, V. V. Speybroeck, J. M. Winne and F. E. Du Prez, Nat. Chem., 2014, 6, 815–821 CrossRef CAS PubMed.
- S. D. Bergman and F. Wudl, J. Mater. Chem., 2008, 18, 41–62 RSC.
- M. J. Barthel, T. Rudolph, A. Teichler, R. M. Paulus, J. Vitz, S. Hoeppener, M. D. Hager, F. H. Schacher and U. S. Schubert, Adv. Funct. Mater., 2013, 23, 4921–4932 CrossRef CAS.
- Q. Tian, Y. C. Yuan, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2009, 19, 1289–1296 RSC.
- J. A. Syrett, G. Mantovani, W. R. S. Barton, D. Pricec and D. M. Haddleton, Polym. Chem., 2010, 1, 102–106 RSC.
- M. J. Joralemon, R. K. O'Reilly, J. B. Matson, A. K. Nugent, C. J. Hawker and K. L. Wooley, Macromolecules, 2005, 38, 5436–5443 CrossRef CAS.
- M. M. Kose, G. Yesilbag and A. Sanyal, Org. Lett., 2008, 10, 2353–2356 CrossRef CAS PubMed.
- M. Li, P. de, S. R. Gondi and B. S. Sumerlin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5093–5100 CrossRef CAS.
- A. Gandini, D. Coelho and A. J. D. Silvestre, Eur. Polym. J., 2008, 44, 4029–4036 CrossRef CAS.
- B. Gacal, H. Durmaz, M. A. Tasdelen, G. Hizal, U. Tunca, Y. Yagci and A. L. Demirel, Macromolecules, 2006, 39, 5330–5336 CrossRef CAS.
- A. Bousquet, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1773–1781 CrossRef CAS.
- H. Durmaz, A. Dag, O. Altintas, T. Erdogan, G. Hizal and U. Tunca, Macromolecules, 2006, 40, 191–198 CrossRef.
- A. Vieyres, T. Lam, R. Gillet, G. Franc, A. Castonguay and A. Kakkar, Chem. Commun., 2010, 46, 1875–1877 RSC.
- H. Durmaz, F. Karatas, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 499–509 CrossRef CAS.
- Z. Shi, S. Hau, J. Luo, T.-D. Kim, N. M. Tucker, J.-W. Ka, H. Sun, A. Pyajt, L. Dalton, A. Chen and A. K.-Y. Jen, Adv. Funct. Mater., 2007, 17, 2557–2563 CrossRef CAS.
- A. J. Inglis, L. Nebhani, O. Altintas, F. G. Schmidt and C. Barner-Kowollik, Macromolecules, 2010, 43, 5515–5520 CrossRef CAS.
- J. P. Swanson, S. Rozvadovsky, J. E. Seppala, M. E. Mackay, R. E. Jensen and P. J. Costanzo, Macromolecules, 2010, 43, 6135–6141 CrossRef CAS.
- K. K. Oehlenschlaeger, J. O. Mueller, J. Brandt, S. Hilf, A. Lederer, M. Wilhelm, R. Graf, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Adv. Mater., 2014, 26, 3561–3566 CrossRef CAS PubMed.
- A. J. Inglis, T. Paulöhrl and C. Barner-Kowollik, Macromolecules, 2010, 43, 33–36 CrossRef CAS.
- T. Paulöhrl, A. J. Inglis and C. Barner-Kowollik, Adv. Mater., 2010, 22, 2788–2791 CrossRef PubMed.
- M. J. Barthel, T. Rudolph, S. Crotty, F. H. Schacher and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4958–4965 CrossRef CAS.
- F. Schacher, A. Walther, M. Ruppel, M. Drechsler and A. H. E. Müller, Macromolecules, 2009, 42, 3540–3548 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra22476d |
| This journal is © The Royal Society of Chemistry 2016 |