
Unconventional synthesis of Cu–Au dendritic nanowires with enhanced electrochemical activity†
Yuan Chena,
Qingchi Xuab,
Bo Hua,
Jun Xu*ab and
Jian Wenga
aDepartment of Biomaterials and Department of Physics, Xiamen University, Xiamen, 361005, P. R. China. E-mail: xujun@xmu.edu.cn
bResearch Institute for Biomimetics and Soft Matter, Xiamen University, Xiamen, 361005, P. R. China
First published on 18th December 2015
Abstract
Cu–Au dendritic nanowires (NWs) were synthesized by heating pre-synthesized Cu NWs and Au ultrathin NWs in a THF/H2O mixture. The obtained dendritic NWs were composed of a Cu/CuO stem and distributed by Au multi-branches throughout the whole structure. The formation of a dendritic structure was initiated from oxidation of Cu to CuO during the phase transition stage, giving an anisotropic dendritic precursor. Then a galvanic replacement reaction between Cu and Au ions created abundant anchor sites, facilitating Au branch formation via self-deformation of ultrathin Au NWs around the Cu NWs surface. The synthesized Cu–Au dendritic NWs could be potentially used as biosensors in detecting glucose for its high sensitivity and excellent performances, due to the demonstration of their enhanced electrochemical activity for oxidation of glucose.
Introduction
One-dimensional metal nanowires (NWs), especially Cu and Au, have received intensive attention for their wide applications owing to their unique optical, electronic and catalytic properties. Synthesis of well-defined Cu or Au NWs has been successfully developed with various methods.1–6 However, currently studies on this topic are restricted to the smooth-surface NWs with limited surface areas. Therefore, increasing the specific surface area of such nanowires becomes an effective way to improve the performance in real applications.On the other hand, nanomaterials in dendritic morphology, which have a plenty of edges, corners, or stepped atoms located on their tips, normally possess high surface areas over other shaped nanostructures. Thus, they enormously increase surface to volume ratio of the original structures, providing quantities of active sites and high efficiency when performing surface related activities such as photoactivity7,8 superhydrophobicity,9,10 catalysis,11–16 biological labelling,17–19 optoelectronics and surface plasmons.19–26 Attribute to the intrinsic properties of Cu and Au nanostructures, researchers have devoted great efforts to morphology-controlled synthesis of Cu and Au dendritic nanostructures, and further explore their potential applications over the past few years. For example, dendritic Cu nanostructures were usually synthesized via template-free aqueous electroless deposition, electrochemical deposition,21,27 hydrothermal approaches,28 or solution-phase chemical synthesis.29,30 Similarly, template-free colloidal synthesis,15,31,32 electrochemical deposition33 and soft template (e.g. polymer)19,25 assistant fabrication were also reported to obtain dendritic Au nanostructures.
Dendritic NWs, with combination of the two unique characters of NW and dendritic structure, is conceivable to have much more improved surface area and enhanced performance. However, this idea could be hardly realized in practice. In colloidal system, synthesis of metal dendritic nanoparticles (e.g. Cu, Au) is usually based on classical capping agents induced anisotropic growth. Nevertheless, this approach is not applicable to dendritic NWs, for dendritic NWs not only displayed similar characters with dendritic nanoparticles, but also retain linear structure. In this case, anisotropic growth of wire and dendritic structure is supposed to be conducted simultaneously, which, however, could not be realized by capping agents. Although a few cases of colloidal Cu or Au dendritic NWs are reported,14,34 the synthetic methodology for dendritic NWs still remains great challenge.
Thanks to excellent electrochemical performance of the dendritic nanostructures over other shapes, electrochemistry becomes the first choice when scientists look for potential applications of this nanostructure.29,35 Previous investigations provide factual evidence that electrochemical activity could be immensely improved when meshing multi noble metal into one nanostructure, especially into dendritic nanostructures.36,37 Therefore, combination of Au and Cu into Cu–Au heterogeneous dendritic nanostructures, especially into Cu–Au dendritic NWs, is a promising way to realize high electrochemical performance other than the individual metal NWs or dendrimers.
Here we report an unconventional approach for the synthesis of Cu–Au dendritic NWs by heating pre-synthesized Cu NWs and ultrathin Au NWs in colloidal THF/H2O solution (as shown in Fig. 1a). Although simultaneous anisotropic growth in two-dimension is a real hindrance, this approach provides a fixed template in one dimension and then introduces desired anisotropic growth in another dimension. Cu NWs served as the stem are decorated with multi-branches evolved from Au NWs, yielding highly uniform dendritic nanostructures with superior electrochemical activity for glucose electro-oxidation.
 | ||
| Fig. 1 (a) Schematic illustration of Cu–Au dendritic NWs formation from Cu NW and ultrathin Au NW. (b) SEM image of as-synthesized Cu NWs with a high-magnification of single wire inserted; (c) TEM image of as-synthesized Au NWs. (d) Low-magnification, and (e) high-magnification SEM images of Cu–Au dendritic NWs. | ||
Experimental
Materials
All chemicals are AR grade, and were used without further purification. Gold(III) chloride trihydrate was purchased from Sigma Aldrich. CuCl2·2H2O and KH2PO4 were purchased from Sinopharm Chemical Reagent Co., Ltd. Oleylamine (OM) was purchased from J&K Scientific Co., Ltd (Beijing, China). Polyvinylpyrrolidone (PVP) (linear, Mw = 8000) and triisopropylsilane (TIPS) was purchased from Alfa Aesar. Other chemicals were purchased from Xilong Chemical Co., Ltd (Shantou, China).Characterization
Scanning electron microscope (SEM) images were obtained on SU-70 with the accelerating voltage of 20 kV. The samples were prepared by dropping 20 μL solution of the products on silicon wafer. Transmission electron microscope (TEM) images were taken from and JEM-2100 operated at 200 kV. HRTEM, high angle annular dark field scanning transmission electron microscopy (HAADF-STEM) and elemental mapping were collected from FEI F30 operated at 300 kV. X-ray diffraction (XRD) pattern was carried out using a Philips Panalytical X'pert PRO with Cu Kα radiation at λ = 1.542 Å over the 2θ range of 10–90°. X-ray photoelectron spectroscopy (XPS) measurements were performed on a PHI Quantum 2000 (USA).Preparation of ultrathin Au NWs
Ultrathin Au NWs (Au NWs) were prepared following a previously reported method.38,39 100 μL OM and 3 mg HAuCl4·3H2O were added into 2.5 mL hexane to form a yellow solution. Then 150 μL TIPS was added to the mixture and this solution were incubated at room temperature for 4–5 h.Preparation of Cu NWs
Cu NWs were prepared according to previous literature.40 CuCl2·2H2O (1.7 g) and glucose (1.93 g) were dissolved in 200 mL of H2O under magnetic stirring. On the other hand, 20 mL of OM, 0.2 mL of oleic acid (OA), were added into 35 mL of ethanol under magnetic stirring. Then, these two solutions were mixed and diluted to 1 L with water, followed by magnetic stirring for 12 h in a 50 °C water bath. The mixture was transferred into a Teflon-lined stainless-steel autoclave with a capacity of 50 mL for hydrothermal treatment at 120 °C for 12 h. After cooled to room temperature, the crude solution of Cu NWs was centrifuged for 10 min at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, and then the precipitate was sonicated and centrifuged twice in an ethanol solution containing 4.0 wt% PVP. Finally, Cu NWs were centrifuged twice again in ethanol solution to remove the free PVP.
000 rpm, and then the precipitate was sonicated and centrifuged twice in an ethanol solution containing 4.0 wt% PVP. Finally, Cu NWs were centrifuged twice again in ethanol solution to remove the free PVP.
Preparation of Cu–Au dendritic NWs
50 μL as-synthesized Cu NWs and 200 μL as-synthesized Au NWs were mixed, purified with ethanol, and then dispersed in 400 μL THF. 1 mL TritonX-100 (8 mM) aqueous solution was quickly injected under vigorous stirring. The resulted mixture was heated at 60 °C for 2 h to evaporate THF. Final products were collected by repeated centrifuging and thorough washing by deionized water and re-dispersed in H2O.Electrochemical experiments
Electrochemical measurements were carried out using an electrochemical workstation (CHI660C) and the three-electrode system. In this system, a platinum wire as auxiliary electrode, an Ag/AgCl (saturated KCl) as reference and working electrode is the Glassy Carbon (GC) electrode (Φ = 2 mm) modified by nanomaterials being examined. Prior to modification, the GC electrode was polished and dried with N2. Then the electrode was covered with 10 μL of solution under test. After dried naturally overnight, the electrode was deposited with 1 μL of 2-propanol containing Nafion solution (5 wt%) to maintain the film on the electrode stable. In the detection process, the modified electrode was immersed in 0.1 M NaOH or 0.1 M NaOH with desired concentration of glucose. All electrochemical experiments were performed at room temperature and ambient pressure.Results and discussion
To prepare Cu–Au dendritic NWs, Cu NWs and ultrathin Au NWs were synthesized and purified according to literatures respectively at first step. The obtained Cu NWs and Au NWs were mixed in THF, followed by addition of large amount of TritonX-100 aqueous solution. The mixture was subsequently heated to 60 °C for 2 h to evaporate THF, and the final products were collected via centrifugation.As showed in Fig. 1b, the as-synthesized Cu NWs presented smooth surface with several μm in length and 50–60 nm in diameter. While the as-synthesized Au NWs were several μm in length as well, but 2–3 nm in diameter (Fig. 1c). However, heating after mixing Cu NWs and Au NWs in THF/TritonX-100 solution gave products with dramatically different morphology. Typical SEM image of such products (Fig. 1d) showed 3D dendritic morphology for most nanostructures, which displayed in 1D NW profile. The length of as-synthesized dendritic NWs were several μm, which was really close to the length of original Cu NWs. Close inspection of the dendritic nanostructures (high magnification SEM Fig. 1e) revealed rough surface on account of abundant branches. The diameter of the dendritic NWs was about 100 nm, which is slightly increased in comparison with the original Cu NWs.
Fig. 2 showed the representative TEM images of Cu–Au dendritic NWs at different magnifications. Low magnification image provided a clear view of the dendritic structures (Fig. 2a), which was essentially in agreement with SEM observation. High magnification image further suggested continuous growth of branches from the original Cu NWs, other than nanoparticle aggregation (Fig. 2b). HRTEM (Fig. 2c and d) observation demonstrated that the as-synthesized dendritic structures were heterogeneous, which consisted of Cu, CuO and Au. In the dendritic structure, the stem domain, whose lattice space was measured to be 0.21 nm, corresponded to fcc (111) plane of Cu. On the other side, branched part of the dendritic structure indicated dominant formation of fcc Au (111) plane inlaid with small amount of CuO (111) plane. Few Cu–Au alloy phase was also observed at branch part (Fig. 2c), where the lattice spacing of 0.29 nm could be index to (110) plane of Au3Cu.41,42 To unpuzzle the distributions of Cu and Au, the dendritic structure were checked by HDDAD-STEM element mapping. As shown in Fig. 2e, both Cu and Au element were detected. Cu and O were found over the whole dendritic structure, while Au allocated uniformly along the surface of NW stem.
 | ||
| Fig. 2 (a and b) Typical TEM images of as-synthesized Cu–Au dendritic NWs with different magnification. (c and d) HRTEM images of corresponded areas in (b). (e) HAADF-STEM and elemental mapping of as-synthesized Cu–Au dendritic NWs. | ||
XRD pattern also established corroboration of Cu–Au heterogeneous structures. Besides Au and Cu, the typical CuO signal could be individually collected (Fig. 3a), indicating CuO was presented in Cu–Au dendritic NWs. The chemical composition of the as-synthesized dendritic nanostructure was subsequently determined by XPS. The full-scan XPS spectrum (Fig. 3b) illustrated that the dendritic nanostructure was composed of C, O, Cu, and Au. C element was probably coming from the ligand residue on surface (oleylamine). The two peaks of Cu 2p3/2 (932.2 eV) and Cu 2p1/2 (952 eV) were associated with zero-valent Cu as shown in Fig. 3c. The shake-up satellite peaks located at 941.2 eV, 944.1 eV were ascribed to Cu2+ (Fig. S1†).43,44 From systematic characterization, it could be concluded that the dendritic nanostructure was heterogeneously built up with Cu/CuO stem and Au branches.
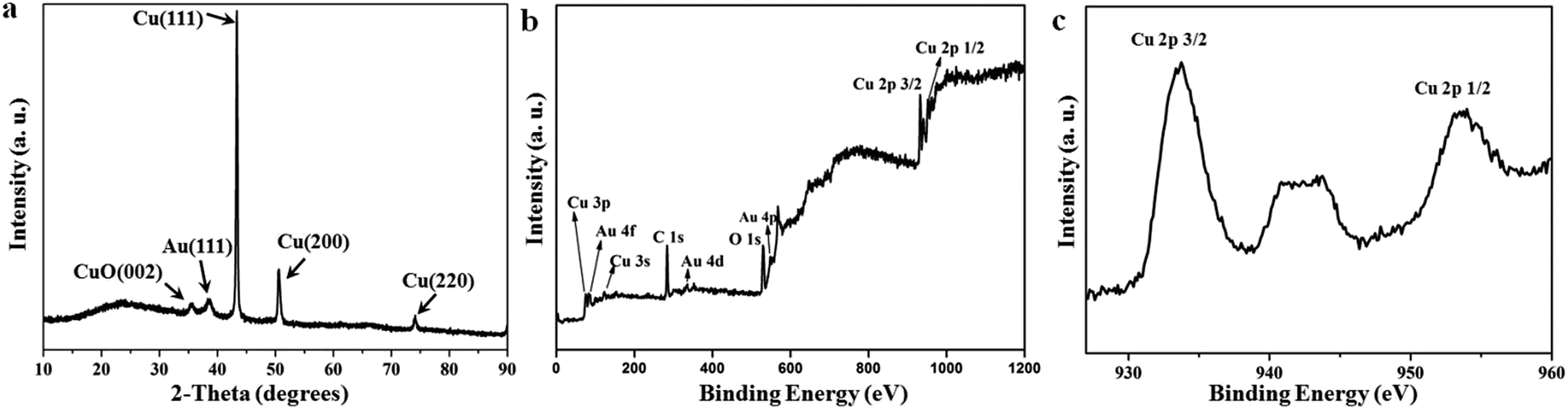 | ||
| Fig. 3 (a) XRD patterns and (b and c) XPS of the as-synthesized Cu–Au dendritic NWs. | ||
To clarify the formation mechanism of the Cu–Au dendritic NWs, we further re-studied the process of synthesis. The whole process involved heating pre-synthesized Cu NWs and Au NWs in the mixture of THF and TritonX-100 aqueous solution. Therefore, in the absence of Cu and Au precursors, capping agent induced anisotropic growth was not applicable to these dendritic structures. In addition, evidence also showed that synthesis process did not request for a particular capping agent, as the dendritic nanostructures could be obtained if a wide range of surfactants were applied, including anionic surfactant sodium dodecyl sulfate (SDS), polymer surfactant poly acrylic acid, and neutralize surfactant PVP and TritonX-100 (Fig. S2†).
Subsequently, in order to understand the mechanism, we separated the process into two steps and tried to address two issues: (1) dendritic structure formation, and (2) Cu–Au heterogeneous formation respectively.
For the first issue, dendritic structures were attributed to oxidation of Cu during phase transition process. We found that without addition of Au NWs, heating Cu NWs in the mixture of THF and TritonX-100 aqueous solution also gave dendritic structures. The obtained dendritic NWs have similar morphology and size distribution with Cu–Au dendritic NWs (as shown in Fig. 4a). As the heating process only involved accelerated oxidation of Cu (Cu nanostructures could be easily oxidized when exposed to air due to the high reactivity of Cu toward oxygen), the resulted dendritic structure must be originated from oxidization of Cu NWs during phase transition process. XRD pattern and XPS (Fig. S3†) confirmed the presence of CuO in comparison with as-synthesized Cu NWs, and suggested the dendritic structures were composed of Cu/CuO (Fig. S3†).
 | ||
| Fig. 4 (a) SEM image of typical Cu dendritic NWs with high-magnification of single Cu dendritic NW inserted. TEM images of as-synthesized products of Cu–Au heterogeneous nanostructures by using (b) 100 μL; (c) 300 μL; (d) 400 μL of ultrathin Au NWs. The typical hollowed Cu–Au dendritic NW was indicated in (c). | ||
Although the detailed mechanism of dendritic CuO formation was not identified, different Cu oxidation rate at diverse reaction sites and defects between hydrophilic CuO and hydrophobic Cu NW were probably the two key factors determining anisotropic growth.45
For the second issue, Cu–Au heterogeneous growth involved galvanic replacement reaction between Cu and Au. As shown in Fig. S4,† a small amount of Au+ and Au3+ were detected on Au NWs by XPS. This was consistent with the fact demonstrated by previous study that a little bit Au+/Au3+–oleylamine would remain on the AuNWs surface as ultrathin Au NWs formed from reduction of Au3+ by amine (oleylamine).46 When Cu NW and Au NW came to a collision during the phase transition (from THF to aqueous solution), trace amount of Au+ and Au3+ triggered galvanic replacement reaction, where Au+/Au3+ reacted with Cu to give Au and Cu2+. The galvanic reaction between Cu and Au ions created active site for Au NW anchoring. Therefore, the attachment of Au NWs to Cu NW surface could be through the process described above. Anchor sites for Au NWs could emerge in multitude throughout one single Cu NW. During phase transition, polarity change and heating of the solution could induce the break of ultrathin Au NWs and succeeding internanowire coalescence.46 Once Au NWs broke, the fragments could fuse to sharp corners in tandem with anchor sites formation. This proposal was strongly supported by the rare residual intermediates found in final products. Fig. S5† provided an example of the coexistence of trace remaining broken Au NW bundles and fused Au NWs along Cu–Au dendritic NWs. It could be clearly concluded that formation of Cu–Au dendritic NW started with Au–Cu inter-nanowire attachment driven by galvanic replacement, and followed by branches evolvement via Au NW self-deformation. Particularly, Au NWs were supposed to be parallel packed with Cu NWs after being transferred into aqueous solution (Fig. S6†), otherwise Au NWs could not uniformly distribute over the whole Cu NWs.
Amount of Au NWs in the system played a crucial role to affect the morphology of Cu–Au dendritic nanostructures. Cu consumption is proportional to Au NW amount. If the volume of Au NWs used was set to be constant, the more concentrated Au NW solution added, the more Cu should be replaced. Decreasing amount of Au NWs did not induced morphology changes for Cu–Au dendritic NWs (Fig. 4b). However, Cu–Au dendritic NWs with hollowed Cu stem were obtained when slightly increasing the amount of Au NWs from 200 μL to 300 μL (Fig. 4c). Further increment of Au NW amount led to expansion of the hollow part, until Cu–Au irregular nanoparticles (Fig. 4d) instead of dendritic nanostructures appeared as the highly hollowed stem could not support the overloaded branches.
Previous studies have demonstrated that Au or Cu dendritic nanostructures could provide high electrochemical active materials to fabricate sensor electrode for glucose.47 Therefore, our Cu–Au dendritic NWs could be a promising active material for electrochemical sensing of glucose. As a proof-of-concept application, a Cu–Au dendritic NW modified GC electrode together with other material (Au NWs, Cu NWs or Cu dendritic NWs) modified electrodes were prepared (see details in Experimental Section) and electro-oxidation ability of glucose on each electrode was studied. The cyclic voltammetry (CVs) were recorded to examine the catalytic activity of these electrodes in 0.1 M NaOH at 50 mV s−1. No obvious current response in the absence of glucose for all modified electrodes in the voltage window between −0.2–0.4 V (Fig. 5a). After glucose (10 mM) was added, no obvious changes happened to other electrodes, except that the oxidation peak appeared around 0.2 V when applied to Cu–Au dendritic NWs modified electrode (Fig. 5b). According to above research (Fig. S3†), Cu dendritic NWs are primarily composed of CuO, whose conductivity is relatively weaker than original NWs. Therefore, the current of Cu NWs is higher than Cu dendritic NWs at the 0.2 V (Fig. 5b). The results showed that Au NWs, Cu NWs, Cu dendritic NWs modified electrodes were all less active than the Cu–Au NWs modified one, suggesting Cu–Au dendritic NWs have high electrochemical sensitivity in glucose detection. From above results, obviously, the enhanced performances must attribute to the Cu–Au heterogeneous dendritic structure rendered large surface areas, as well as the polarization provided by Au branches.48
 | ||
| Fig. 5 CVs for different nanomaterials modified electrodes (a) in the absence of and (b) in the presence of 10 mM glucose in 0.1 M NaOH solution at a scan rate of 50 mV s−1. (c) Typical amperometric response of Cu–Au dendritic NWs modified electrode to successive addition of glucose into 0.1 M NaOH, with applied potential of 0.2 V. (d) Chronoamperometric current vs. glucose concentration plot for Cu–Au dendritic NWs modified electrode. Linear fitting curve with different glucose concentrations was inserted in (d). | ||
We further investigated the potential usage of Cu–Au dendritic NWs as biosensor to detect the concentration of glucose by I–t technique. Fig. 5c showed amperometric responses at Cu–Au dendritic NWs modified electrode to successive additions of glucose at 0.2 V. Upon addition of glucose into the 0.1 M NaOH solution, Cu–Au dendritic NWs modified electrodes could respond fast (within 2 s) and provide steady state signals within 2 s. The corresponding catalytic current showed linear response in a wide linear range of 0.025–0.43 mM with the sensitivity of 32.18 μA mM−1 (see details in ESI†). Therefore, the Cu–Au dendritic NWs have shown significantly enhanced electrochemical activity, with potential application in electrochemical detection of glucose.
Conclusions
In summary, we developed an unconventional method for synthesis of Cu–Au dendritic NWs from pre-synthesized Cu NWs and Au ultrathin NWs. The oxidation of Cu to CuO during phase transition stage resulted anisotropic dendritic precursors. The galvanic replacement reaction between Cu and Au ions introduced Au/Cu anchor sites, which allowed Au NWs to form branches on the surface of dendritic precursors via NW self-deformation. The obtained Cu–Au dendritic NWs showed enhanced electrochemical activity for oxidation of glucose owing to their unique dendritic morphology and the uniformly distributed Au branches in such hierarchical structures. It is expected that the excellent activity of Cu–Au dendritic NWs in glucose detection making it potentially applied as glucose biosensor.Acknowledgements
This study is financially supported by National Nature Science Foundation of China (21401153), and the Fundamental Research Funds for the Central Universities (20720150016, 20720150017).References
- X. Lu, M. S. Yavuz, H.-Y. Tuan, B. A. Korgel and Y. Xia, J. Am. Chem. Soc., 2008, 130, 8900–8901 CrossRef CAS PubMed.
- C. Wang, Y. Hu, C. M. Lieber and S. Sun, J. Am. Chem. Soc., 2008, 130, 8902–8903 CrossRef CAS PubMed.
- F. Kim, K. Sohn, J. Wu and J. Huang, J. Am. Chem. Soc., 2008, 130, 14442–14443 CrossRef CAS PubMed.
- J. Zhang, J. Du, B. Han, Z. Liu, T. Jiang and Z. Zhang, Angew. Chem., Int. Ed., 2006, 45, 1116–1119 CrossRef CAS PubMed.
- D. Zhang, R. Wang, M. Wen, D. Weng, X. Cui, J. Sun, H. Li and Y. Lu, J. Am. Chem. Soc., 2012, 134, 14283–14286 CrossRef CAS PubMed.
- S. Ye, A. R. Rathmell, I. E. Stewart, Y.-C. Ha, A. R. Wilson, Z. Chen and B. J. Wiley, Chem. Commun., 2014, 50, 2562–2564 RSC.
- H. Yan, R. He, J. Johnson, M. Law, R. J. Saykally and P. Yang, J. Am. Chem. Soc., 2003, 125, 4728–4729 CrossRef CAS PubMed.
- P. S. Kumar, I. Pastoriza-Santos, B. Rodriguez-Gonzalez, F. J. G. de Abajo and L. M. Liz-Marzan, Nanotechnology, 2008, 19, 015606 CrossRef PubMed.
- C. Gu and T.-Y. Zhang, Langmuir, 2008, 24, 12010–12016 CrossRef CAS PubMed.
- M. Zhu and G. Diao, Nanoscale, 2011, 3, 2748–2767 RSC.
- S. S. Li, J. N. Zheng, X. H. Ma, Y. Y. Hu, A. J. Wang, J. R. Chen and J. J. Feng, Nanoscale, 2014, 6, 5708–5713 RSC.
- M. D. Ye, M. Y. Wang, D. J. Zheng, N. Zhang, C. J. Lin and Z. Q. Lin, Nanoscale, 2014, 6, 3576–3584 RSC.
- M. H. Rashid and T. K. Mandal, J. Phys. Chem. C, 2007, 111, 16750–16760 CAS.
- W. Hong, J. Wang and E. Wang, Small, 2014, 10, 3262–3265 CrossRef CAS PubMed.
- Y. Qin, Y. Song, N. Sun, N. Zhao, M. Li and L. Qi, Chem. Mater., 2008, 20, 3965–3972 CrossRef CAS.
- S. Lai, C. Fu, Y. Chen, X. Yu, X. Lai, C. Ye and J. Hu, J. Power Sources, 2015, 274, 604–610 CrossRef CAS.
- J. Khandare and M. Calderon, Nanoscale, 2015, 7, 3806–3807 RSC.
- M. Cao, T. Liu, S. Gao, G. Sun, X. Wu, C. Hu and Z. L. Wang, Angew. Chem., Int. Ed., 2005, 44, 4197–4201 CrossRef CAS PubMed.
- P. Sajanlal and T. Pradeep, Nano Res., 2009, 2, 306–320 CrossRef CAS.
- W. Ren, S. Guo, S. Dong and E. Wang, J. Phys. Chem. C, 2011, 115, 10315–10320 CAS.
- R. Qiu, H. G. Cha, H. B. Noh, Y. B. Shim, X. L. Zhang, R. Qiao, D. Zhang, Y. I. Kim, U. Pal and Y. S. Kang, J. Phys. Chem. C, 2009, 113, 15891–15896 CAS.
- A. Gutés, C. Carraro and R. Maboudian, J. Am. Chem. Soc., 2010, 132, 1476–1477 CrossRef PubMed.
- M. Guan, Z. Zhou, R. Duan, B. Du, X. Li, L. Liu and Q. Zhang, RSC Adv., 2015, 5, 26540–26542 RSC.
- S. Barbosa, A. Agrawal, L. Rodríguez-Lorenzo, I. Pastoriza-Santos, R. A. Alvarez-Puebla, A. Kornowski, H. Weller and L. M. Liz-Marzán, Langmuir, 2010, 26, 14943–14950 CrossRef CAS PubMed.
- T. Huang, F. Meng and L. Qi, Langmuir, 2010, 26, 7582–7589 CrossRef CAS PubMed.
- N. Pazos-Pérez, S. Barbosa, L. Rodríguez-Lorenzo, P. Aldeanueva-Potel, J. Pérez-Juste, I. Pastoriza-Santos, R. A. Alvarez-Puebla and L. M. Liz-Marzán, J. Phys. Chem. Lett., 2010, 1, 24–27 CrossRef PubMed.
- H. C. Shin, J. Dong and M. Liu, Adv. Mater., 2003, 15, 1610–1614 CrossRef CAS.
- W. Shao and G. Zangari, J. Phys. Chem. C, 2009, 113, 10097–10102 CAS.
- Z. Liu, Y. Chen and Y. Zheng, CrystEngComm, 2014, 16, 9054–9062 RSC.
- Z. Zhang, X. Shao, H. Yu, Y. Wang and M. Han, Chem. Mater., 2005, 17, 332–336 CrossRef CAS.
- X. Sun and M. Hagner, Langmuir, 2007, 23, 9147–9150 CrossRef CAS PubMed.
- M. Pan, S. Xing, T. Sun, W. Zhou, M. Sindoro, H. H. Teo, Q. Yan and H. Chen, Chem. Commun., 2010, 46, 7112–7114 RSC.
- X. Xu, J. Jia, X. Yang and S. Dong, Langmuir, 2010, 26, 7627–7631 CrossRef CAS PubMed.
- X. Zhang, G. Wang, W. Zhang, N. Hu, H. Wu and B. Fang, J. Phys. Chem. C, 2008, 112, 8856–8862 CAS.
- Y. Chen, S. Lai, S. Jiang, Y. Liu, C. Fu, A. Li, Y. Chen, X. Lai and J. Hu, Mater. Lett., 2015, 157, 15–18 CrossRef CAS.
- H.-l. Liu, F. Nosheen and X. Wang, Chem. Soc. Rev., 2015, 44, 3056–3078 RSC.
- H. You, S. Yang, B. Ding and H. Yang, Chem. Soc. Rev., 2013, 42, 2880–2904 RSC.
- J. Xu, H. Wang, C. Liu, Y. Yang, T. Chen, Y. Wang, F. Wang, X. Liu, B. Xing and H. Chen, J. Am. Chem. Soc., 2010, 132, 11920–11922 CrossRef CAS PubMed.
- H. Feng, Y. Yang, Y. You, G. Li, J. Guo, T. Yu, Z. Shen, T. Wu and B. Xing, Chem. Commun., 2009, 1984–1986 RSC.
- S. Li, Y. Chen, L. Huang and D. Pan, Inorg. Chem., 2014, 53, 4440–4444 CrossRef CAS PubMed.
- W. Zhao, L. Yang, Y. Yin and M. Jin, J. Mater. Chem. A, 2014, 2, 902–906 CAS.
- W. Chen, R. Yu, L. Li, A. Wang, Q. Peng and Y. Li, Angew. Chem., Int. Ed., 2010, 49, 2917–2921 CrossRef CAS PubMed.
- P. Liu and E. J. M. Hensen, J. Am. Chem. Soc., 2013, 135, 14032–14035 CrossRef CAS PubMed.
- I. Platzman, R. Brener, H. Haick and R. Tannenbaum, J. Phys. Chem. C, 2008, 112, 1101–1108 CAS.
- X. Jiang, T. Herricks and Y. Xia, Nano Lett., 2002, 2, 1333–1338 CrossRef CAS.
- J. Xu, Y. Wang, X. Qi, C. Liu, J. He, H. Zhang and H. Chen, Angew. Chem., Int. Ed., 2013, 52, 6019–6023 CrossRef CAS PubMed.
- J. Wang, Chem. Rev., 2008, 108, 814–825 CrossRef CAS PubMed.
- Y. Lee, M. A. Garcia, N. A. Frey Huls and S. Sun, Angew. Chem., Int. Ed., 2010, 49, 1271–1274 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra23362c |
| This journal is © The Royal Society of Chemistry 2016 |