
Phthalate tethered strategy: carbohydrate nitrile oxide cycloaddition to 12–15 member chiral macrocycles with alkenyl chain length controlled orientation of bridged isoxazolines†
Swapan Majumdar*a,
Jewel Hossaina,
Ramalingam Natarajanb,
Ashish K. Banerjeeb and
Dilip K. Maiti*c
aDepartment of Chemistry, Tripura University, Suryamaninagar 799 022, India. E-mail: smajumdar@tripurauniv.in; Fax: +91-381-2374802; Tel: +91-381-237-9070
bIndian Institute of Chemical Biology, 4, Raja S. C. Mullick Road, Kolkata 700 032, India
cDepartment of Chemistry, University of Calcutta, 92 A. P. C. Raod, Kolkata-700009, India. E-mail: dkmchem@caluniv.ac.in; Fax: +91-33-2351-9755; Tel: +91-33-2350-9937
First published on 3rd December 2015
Abstract
The highly selective synthesis of phthalate templated bridged isoxazoline macrocyclic lactones is demonstrated using readily accessible carbohydrate precursors via intramolecular nitrile oxide–alkene cycloaddition. The structures of the macrocycles were established by 2D NMR as well as X-ray diffraction study. The orientation of the bridged isoxazoline ring is dependent on the distance between the 1,3-dipole and dipolarophile. The macrocycles are amenable to further transformations to higher amino sugars by sequential removal of the phthalate template and reductive cleavage of the isoxazoline moiety.
Macrocyclic skeletons are well known as hosts in supramolecular chemistry in recognition of ionic and neutral molecules.1 The importance of macrocyclic structures, including nucleosides as antibacterial agents based on novel targets as gene and drug-delivery systems is well documented.2 Generally, the synthesis of medium and large ring compounds by conventional cyclization methods is difficult to achieve, and only recently has the introduction of efficient ring-closing metathesis led to the easier preparation of such rings. Therefore, the development of strategies for the synthesis of macrocycles remains an ever-important task for synthetic chemists. Of particular importance is the construction of chiral macrocyclic frameworks from chiral precursors, because chirality has a profound influence3 on the biological activity of drugs and related molecules. 1,3-Dipolar cycloaddition of nitrile oxides with olefins is one of the most powerful synthetic tools in organic synthesis4 due to the operational simplicity, atom-economic nature and above all the product cycloadducts are amenable to transformations that can lead to the introduction of extra functionalities. The intermolecular cycloaddition of nitrile oxides with olefins produces isoxazoline derivatives, whereas its intramolecular version furnishes bicyclic fused and/or bridged isoxazolines in a highly regio- and stereoselective fashion.5 The isoxazoline ring has been revealed to be a latent precursor for a variety of corresponding bi-functional compounds such as γ-amino alcohols and β-hydroxy-ketones by reductive cleavage of the N–O bond; therefore the isoxazoline heterocycles has been utilized for construction of natural products having such functionalities.6 In this context, 8 to 12-membered chiral oxa- and azacycles were successfully synthesized via tether or structural constrain based on carbohydrate/amino acid derived nitrile oxide–olefin cycloaddition.7 On the other hand, the amino sugars constitute integral components of many natural products and medicinally relevant compounds.8 They are also important compounds for antibiotic research,9 anticancer therapies,10 nucleic acid research11 and biopolymers.12 Higher amino sugars are interesting targets for synthetic, biological and pharmaceutical research because of their limited availability from natural sources.13 Thus, in order to investigate their biological function, the development of facile and adaptable routes to this class of compounds is of fundamental importance. Our recent research interest is to synthesize chiral isoxazoline derivatives 2 or 3 from the reaction between nitrile oxide (1) and allyl alcohol, because reductive cleavage of resultant cycloadduct isoxazoline leads to construction of important precursors for higher and branched amino sugars 4 or 5 (Scheme 1). It was reported in the literature that the intermolecular nitrile oxide cycloaddition between allyl alcohols and nitrile oxides 1 (R = Bn) or related nitrone produced mixtures of regio- and diastereomeric isoxazolines or isoxazolidines.14 Herein we envisaged that on installation of the phthalate moiety between the alkenyl and carbohydrate moieties, the intramolecular nitrile oxide cycloaddition (INOC) of this rigid phthalate tethered bearing carbohydrate nitrile oxide 6 is expected to construct isoxazoline fused chiral macrolide 7 in a highly regio- and diastereo-selective manner (Scheme 2).
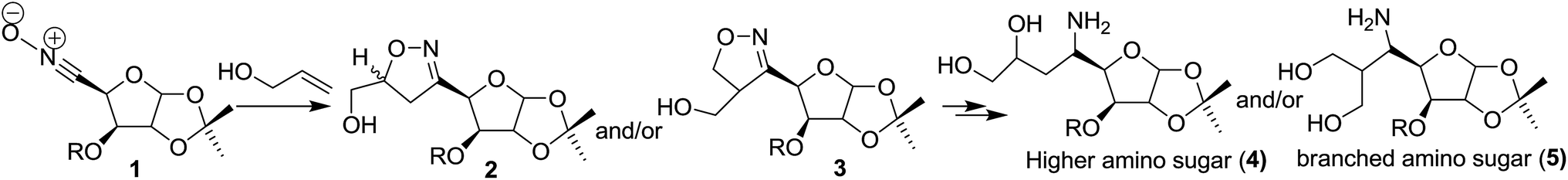 | ||
| Scheme 1 Possible route to higher amino sugar via intermolecular nitrile oxide cycloaddition. | ||
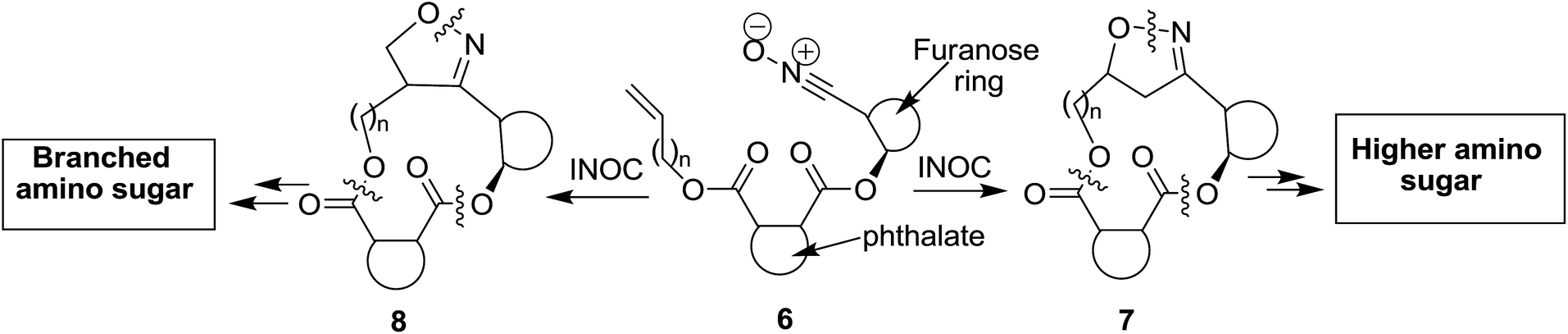 | ||
| Scheme 2 Phthalate tether strategy to chiral macrocycles and higher amino sugar via INOC. | ||
On removal of the tether group leads to exclusive formation of the bridged isoxazoline derivative 2. Of course possibility for the formation of isoxazoline fused macrolide 8 (Scheme 2) cannot be ruled out. The ring size of the core macrolide is dependent on the value of “n”, i.e. the number of intervening carbon atom(s) between olefin and the tether phthalate. To the best of our knowledge, the synthesis of chiral macro-heterocycles via phthalate tethered nitrile oxide cycloaddition and subsequent transformation to higher amino sugar remained unexplored. In this communication paper we describe the synthesis of 12–15 member enantiopure macrocycles based on intramolecular nitrile oxide cycloaddition (INOC) of phthalate tethered carbohydrate substrate and the subsequent transformation into amino sugar derivative.
The phthalate tethered carbohydrate scaffolds 11a–f (Scheme 3) were synthesized in two steps starting from 1,2:5,6-di-O-isopropylidene glucofuranoside (9a) or 1,2:5,6-di-O-isopropylidene allofuranoside (9b). The monocarboxylic acid 10a and 10b were synthesized through the reaction between 9a or 9b with phthalic anhydride in the presence of pyridine in DMF at 110 °C (Scheme 2). Alkylation of monocarboxylic acid 10a or 10b with allyl bromide in the presence of anhydrous K2CO3 in DMF afforded 11a, b, whereas higher analogues 11d–f were achieved from 10a using 4-bromo butene, 5-bromo pentene and 6-bromo hexene respectively. Compound 11c was synthesized through the amidation of 10a with N-allyl-N-benzyl amine using coupling agent EDCI and activator HOBt. The oxime derivatives 13a–f were obtained through sequential transformation of 11a–f involving selective removal15 of 5,6-acetonide moiety to corresponding diols 12a–f and sodium metaperiodate mediated oxidative carbon–carbon bond cleavage16 of the diols (12a–f) to aldehydes followed by treatment of hydroxylamine hydrochloride in the presence of pyridine in ethanol.7d The oxime derivatives 13a–f were found as a mixture of syn and anti-isomers as revealed by NMR spectra, while 1H and 13C NMR spectrum of 11c, 12c and 13c were found to be more complex due to the presence of tertiary amide moiety (rotomers).
 | ||
| Scheme 3 Reagents and condition: aphthalic anhydride, Py; balkenyl DMAP, bromide, K2CO3, DMF for 11a–b, d–f and N-allylbenzylamine, EDCI, HOBt, DCM for 11c, 61–66% (two steps); c75% aqueous acetic acid, rt or Py, HBIm·TFA, water, 70 °C, 1 h, 83–89%; dNaIO4, water–MeOH; eNH2OH·HCl, ethanol, reflux, 79–83% (two steps); f 4% NaOCl (aq.)–DCM, rt, overnight, 52–61%. | ||
The bridged isoxazoline 15a was afforded exclusively via the INOC of the nitrile oxide 14a on treatment of oxime 13a with 4% NaOCl and Et3N in dichloromethane17 as evident from the 1H and 13NMR spectral analyses. Completion of the cycloaddition reaction was monitored by disappearance of the 1H NMR signal of the olefinic proton through analyses of the reaction mixture containing product 15a. The presence of the bridged –CH2– group in the isoxazoline 15a was established by the appearance of two protons multiplet at δ 3.34–3.14 ppm in the 1H NMR spectrum as well as a high field methylene (confirmed by DEPT experiment) carbon signal at δ 36.6. The down field quaternary carbon peak at δ 154.6 in the 13C NMR spectrum of 15a confirm the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N. The structure of 15a was further corroborated by DEPT, 1H–1H COSY and NOESY experiments. In the NOESY spectrum no co-relations were found between either bridgehead proton, or bridged methylene protons with other pre-existing carbohydrate chiral centers. It is possible only if the orientation protons those are located at C-3/C-4 (these protons are α-oriented) and methylene group are opposite to each other. Finally the β-orientation of bridged methylene group was established by the single crystal X-ray diffraction analyses (panel A, Fig. 1).18a
N. The structure of 15a was further corroborated by DEPT, 1H–1H COSY and NOESY experiments. In the NOESY spectrum no co-relations were found between either bridgehead proton, or bridged methylene protons with other pre-existing carbohydrate chiral centers. It is possible only if the orientation protons those are located at C-3/C-4 (these protons are α-oriented) and methylene group are opposite to each other. Finally the β-orientation of bridged methylene group was established by the single crystal X-ray diffraction analyses (panel A, Fig. 1).18a
 | ||
| Fig. 1 Single crystal X-ray structures of 15a and 15f. | ||
A change in the stereochemistry at C-3 of the carbohydrate backbone did not influence changing the regioselectivity in the INOC of 14b, which was derived from oxime 13b on treatment of aqueous NaOCl. The 1H NMR pattern of 15b has shown close similarity to that of 15a. The bridged isoxazoline structure of 15b was consistent with the appearance of two sets of doublet of doublets at δ 3.35 (J = 17.4, 11.4 Hz) and δ 3.00 (J = 17.4, 3.0 Hz) in the proton NMR spectrum as well as a high field –CH2– peak at δ 38.3 and a quaternary carbon peak (CN) at δ 154.4 in the 13C NMR spectrum. The stereochemistry of the newly formed chiral center was established by NOESY experiment. A distinct cross peaks between C3–H and C6–HA and C6–HB strongly supported the α-orientation of bridged methylene moiety in 15b. These two observations revealed that the configuration at C-3 of the carbohydrate backbone had no effect in the controlling of regioselectivity in the cycloaddition process. The macrocycles 15a and 15b are diastereomeric because they were resulted from the cycloaddition of two epimeric nitrile oxides 14a and 14b.
In order to investigate whether a change of allyl ester by allyl amide could bring about any change in the regioselectivity of the cycloaddition, we have synthesized oxime 13c from 10a via coupling with N-allyl benzyl amine using EDCI, selective cleavage of 5,6-isopropylidene followed by oxidative cleavage to aldehyde and oxime formation with hydroxyl amine hydrochloride (Scheme 3). The resulting oxime 13c was subjected to react with NaOCl solution for the generation of nitrile oxide 14c, which again gave rise to 12-membered bridged isoxazoline 15c exclusively. The 1H and 13C NMR spectrum of 15c exhibits all the characteristics features of bridged isoxazoline observed for 15a and 15b. Appearance of two sets of double doublets at δ 3.89 (J = 18.6, 3.0 Hz) and 3.30 (J = 18.6, 12.0 Hz) in the proton NMR spectrum and a methylene carbon signal at δ 38.4 as well as quaternary carbon signal (CN) at δ 158.0 was a clear indication of the bridged isoxazoline nature of 15c. Like the NOESY experiments of 15a, here again we could not see any cross peaks, which could correlate either the bridgehead proton of bridged methylene protons with the protons of carbohydrate chiral centers at C-3 and C-4 (with respect to sugar skeleton) thus the configuration of bridged methylene is assigned as β-oriented. Due to the amorphous nature of the cycloadduct 15c it was not possible to study the structure by single crystal X-ray diffraction experiment. The yield (58–61%) found in these cycloadducts 15a-cis quite impressive considering the sizes of the 12-membered macrocyclic ring. It is worthy to mention that in all cases reactions are highly regio- and diastereoselective in nature, because a single product was isolated in each case.
After developing successful cyclisation approach for 12-membered macrocycles based on phthalate strategy, we turned our attention to higher homologues of these macrocycles through the chain elongation of the alkenyl moiety. Thus sugar derived substrate 11d–f were synthesized using higher homologue of allyl bromide as alkylating agent with subsequent selective de-protection, oxidative cleavage and oxime formation to obtain 13d–f (Scheme 3). The in situ generated sugar-based nitrile oxides 14d–f underwent smooth cyclisation to 13 to 15-membered bridged isoxazoline macrocycles 15d–f in a regio- and diastereoselective manner. The presence of bridged methylene (–CH2–) in 15d–f was established due to appearance of two sets of doublets with a very large coupling constant (Jgem > 16 Hz) in the 1H NMR and very high field methylene carbon signal in the 13C NMR spectrum of the cycloadduct. The stereochemistry of the newly formed chiral center i.e. the orientation of bridged methylene group of the amorphous compound 15d–e was established by NOESY experiment in a similar way that of 15a and 15c. But the stereochemistry of the bridged methylene in 15f was established as α-oriented on the basis of NOE experiment which is actually opposite to that of 15a and 15c–e. Strong NOE was found between protons of bridged –CH2– and the α-oriented protons in C-3 and C-4. Finally the structure of 15f was confirmed by single crystal X-ray analyses (panel B, Fig. 1).18b At present the reason behind this observation is unknown to us but, one possibility could be the flexibility of the longer alkenyl chain that changes the orientation of dipolarophile.
One of the important features of this macro cyclization strategy is that the rigid phthalate template can be removed from the macrocycles by base catalyzed hydrolysis to achieve the corresponding isoxazoline diols of biological importance. Thus, on treatment of 15a and 15f separately with aq. LiOH in water–dioxane at room temperature afforded the desired diols 16 and 17 in 81% and 79% yield respectively (Scheme 4). The isoxazoline moiety bearing diol 16 is a potential candidate for higher amino sugar because isoxazoline ring can easily be cleaved under reductive conditions19 to afford the corresponding amino-alcohol. Thus hydrogenolysis of 16 with H2–Pd/C in ethyl acetate followed by acetylation with acetic anhydride–pyridine in the presence of catalytic amount of DMAP furnished tetraacetate 18 (Scheme 4) with 72% yield as single isomer.
 | ||
| Scheme 4 Easy accesses to valuable bridged isoxazoline and higher amino sugars. | ||
In conclusion, the phthalate tethered INOC strategy described here presents a novel method for the synthesis of 12 to 15-membered chiral macrocycles having bridged isoxazoline moiety in a highly regio- and diastereoselective manner. The ring size and the stereochemistry of the newly created chiral center were established especially by 2D-NMR as well as X-ray crystallographic analyses of 15a and 15f. Removal of phthalate template and cleavage of isoxazoline ring provided an access to higher deoxy amino sugar of biological and pharmaceutical interest. The results from this innovative strategy open up a new avenue towards easy access to valuable chiral macrocycles of different ring sizes and higher amino sugars. Studies on the synthesis and DFT studies of large ring macro-heterocycles by varying the position of alkene with other heteroatoms are underway and will be reported in due course.
Acknowledgements
One of the authors (J.H.) is thankful to Department of Higher Education, Govt. of Tripura, India for his permission to pursue research work. We are also thankful to Dr T. Sarkar, Mr K. Sarkar and S. Chowdhury for recording NMR and mass spectral data respectively.References
- (a) J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VHC, Weinheim, 1995 Search PubMed; (b) C. J. Pedersen, Angew. Chem., 1988, 100, 1053 CrossRef CAS; (c) L. R. Nassimbeni, Acc. Chem. Res., 2003, 36, 631 CrossRef CAS PubMed; (d) J.-M. Lehn, in Separations and Reactions in Organic Supramolecular Chemistry, ed. F. Toda and R. Bishop, Wiley & Sons. Ltd., 2004 Search PubMed.
- (a) J. R. Horton, J. M. Bostock, I. Chopra, L. Hesse, S. E. V. Phillips, D. J. Adams, A. P. Johnson and C. W. G. Fishwick, Bioorg. Med. Chem. Lett., 2003, 13, 1557–1560 CrossRef CAS PubMed; (b) M.-L. Miramaon, N. Mignet and J. Herscovici, J. Org. Chem., 2004, 69, 6949–6952 CrossRef; (c) P. Busca, M. Etheve-Quelquejeu and J. M. Valery, Tetrahedron Lett., 2003, 44, 9131–9134 CrossRef CAS.
- (a) I. K. Reddy and R. Mehvar, in Chirality in Drug Design and Development,Marcel Dekker, Inc., New York, 1994 Search PubMed; (b) R. Crossley, Chirality and the Biological activity of Drugs, CRC Press, Boca Roton, 1995 Search PubMed; (c) I. Agranat, H. Canerand and J. Caldwell, Nat. Rev. Drug Discovery, 2002, 1, 753–768 CrossRef CAS PubMed; (d) N. M. Davies and X. V. Teng, Adv. Pharm., 2003, 1, 242–252 Search PubMed; (e) J. Patocka and A. Dvorak, J. Appl. Med., 2004, 2, 95–100 CAS.
- (a) A. Padwa, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, 1991, vol. 4, p. 1069 Search PubMed; (b) N. Chatterjee, P. Pandit, S. Halder, A. Patra and D. K. Maiti, J. Org. Chem., 2008, 73, 7775–7778 CrossRef CAS PubMed.
- (a) K. V. Gothelf and K. A. Jorgensen, Chem. Rev., 1998, 98, 863–910 CrossRef CAS PubMed; (b) M. Frederickson, Tetrahedron, 1997, 53, 403–425 CrossRef CAS; (c) H. M. I. Osborn, N. Gemmell and L. M. Harwood, J. Chem. Soc., Perkin Trans. 1, 2002, 2419–2438 RSC.
- (a) T. K. M. Shing, W.-C. Fung and C.-H. Wong, J. Chem. Soc., Chem. Commun., 1994, 449–450 RSC; (b) A. Kadowaki, Y. Nagata, H. Unob and A. Kamimurac, Tetrahedron Lett., 2007, 48, 1823–1825 CrossRef CAS; (c) S. Mukherjee, S. B. Mandal and A. Bhattacharjya, RSC Adv., 2012, 2, 8969–8978 RSC.
- (a) J. Sengupta, R. Mukhopadhyay, A. Bhattacharjya, M. M. Bhadbhade and G. V. Bhosekar, J. Org. Chem., 2005, 70, 8579–8582 CrossRef CAS PubMed; (b) Y. Liu, A. Maden and W. V. Murray, Tetrahedron, 2002, 58, 3159–3170 CrossRef CAS; (c) T. K. M. Shing and Y.-L. Zhong, Synlett, 2006, 1205–1208 CrossRef CAS; (d) S. Majumdar, R. Mukhopadhyay and A. Bhattacharjya, Tetrahedron, 2000, 56, 8945–8951 CrossRef CAS.
- (a) M. Chaterjee, P. J. Smith and C. A. Townsend, J. Am. Chem. Soc., 1996, 118, 1938–1948 CrossRef; (b) R. J. Ferrier, Carbohydr. Chem., 2006, 31, 123–141 Search PubMed; (c) E. J. Reist, R. R. Spencer and B. R. Baker, J. Am. Chem. Soc., 1960, 82, 2025–2029 CrossRef CAS; (d) X. M. Ji, J. Mo, H. Liu and H. P. Sun, Carbohydr. Res., 2006, 341, 2312–2320 CrossRef CAS PubMed; (e) S. Knapp, Chem. Soc. Rev., 1999, 28, 61–72 RSC; (f) J. J. Xie, Carbohydr. Res., 2003, 338, 399–406 CrossRef CAS PubMed; (g) L. Ermolenko, N. A. Sasaki and P. Potier, Tetrahedron Lett., 1999, 40, 5187–5190 CrossRef CAS.
- (a) S. F. Kuan, J. C. Byrd, C. Basbaum and Y. S. Kim, J. Biol. Chem., 1989, 264, 19271–19277 CAS; (b) J. Wang, J. Li, D. Tuttle, J. Y. Takemoto and C. W. T. Chang, Org. Lett., 2002, 4, 3997–4000 CrossRef CAS PubMed; (c) T. Nishiyama, M. Isobe and Y. Ichikawa, Angew. Chem., Int. Ed., 2005, 117, 4446–4449 CrossRef.
- (a) D. Kahne, C. Leimkuhler, W. Lu and C. Walsh, Chem. Rev., 2005, 105, 425–448 CrossRef CAS PubMed; (b) S. C. Timmons and J. S. Thorson, Curr. Opin. Chem. Biol., 2008, 12, 297–305 CrossRef CAS; (c) B. Wu, J. Y. He and E. E. Swayze, Org. Lett., 2002, 4, 3455–3458 CrossRef CAS PubMed.
- (a) B. R. Griffith, C. Krepel, X. Fu, S. Blanchard, A. Ahmed, C. E. Edmiston and J. S. Thorson, J. Am. Chem. Soc., 2007, 129, 8150–8155 CrossRef CAS PubMed; (b) R. SanMartin, B. Tavassoli, K. E. Walsh, D. S. Walter and T. Gallagher, Org. Lett., 2000, 2, 4051–4054 CrossRef CAS.
- M. J. Han, K. S. Yoo, Y. H. Kim and J. Y. Chang, Tetrahedron Lett., 2002, 43, 5597–5600 CrossRef CAS.
- (a) C. Liang, X. D. Zhang, K. F. Rubert and T. C. Balser, Biol. Fertil. Soils, 2007, 43, 631–639 CrossRef; (b) R. Benner and K. Kaiser, Limnol. Oceanogr., 2003, 48, 118–128 CrossRef CAS.
- (a) S. Chandrasekhar, C. L. Rao, C. Nagesh, C. R. Reddy and B. Sridhar, Tetrahedron Lett., 2007, 48, 5869–5873 CrossRef CAS; (b) R. C. Mishra, N. Tewari, S. S. Verma, R. P. Tripathi, M. Kumar and P. K. Shukla, J. Carbohydr. Chem., 2004, 23, 353–374 CrossRef CAS; (c) N. S. Karanjule, S. D. Markad, T. Sharma, S. G. Sabharwal, V. G. Puranik and D. D. Dhavale, J. Org. Chem., 2005, 70, 1356–1363 CrossRef CAS PubMed.
- (a) S. Majumdar, M. Chakraborty, D. K. Maiti, S. Chowdhury and J. Hossain, RSC Adv., 2014, 4, 16497–16502 RSC; (b) J. S. Yadav, M. C. Chander and K. K. Reddy, Tetrahedron Lett., 1992, 33, 135–138 CrossRef CAS; (c) K. H. Park, Y. J. Yoon and S. G. Lee, Tetrahedron Lett., 1994, 35, 9737–9740 CrossRef CAS; (d) K. S. Kim, Y. H. Song, B. H. Lee and C. S. Hahn, J. Org. Chem., 1986, 51, 404–407 CrossRef CAS.
- (a) S. Majumdar, A. Bhattacharjya and A. Patra, Tetrahedron, 1999, 55, 12157–12174 CrossRef CAS; (b) S. Tripathi, J. K. Maity, B. Achari and S. B. Mandal, Carbohydr. Res., 2005, 340, 1081–1087 CrossRef CAS PubMed; (c) Y.-L. Zhong and T. K. M. Shing, J. Org. Chem., 1997, 62, 2622–2624 CrossRef CAS PubMed.
- (a) S. Kanemasa, Y. Norisue, H. Suga and O. Tsuge, Bull. Chem. Soc. Jpn., 1988, 61, 3973–3982 CrossRef CAS; (b) B. Jiang, Y. Liu and W.-S. Zhou, J. Org. Chem., 2000, 65, 6231–6236 CrossRef CAS PubMed; (c) J. W. Bode and E. M. Carreira, Org. Lett., 2001, 3, 1587–1590 CrossRef CAS PubMed; (d) A. Fernandez-Mateos, G. P. Coca and R. Rubio Gonzalez, Tetrahedron, 2005, 61, 8699–8704 CrossRef CAS.
- (a) Crystallographic data for 15a have been deposited with the Cambridge Crystallographic Data Centre (CCDC) as supplementary publication number CCDC 1418502; (b) CCDC deposition number of 15f is 1418503.
- (a) S. Kanemasa and O. Tsuge, Heterocycles, 1990, 30, 719–736 CrossRef CAS; (b) D. P. Curran, J. Am. Chem. Soc., 1983, 105, 5826–5833 CrossRef CAS; (c) B. Cacciari, C. Caldari, S. Manfredini and G. Spalluto, Synthesis, 1994, 1158–1162 Search PubMed; (d) A. Barco, S. Benetti, C. De Risi, G. P. Pollini and V. Zanirato, Tetrahedron, 1995, 51, 7721–7726 CrossRef CAS; (e) J. R. Nagireddy, G. K. Tranmer, E. Carlson and W. Tam, Beilstein J. Org. Chem., 2014, 10, 2200–2205 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details experimental procedure, physical and spectroscopic characterization data,1H and 13CNMR spectra for all compounds and CIF files of 15a and 15f. CCDC 1418502 and 1418503. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra22436e |
| This journal is © The Royal Society of Chemistry 2015 |