DOI:
10.1039/C5RA21602H
(Paper)
RSC Adv., 2015,
5, 106044-106052
Effects of conformational symmetry in conjugated side chains on intermolecular packing of conjugated polymers and photovoltaic properties†
Received
16th October 2015
, Accepted 7th December 2015
First published on 8th December 2015
Abstract
Introducing conjugated side chains onto the backbone of two-dimensionally (2D) conjugated polymers has been utilized for tuning the optoelectronic characteristics of the polymer and the morphological properties of organic photovoltaics. To investigate the effects of conformational symmetry of conjugated side chains, two benzo[1,2-b:3,4-b′]dithiophene (BDT)-based derivatives, one with the asymmetric alkoxythienyl (Th) side chain and the other with the symmetric alkoxyphenyl (Ph) side chain, were synthesized as donor units and copolymerized with fluorinated benzothiadiazole (2FBT). These two side chains were selected for the distinct differences between their structures, and were found to affect the intrinsic characteristics of these BDT polymers. The introduction of the symmetric conjugated side chain to the conjugated backbone of the polymer was observed to improve both light harvesting and the charge carrier mobility, apparently by increasing the extent of packing between the polymer chains. Power conversion efficiency (PCE) values of photovoltaic devices fabricated using these conjugated polymers were strongly related to the light absorbance and crystallinity in a film of the blend of polymer and [6,6]-phenyl C71-butyric acid methyl ester (PC71BM). PBDT2FBT-Ph showed effective light absorption, an optimum morphology that we argue is due to the symmetry of the conjugated Ph side chain, and a maximum PCE of 6.23%, with an open-circuit voltage of 0.83 V, a short-circuit current density of 11.33 mA cm−2, and a fill factor of 66.3%. These results demonstrate that symmetric conjugated side chains are promising groups to produce 2D-conjugated polymers for high-performance photovoltaics. This systematic study of side chain engineering provides a valuable strategy to synthesize 2D conjugated polymers and to achieve high PCE values in organic photovoltaics.
1. Introduction
There has been significant progress in polymer solar cells (PSCs) based on a bulk heterojunction structure consisting of a conjugated polymer and a fullerene derivative as an electron donor and acceptor, respectively, and this progress has led to a rapid improvement in solar cell performance.1–3 Recently, organic solar cells comprised of this donor and acceptor have been shown to display a power conversion efficiency (PCE) exceeding 10%,4–7 and bulk heterojunction (BHJ) PSCs have thus become potential alternative renewable energy devices.8–10 Tremendous efforts have been devoted to optimizing and developing new materials for such cells, and consequently several rules for designing such materials have been proposed. Various design strategies have also been suggested for increasing the short-circuit current (Jsc), open-circuit voltage (Voc), and fill factor (FF) of PSCs since these three measures are highly associated with the PCE of the cell.11–13 The approach of designing donor–acceptor (D–A)-type conjugated polymers as the donor material, by focusing on developing new designs for the backbones of these conjugated polymers, has been considered as an effective way of using these polymers to achieve highly efficient organic photovoltaics.14–16,18 Moreover, attaching conjugated side chains to the backbone of the D–A polymer has been studied and considered as an important way to modify the polymer and hence tune its optoelectronic characteristics and the morphological properties of organic photovoltaics. Many recent studies have reported on the effects of such side chains, but have not fully provided systematic investigations to understand the correlations between the side chains of conjugated polymers and their photovoltaic performances. Conjugation originates from the overlap of π orbitals in alternating single and double bonds. Attaching conjugated side chains to the backbone increases the extent of the conjugation in directions perpendicular to that of the backbone and thereby broadens the π orbital on the polymer chain; such effects can in turn promote effective transport of charges between polymer chains. Specifically, the attachment of these side chains results in a two-dimensional (2D) conjugated polymer that can improve charge transport by increasing π–π overlap and thereby maximizing π–π interaction. When conjugated side chains are attached to a polymer backbone, the absorption peak in the ultraviolet and visible (UV-Vis) range becomes slightly red-shifted, the energy levels change slightly, and Jsc increases.19 Furthermore, Voc can be easily controlled by choosing appropriate side chains with specific electron affinities to affect the energy levels. However, the influences of the regularity of the symmetric conjugated side chains on the polymer backbone, and on the light absorption of the resulting 2D-conjugated structure, have not been studied sufficiently.
In this study, we synthesized two types of polymers in which BDT was the donating unit and fluorinated benzothiadiazole (2FBT)17 was accepting unit (Scheme 1). Then, we fabricated solar cells that incorporated one of two conjugated polymers as the donor material, and [6,6]-phenyl C71-butyric acid methyl ester (PC71BM) as the acceptor material. The donor polymers that we synthesized both included benzo[1,2-b:3,4-b′]dithiophene (BDT) backbones because BDT has been used broadly as a 2-D conjugated electron-donating moiety and because various side chains can be attached to it easily. We tested two types of side chains, the alkoxyphenyl (“Ph”) group and the alkoxythienyl (“Th”) group, in order to investigate the effects of conformational symmetry of conjugated side chains. These two side chains were also selected because of the distinct differences between their structures, which affect the intrinsic characteristics of the polymers. While the alkoxythienyl and alkoxyphenyl groups have similar structures with aromatic rings, the alkoxythienyl ring includes a sulfur atom and is asymmetric whereas the alkoxyphenyl ring lacks this sulfur atom and is symmetric. The effects of the conformational symmetry in these conjugated side groups on light absorption and conjugation were measured. The BDT–2FBT polymer with the alkoxyphenyl chain displayed high crystallinity as a result of effective polymer chain packing, apparently due to this polymer having a predominantly planar conformation. In the solar cell, the symmetric side chains induced intense light absorption and facilitated intramolecular and intermolecular charge transport, and thereby yielded a high Jsc. Increasing the symmetry of the substituents on the backbone resulted in an increase in the regioregularity of the conjugated backbone as well as a broadening of the absorption range and increase of the absorption intensity; these changes combined to increase the external quantum efficiency (EQE) values and Jsc.
 |
| | Scheme 1 Molecular design and synthesis of conjugated polymers. | |
2. Results and discussion
2.1. Design, synthesis and characterization
To investigate the effects of conformational symmetry in conjugated side chains on the photovoltaic properties of PSCs, two corresponding polymers based on alkoxythienyl- or alkoxyphenyl-substituted BDT were prepared according to a modification of a previously reported synthetic pathway. To enhance the solubilities of 2,6-bis(trimethyltin)-4,8-bis(5-(2-butyloctyloxy)thiophene-2-yl)benzo[1,2-b:4,5-b′]dithiophene (M1) and 2,6-bis(trimethyltin)-4,8-bis(4-butyloctyloxy-1-phenyl)-benzo[1,2-b:4,5-b′]-dithiophene (M2), a butyloctyl alkyl branched chain was attached to an alkoxyaromatic side chain (Scheme 2).18 We also used previously reported methods to synthesize 5,6-difluoro-4,7-bis(5-bromo-4-(2-ethylhexyl)-2-thienyl)-2,1,3-benzothiadiazole (M3).17 PBDT2FBT-Th and PBDT2FBT-Ph were polymerized by applying the Stille coupling polycondensation reaction. The detailed procedures for synthesizing the monomers are presented in the Experimental section. The resulting polymers were carefully purified through a Soxhlet extraction using methanol, acetone, hexane, and chloroform. Note that PBDT2FBT-Ph was finally extracted by using chlorobenzene (CB) because of its lower solubility. PBDT2FBT-Th showed good solubility at room temperature in chlorinated organic solvents such as chloroform, CB, and o-dichlorobenzene (DCB). However, PBDT2FBT-Ph was only moderately soluble in organic solvents such as CB and DCB. The molecular weights of the polymers were determined by using high-temperature gel permeation chromatography with DCB as the eluent and polystyrene as the standard. The average molecular weights of PBDT2FBT-Th, PBDT2FBT-Ph were 17.8 kDa and 14.1 kDa with polydispersity indices (PDIs) of 1.63 and 1.17, respectively. Although the two polymers showed opposite trends with regards to solubility and degree of aggregation, they did not differ much in molecular weight. The thermal stability of these two polymers was investigated using thermo-gravimetric analysis (TGA) (Fig. S11a†). Both PBDT2FBT-Th and PBDT2FBT-Ph exhibited good thermal stabilities, with onset decomposition temperatures (5% weight loss) at 302 °C and 406 °C in an inert atmosphere. Neither clear transitions nor endothermic or exothermic behaviors were observed between 30 °C and 250 °C in differential scanning calorimetry (DSC) experiments, and no melting was observed under an optical microscope upon heating the samples to 300 °C.
 |
| | Scheme 2 Synthetic routes of monomers. | |
2.2. Optical and electrochemical properties
Normalized optical UV-Vis absorption spectra of the polymers in CB solutions and solid films are shown in Fig. 1. The absorption spectra of the solutions of PBDT2FBT-Th and PBDT2FBT-Ph in CB were measured at RT. Both polymers yielded spectra featuring two absorption bands, one at a short wavelength (350 ≤ λ ≤ 450 nm), which originated from π–π* transitions, and another broad band at 550 ≤ λ ≤ 650 nm, caused by intramolecular charge transfer as has been observed for other D–A-type conjugated copolymers.20 In the solid state, the main absorption bands of the polymer films were red-shifted by 40–70 nm relative to those of the solutions, mainly due to an increased interaction between the polymer chains.21 The highest wavelengths of light absorbed by the polymers with thienyl and phenyl side chains on the backbone were, respectively, λ = 730 nm and λ = 723 nm, which correspond to 1.70 eV and 1.72 eV, respectively. Such variations of the band edges may have been affected by the electron affinities of the substituents in the side chains.22,23 Especially at 300 ≤ λ ≤ 500 nm, PBDT2FBT-Th showed broad indistinct peaks, apparently as a result of the short conjugation length in the fused ring attached to BDT, whereas PBDT2FBT-Ph showed distinct peaks in this wavelength range. We suggest that the difference between the absorption spectra of PBDT2FBT-Th and PBDT2FBT-Ph in the 300 to 500 nm wavelength range ultimately arose from the different extents of symmetry in their side groups. As indicated above, the hexagonal phenyl ring is symmetrical, but the five-member thienyl ring, which contains one sulfur, is asymmetric. The asymmetry of the thienyl ring should yield a greater diversity of conformations in the PBDT2FBT-Th repeating unit than the symmetrical phenyl side group would produce for the PBDT2FBT-Ph unit (Fig. 2). Moreover, for a polymer, i.e., with more than one repeating unit, the number of possible conformations increases exponentially, thus yielding that many more possible conformations for the PBDT2FBT-Th polymer than for the PBDT2FBT-Ph polymer. Such greater conformational diversity in the PBDT2FBT-Th polymer in turn explains the broadening of its absorption peaks in the short-wavelength range relative to those of PBDT2FBT-Ph.
 |
| | Fig. 1 Normalized UV-Vis absorption spectra of the polymers. (a) in DCB at a concentration of 0.025 g L−1. (b) Spin-coated thin films prepared by DCB. | |
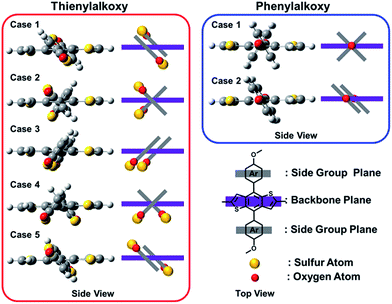 |
| | Fig. 2 Possible cases of conformations for thienylalkoxy side chains and phenylalkoxy side chains per a repeating unit of donor in polymer. | |
The solution spectra for both polymers showed shoulders for the peaks at long wavelengths, which are ascribed to strong aggregation related to polymer interaction. Such aggregation may in turn have been caused by planar backbone conformations leading to effective intermolecular interactions.24 Furthermore, PBDT2FBT-Ph showed a very strong vibronic structure next to the absorption maximum in the solid-state spectrum, while PBDT2FBT-Th produced a weak shoulder here. The weakness of this PBDT2FBT-Th shoulder may have been due to conformational distortion enhanced by intramolecular repulsion between the sulfur atoms in the side chains and in the neighboring BDT units, resulting in restricted intermolecular chain packing and enhanced solubility. Also, as outlined above, as the number of repeating units increase, so does the number of possible conformations of the polymer, especially for a polymer such as PBDT2FBT-Th for which the asymmetry of the side chain already yields conformational diversity in the repeating unit (Fig. 2). These factors taken together would appear to contribute to the decreased intermolecular packing for PBDT2FBT-Th. It is noteworthy that the measured absorption coefficient values of PBDT2FBT-Th were lower than those of PBDT2FBT-Ph for the entire UV-Vis range (Fig. S12†). The molar absorptivity also showed the same trend as did the absorption coefficients (Fig. S13†). This trend is ascribed to backbone tilting from intramolecular repulsion between the sulfur atoms in thienyl side chains and in the neighboring BDT units causing a decrease in the chain planarity and hence restricting intermolecular chain packing. In contrast, we suggest that the push–pull effect in PBDT2FBT-Ph is maximized by its maintaining a flattened plane along the donor and acceptor backbone to induce more effective charge transport than for the polymer with the thienyl side group. These proposals are supported by simulations using theoretical calculations to show that the dihedral angle between the donor and acceptor units is smaller in the phenyl-substituted polymer than in the thienyl-substituted polymer (Fig. 3). Consequently, formation of well-developed polymer crystals may be possible when the polymer has a phenyl side chain; and this good planarity apparently increased the intensities of the peaks in the UV-Vis absorption spectrum and may improve charge transport. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels of the two polymers (Fig. S11b†) were derived from electrochemical cyclic voltammetry (CV) plots obtained using a glassy carbon electrode (Table 1). The HOMO energy levels were −5.12 eV for PBDT2FBT-Th and −5.23 eV for PBDT2FBT-Ph. The introduction of the phenyl ring as the side group decreased the HOMO energy level compared to the introduction of the thienyl ring; this difference implies weaker electron donation in PBDT2FBT-Ph than in PBDT2FBT-Ph, consistent with the lower electron-donating propensity of the phenyl ring compared to that of the thienyl ring.25 Clearly, a substituent can affect the energy level of a polymer backbone, and replacing a thienyl moiety with a phenyl moiety in the side group would in general be expected to reduce the HOMO values of the resulting polymer. Such a reduction can increase the Voc in photovoltaic devices that use the polymer.
 |
| | Fig. 3 Calculated optimized geometries for trimers and energy-minimized structure (B3LYP/6-31G*) of the HOMO and LUMO of the model compounds, (a) PBDT2FBT-Th and (b) PBDT2FBT-Ph. The yellow, red, blue and sky blue symbols represent sulfur, oxygen, nitrogen, and fluorine atoms, respectively. | |
Table 1 Intrinsic properties of polymers
| Polymers |
Mn (kDa)/PDIa |
Td (°C) b |
λsolmaxc (nm) |
λfilmmaxd (nm) |
λfilmonsetd (nm) |
HOMO/LUMOe (eV) |
HOMO/LUMOf (eV) |
Eoptgg (eV) |
Ecalgh (eV) |
| Determined by GPC using polystyrene standards and DCB as eluent. 5% weight loss temperatures measured by TGA under nitrogen atmosphere. Measured in dilute chlorobenzene solution at a concentration of 0.05 g L−1. Spin-cast from 10 mg mL−1 chlorobenzene solution. Measured by cyclic voltammetry. Calculated by DFT. Estimated from the onset of the UV-Vis spectra measured from thin films. Calculated by DFT. |
| PBDT2FBT-Th |
17.8/1.63 |
302 |
569 |
608 |
730 |
−5.12/−3.42 |
−4.69/−2.82 |
1.70 |
1.87 |
| PBDT2FBT-Ph |
14.1/1.17 |
406 |
577 |
654 |
723 |
−5.23/−3.51 |
−4.71/−2.83 |
1.72 |
1.88 |
2.3. Theoretical calculations
To gain deeper insight into the effect of the symmetry in conjugated side chains on the molecular structure and electronic properties of the polymers, a theoretical calculation was performed (Fig. 3). Energy levels and gaps of the two model polymers were calculated using density functional theory (DFT) calculations. These calculations used a Gaussian simulation and a B3LYP/6-31G model,26–30 which employs dihedral angle data to explain the electronic structure and conformation of polymer backbone, and which therefore helps in our case determine the degree of planarity in the repeating units. The calculations were performed with optimized moieties and three repeating units (i.e., n = 3) to predict the energy levels, energy gaps, and conformations. The theoretically calculated energy levels and energy gaps of the model structures were larger than those determined experimentally, and this difference is attributed to the limitations of the DFT model for low-bandgap semiconducting polymers.31 Fig. 3 also shows a superposition of the HOMOs and LUMOs on the repeating units of the polymers to illustrate the difference in the electrochemical properties caused by the difference in the shapes of the conjugated side chains. The dihedral angle in the backbone bridge joint (θ2) between the BDT donating unit and the 2FBT accepting unit was 17.0° for the PBDT2FBT-Ph model but 20.7° for the PBDT2FBT-Th model, indicative of the backbone conformation induced by the phenyl side group being more planar than that induced by the thienyl side group. Interestingly, the bond angle difference between BDT unit and the 2FBT unit for both polymers was calculated to be relatively small, but as the number of monomers is increased, the rotation angle along the polymer backbone increases greatly. This result is different from the other results using only alkyl or alkoxy chains without any aromatic ring in the side chain. Generally, in such polymers, the alkyl chain length and type of side chain determine the orientation of the polymer chain, which strongly influences the intermolecular packing. In this way, side chains of polymers have a great influence on their crystalline properties because differences in the bulkiness and mobility of side chains can alter the intermolecular packing of conjugated backbones.32–34 To explain why the rotational angle (θ2) of PBDT2FBT-Th was found to be greater along the direction of chain growth than was that of PBDT2FBT-Ph (Fig. 3), model calculations were performed for the optimized conformation of the donor unit linked with side chains but without an acceptor unit (Fig. S14†). The planes of the thienyl chain and the phenyl chain showed similar dihedral angles of 53.4° and 58.9° with respect to the BDT plane. These results suggest that a 2D conjugated system with thienyl or phenyl side chains can affect the planarity of the BDT unit. In the model containing the thienyl ring, the sulfur atom of the thienyl ring was observed to be close to the sulfur atom or the carbon atom of the BDT, so avoiding steric hindrance and minimizing the repulsive forces can affect the optimized conformation. In contrast, the symmetrical phenyl side chain has no sulfur atom and was observed to be distant from the sulfur or the carbon of BDT with no tilt in the bond. Then, the fused ring plane for BDT is twisted differently by each side chains along the direction of minimized steric hindrance. The dihedral angles for the twisted BDT planes (θ4) in thienyl- and phenyl-containing polymers were measured to be 1.43° and 0.88°, respectively (Fig. S14†). The effect of conjugated side chains on the BDT unit was found to be related to polymer crystallinity, which is discussed below; this was inferred from UV-Vis data and supported by the calculated bond angle and planarity of the geometrically optimized structures. In addition, note that as the planarity of the polymer chain increased along the backbone direction, the push–pull effect between donor and acceptor units also increased – and this relationship may explain the peak intensity of the UV-Vis absorption of PBDT2FBT-Ph being measured higher than that of PBDT2FBT-Th.
2.4. Crystalline properties
To obtain an in-depth understanding of the relation of planarity and crystalline property upon the side chain effects in the thin films, atomic force microscopy (AFM) and grazing-incidence wide-angle X-ray scattering (GIWAXS) measurements were taken.35 As the coplanarity of the polymers increased, the crystalline features of the polymers in the pure polymer films became distinct (Fig. 4a). The out-of-plane (OOP) GIWAXS profile of PBDT2FBT-Ph film displayed pronounced lamellar stacking (h00) peaks at 0.27 Å−1, 0.52 Å−1 and 0.76 Å−1 arising from chain packing (Fig. 4b). In contrast, the OOP profile collected from the PBDT2FBT-Th film showed a weak lamellar stacking (h00) peak at 0.29 Å−1 and a pronounced π–π stacking (010) peak at 1.74 Å−1. The in-plane (IP) GIXAXS profile of the PBDT2FBT-Ph film displayed a pronounced lamellar stacking (h00) peak at 0.25 Å−1 and a pronounced π–π stacking (010) peak at 1.68 Å−1 (Fig. S15†). In contrast, the IP profile collected from the PBDT2FBT-Th film showed a weak lamellar stacking (h00) peak at 0.24 Å−1 (Fig. S15†). The GIWAXS pattern of PBDT2FBT-Ph exhibited a strong (010) diffraction peak and higher-order peaks at (200) and (300) due to the planarity of the polymer chain. We also prepared films of PBDT2FBT-Ph:PC71BM and PBDT2FBT-Th:PC71BM blends by using 1,8-diiodooctane (DIO), and investigated their crystalline properties as well. The PBDT2FBT-Ph:PC71BM film also yielded higher-order (200) and (300) peaks. Also, both polymers predominantly showed edge-on orientations and crystallinity in the neat polymer and the blend films. In the blend systems, PBDT2FBT-Th and PBDT2FBT-Ph yielded weak crystal diffraction peaks, indicating that the polymer and PC71BM mixed well. The relatively low intensity of the PCBM ring in the PBDT2FBT-Ph blend suggests that the fullerenes did not easily crystallize out during the film drying process, perhaps due to the low solubility of PBDT2FBT-Ph compared to that of PBDT2FBT-Th. When the polymers were mixed with fullerene, PBDT2FBT-Ph may not have fully dissolved, and thus the fullerenes may have dissolved well in the blend, hence yielding a weaker fullerene-derived crystalline peak in the blend film.36,37 GIWAXS analysis of PBDT2FBT-Ph in blend system exhibited prominent higher-order (h00) diffraction peaks that were attributed to effective intermolecular packing due to the enhanced planarity of the polymers compared to the case for PBDT2FBT-Th. The high crystallinity of PBDT2FBT-Ph was beneficial for charge transport in the photovoltaic devices. The corresponding AFM height images (shown in the insets of Fig. 4a) of the pure polymers showed RMS roughness values of 0.56 nm for PBDT2FBT-Th and 1.46 nm for PBDT2FBT-Ph. The greater roughness of PBDT2FBT-Ph was probably due to aggregation of this polymer caused by its low solubility.
 |
| | Fig. 4 (a) GIWAXS data of polymer films (top) homogeneous polymer and (bottom) polymers/PC71BM (DIO) blend films. (b) The corresponding 1D GIWAXS diffractogram profiles along the out-of-plane direction. | |
Hole mobility (μh) values of the polymers were investigated by using the space-charge-limited current (SCLC) method to measure their charge transport properties. Both μh and the electron mobility μe were lower for the PBDT2FBT-Th polymer than for the PBDT2FBT-Th polymer (Table 2). According to the UV-Vis results and DFT calculations, PBDT2FBT-Ph has greater planarity than PBDT2FBT-Th in the backbone chain, which would yield faster charge transport in the PBDT2FBT-Ph chain. This analysis is consistent with the relatively high crystallinity of PBDT2FBT-Ph in both the homogeneous polymer and blend films according to the high-order GIWAXS peaks described above. The relative degree of crystallinity (rDoC) of the PBDT2FBT crystals in the two polymer–fullerene blend films were calculated and compared, and showed more than two-fold greater crystallinity of PBDT2FBT in the PBDT2FBT-Ph:PC71BM blend film (0.65) than in the PBDT2FBT-Th:PC71BM blend film (0.29).38 This result indicates that high crystallinity resulting from effective intermolecular packing via chain–chain interactions with a planar structure helped to increase μh.39,40
Table 2 SCLC results measured by hole-only device with ITO/MoO3 (9 nm)/PBDT2FBTs:PC71BM/MoO3 (9 nm)/Ag (100 nm) architecture
| Polymers |
Hole mobility μh (cm2 V−1 s−1) |
Electron mobility μe (cm2 V−1 s−1) |
μh/μe |
| PBDT2FBT-Th |
3.91 × 10−5 |
5.01 × 10−4 |
0.078 |
| PBDT2FBT-Ph |
2.50 × 10−4 |
5.64 × 10−4 |
0.44 |
2.5. Photovoltaic performances
Photovoltaic devices were fabricated with PBDT2FBTs as the electron donor and PC71BM as the electron acceptor. The device architecture was ITO/MoO3/polymers![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PC71BM/LiF/Al, and the device area was 0.055 cm2. The performance of the organic solar cells composed of these polymers was very sensitive to the film morphology, so the solvents were selected according to how well they dissolved the two polymers. PBDT2FBT-Th has good solubility in common organic solvents, including CF, CB, and DCB whereas PBDT2FBT-Ph dissolves only at high temperatures in organic solvents. This relatively poor solubility limits the ability to process PBDT2FBT-Ph; hence DCB was chosen the solvent because of its high boiling point. Current–voltage (J–V) characteristics (Fig. 5a) of the devices were obtained under AM 1.5 illumination conditions with an intensity of 100 mW cm−2. To optimize the device performance, we fabricated these devices under various weight ratios and thicknesses of polymers/PC71BM using DIO as a solvent additive.41 The corresponding Jsc, Voc, FF and PCEs values derived from the J–V curves and all of the relevant parameters are summarized in Table 3. The photovoltaic performances of the devices depended significantly on which polymer was included, and hence on the type of side chain. The best device performance was obtained for a 1:1.2 weight ratio of PBDT2FBT-Ph to PC71BM that was spin cast using DCB with DIO; this device yielded a Jsc = 11.33 mA cm−2, FF = 66.3%, Voc = 0.83 V, and PCE = 6.23%. A lower device performance was measured for a 1:1.2 weight ratio of PBDT2FBT-Th to PC71BM spin cast using DCB with DIO; this device displayed a Jsc = 8.61 mA cm−2, FF = 42.6%, Voc = 0.82 V, and PCE = 3.04%. Also note that inclusion of DIO increased the PCEs of both devices, and these increases appear to be closely related to morphological changes in their photoactive layers and improved contact between their active layers and electrodes.42–45 Just as PBDT2FBT-Ph showed a lower HOMO energy level than did PBDT2FBT-Th according to the CV experiments, the Voc of the device with PBDT2FBT-Ph was higher than that of PBDT2FBT-Th. These two devices fabricated under optimized conditions showed different EQEs in the range 300 ≤ λ ≤ 700 nm. The device with PBDT2FBT-Ph exhibited higher EQE peaks than the device with PBDT2FBT-Th throughout this range of wavelengths (Fig. 5b). The device with PBDT2FBT-Ph achieved a maximum EQE of more than 60% (at λ = 500 nm), and therefore displayed a higher Jsc than did the device with PBDT2FBT-Th. The Jsc and FF values of the PBDT2FBT-Ph-based devices were found to be much higher than those of the PBDT2FBT-Th-based devices, which is attributed to differences between their light absorption strengths, charge carrier mobilities, and morphological structures of the active layers. To understand the photovoltaic properties of the two polymers, the morphologies of films of blends of each of the polymers with PC70BM were investigated using tapping-mode AFM and TEM (Fig. 6a and b and S16†). All such films were prepared by spin coating their DCB solution on top of a silicon wafer. Note that a phase separation occurred at the surface of the PBDT2FBT-Th:PC71BM film, which was caused by the intermolecular interaction between PBDT2FBT-Th and PC71BM during the formation of this film. The roughness of the blend films was also increased by addition of DIO additive (Fig. 6c and d and S16†). The bi-continuous interpenetrating networks of the PBDT2FBT-Th:PC71BM and PBDT2FBT-Ph:PC71BM blends prepared with DIO produced efficient exciton diffusion, dissociation, and charge transport.38–42 Increased surface roughness can increase internal light scattering and enhance light absorption (Fig. S12†). TEM images showed fibrillar structures in the PBDT2FBT-Ph:PC71BM blend systems. These fibrils arose from the highly crystalline features of the PBDT2FBT-Ph polymer indicated by the GIWAXS measurements (Fig. 4). Both the PBDT2FBT-Th:PC71BM and PBDT2FBT-Ph:PC71BM blend systems prepared with DIO yielded TEM images that revealed more distinct and dense nanofibrillar domains compared with the corresponding systems prepared without DIO (Fig. 6c and d). The strongly developed fibrillar structures of PBDT2FBT-Ph in the blend film enhanced the charge transport.33,46 The blend film containing PBDT2FBT-Th showed a relatively large phase separation on the micrometer length scale, as indicated by a bright region with a darker PC71BM region. In contrast, for the film containing PBDT2FBT-Ph and PC71BM, these two components were well distributed and showed an interpenetrated in fibril shapes, and this film displayed slightly longer wire-like forms and clearer shapes than did the blend film containing PBDT2FBT-Th. This fibril structure is desirable because it fosters efficient exciton diffusion and exciton dissociation, and thereby leads to increases in Jsc and FF.47 The increased efficiency of the PBDT2FBT-Ph blend may be attributed due to the presence of wire-like networks in the film.
:PC71BM/LiF/Al, and the device area was 0.055 cm2. The performance of the organic solar cells composed of these polymers was very sensitive to the film morphology, so the solvents were selected according to how well they dissolved the two polymers. PBDT2FBT-Th has good solubility in common organic solvents, including CF, CB, and DCB whereas PBDT2FBT-Ph dissolves only at high temperatures in organic solvents. This relatively poor solubility limits the ability to process PBDT2FBT-Ph; hence DCB was chosen the solvent because of its high boiling point. Current–voltage (J–V) characteristics (Fig. 5a) of the devices were obtained under AM 1.5 illumination conditions with an intensity of 100 mW cm−2. To optimize the device performance, we fabricated these devices under various weight ratios and thicknesses of polymers/PC71BM using DIO as a solvent additive.41 The corresponding Jsc, Voc, FF and PCEs values derived from the J–V curves and all of the relevant parameters are summarized in Table 3. The photovoltaic performances of the devices depended significantly on which polymer was included, and hence on the type of side chain. The best device performance was obtained for a 1:1.2 weight ratio of PBDT2FBT-Ph to PC71BM that was spin cast using DCB with DIO; this device yielded a Jsc = 11.33 mA cm−2, FF = 66.3%, Voc = 0.83 V, and PCE = 6.23%. A lower device performance was measured for a 1:1.2 weight ratio of PBDT2FBT-Th to PC71BM spin cast using DCB with DIO; this device displayed a Jsc = 8.61 mA cm−2, FF = 42.6%, Voc = 0.82 V, and PCE = 3.04%. Also note that inclusion of DIO increased the PCEs of both devices, and these increases appear to be closely related to morphological changes in their photoactive layers and improved contact between their active layers and electrodes.42–45 Just as PBDT2FBT-Ph showed a lower HOMO energy level than did PBDT2FBT-Th according to the CV experiments, the Voc of the device with PBDT2FBT-Ph was higher than that of PBDT2FBT-Th. These two devices fabricated under optimized conditions showed different EQEs in the range 300 ≤ λ ≤ 700 nm. The device with PBDT2FBT-Ph exhibited higher EQE peaks than the device with PBDT2FBT-Th throughout this range of wavelengths (Fig. 5b). The device with PBDT2FBT-Ph achieved a maximum EQE of more than 60% (at λ = 500 nm), and therefore displayed a higher Jsc than did the device with PBDT2FBT-Th. The Jsc and FF values of the PBDT2FBT-Ph-based devices were found to be much higher than those of the PBDT2FBT-Th-based devices, which is attributed to differences between their light absorption strengths, charge carrier mobilities, and morphological structures of the active layers. To understand the photovoltaic properties of the two polymers, the morphologies of films of blends of each of the polymers with PC70BM were investigated using tapping-mode AFM and TEM (Fig. 6a and b and S16†). All such films were prepared by spin coating their DCB solution on top of a silicon wafer. Note that a phase separation occurred at the surface of the PBDT2FBT-Th:PC71BM film, which was caused by the intermolecular interaction between PBDT2FBT-Th and PC71BM during the formation of this film. The roughness of the blend films was also increased by addition of DIO additive (Fig. 6c and d and S16†). The bi-continuous interpenetrating networks of the PBDT2FBT-Th:PC71BM and PBDT2FBT-Ph:PC71BM blends prepared with DIO produced efficient exciton diffusion, dissociation, and charge transport.38–42 Increased surface roughness can increase internal light scattering and enhance light absorption (Fig. S12†). TEM images showed fibrillar structures in the PBDT2FBT-Ph:PC71BM blend systems. These fibrils arose from the highly crystalline features of the PBDT2FBT-Ph polymer indicated by the GIWAXS measurements (Fig. 4). Both the PBDT2FBT-Th:PC71BM and PBDT2FBT-Ph:PC71BM blend systems prepared with DIO yielded TEM images that revealed more distinct and dense nanofibrillar domains compared with the corresponding systems prepared without DIO (Fig. 6c and d). The strongly developed fibrillar structures of PBDT2FBT-Ph in the blend film enhanced the charge transport.33,46 The blend film containing PBDT2FBT-Th showed a relatively large phase separation on the micrometer length scale, as indicated by a bright region with a darker PC71BM region. In contrast, for the film containing PBDT2FBT-Ph and PC71BM, these two components were well distributed and showed an interpenetrated in fibril shapes, and this film displayed slightly longer wire-like forms and clearer shapes than did the blend film containing PBDT2FBT-Th. This fibril structure is desirable because it fosters efficient exciton diffusion and exciton dissociation, and thereby leads to increases in Jsc and FF.47 The increased efficiency of the PBDT2FBT-Ph blend may be attributed due to the presence of wire-like networks in the film.
 |
| | Fig. 5 (a) J–V characteristics of PBDT2FBT polymers/PC71BM solar cell prepared by DCB under illumination of AM 1.5G, 100 mW cm−2 and (b) EQE curves of the corresponding polymer solar cell. | |
Table 3 Performance metrics are average numbers from eight devices of each type under the illumination condition of AM 1.5G, 100 mW cm−2. Weight ratio of polymer:PC71BM was 1:1.2. DIO was added 1.5 v%
| Polymers |
Solvent additive |
Voc (V) |
Jsc (mA cm−2) |
FF (%) |
PCEmax (avg.) (%) |
| PBDT2FBT-Th |
Without |
0.82 |
8.28 |
39.1 |
2.69 (2.45) |
| DIO |
0.82 |
8.61 |
42.6 |
3.04 (2.98) |
| PBDT2FBT-Ph |
Without |
0.81 |
10.08 |
40.6 |
3.31 (3.19) |
| DIO |
0.83 |
11.33 |
66.3 |
6.23 (6.02) |
 |
| | Fig. 6 (a and b) AFM height images and TEM images of blend films prepared by DCB for PBDT2FBT-Th:PC71BM and PBDT2FBT-Ph:PC71BM. (c and d) AFM height images and TEM images of blend films prepared by DCB with DIO for PBDT2FBT-Th:PC71BM and PBDT2FBT-Ph:PC71BM. | |
3. Conclusions
In summary, two polymers, PBDT2FBT-Th and PBDT2FBT-Ph, with asymmetric and symmetric conjugated side chains, respectively, have been designed and synthesized. We compared these two conjugated polymers to study how the conformational symmetry of a conjugated side chain attached to a polymer backbone based on BDT–2FBT affects the optoelectronic and relevant photovoltaic properties of this polymer. The side chains were the asymmetric alkoxythienyl group and the symmetric alkoxyphenyl group. The optoelectronic characteristics of the two polymers, including hole mobility and light absorption, critically influenced their photovoltaic performances. Introduction of the symmetric conjugated side chain to the conjugated backbone of the polymer was found to improve both the light-harvesting ability of the polymer and its charge carrier mobility, apparently by increasing the packing between the polymer chains. The asymmetric thienyl side chain caused a dent in the fused ring and torsion in the backbone due to steric hindrance between neighboring atoms. The structure with the symmetric phenyl side chain attached to the backbone absorbed more light over a wider wavelength range than did the structure with the asymmetric thienyl chain. Compared to the polymer containing the thienyl side chains, that containing the phenyl side chains displayed higher crystallinity, probably due to the more planar structure of this polymer and its more extensive intermolecular packing. This higher crystallinity apparently resulted in a higher absorption coefficient and a well-formed wire-like morphology in a film of a blend of this polymer with PC71BM. Moreover, the Jsc and EQE values of the structure containing the phenyl side chains were also found to be much higher than those of the structure containing the thienyl side chain, and yielded a higher photovoltaic efficiency that was measured as 6.23%. Based on our results, we suggest that introduction of symmetrical side chains onto a polymer backbone can increase the amount of light absorbed by the molecule by generating a more regular conformation. Such an introduction can also, by promoting a planar chain, induce effective intermolecular packing. This strategy may help improve the performance of optoelectronic devices.
4. Experimental section
4.1. Materials
All materials including chemicals and solvents from commercial sources were used without further purification. 2-(2-Butyloctyloxy)thiophene, and 5,6-difluoro-4,7-bis(5-bromo-4-(2-ethylhexyl)-2-thienyl)-2,1,3-benzothiadiazole, were synthesized according to modified literature procedures. To probe the effects of symmetry in side groups attached to the polymer backbone, two polymers, i.e., alkoxythienyl-substituted and alkoxyphenyl-substituted benzo[1,2-b:3,4-b′]dithiophene (BDT) polymers, were synthesized as shown in Scheme 2. Experimental details of this synthesis and of the synthesis of the monomers are described in ESI.†
4.2. Synthesis of polymers
PBDT2FBT-Th. M1 (321.7 mg, 0.3 mmol) and M3 (220.4 mg, 0.3 mmol) in 9 mL of anhydrous toluene and 1 mL of DMF were put into a two-necked flask. The solution was refluxed at 110 °C for 48 h under nitrogen, followed by end-capping using 2-tributyltin thiophene and then 2-bromothiophene. After being allowed to cool down to room temperature, the solution was quenched in methanol, filtered, and then purified by using the Soxhlet extraction successively with methanol, acetone, hexane, chloroform and CB. Finally, the CB fraction was concentrated, and then the concentrate was precipitated in methanol. A dark blue solid was obtained after filtering and dried overnight with a yield of 67%.
PBDT2FBT-Ph. M2 (311.0 mg, 0.3 mmol) and M3 (215.6 mg, 0.3 mmol) in 9 mL of anhydrous toluene and 1 mL of DMF were put into a two-necked flask. The solution was refluxed at 110 °C for 48 h under nitrogen, followed by end-capping using 2-tributyltinthiophene and then 2-bromothiophene. After being allowed to cool down to room temperature, the solution was quenched in methanol, filtered, and then purified by using the Soxhlet extraction successively with methanol, acetone, hexane, chloroform and CB. Finally, the CB fraction was concentrated, and then the concentrate was precipitated in methanol. A dark blue solid was obtained after filtering and dried overnight with a yield of 70%.
4.3. Fabrication and characterization of organic solar cells
ITO glass was used as the anodes and cleaned stepwise in an aqueous detergent solution, deionized water, isopropyl alcohol, and acetone. The cleaned ITO glass was then exposed to UV-ozone. PEDOT:PSS was spin-coated on this substrate and baked at 120 °C for 30 min in a convection oven. For making photovoltaic devices with an MoO3 interlayer, an MoO3 film (9 nm) was thermally evaporated at a rate of 0.1 nm s−1 on the UV-treated ITO substrates. PBDT2FBT-Th, PBDT2FBT-Ph, and PC71BM were separately dissolved in o-DCB and the blend solutions were kept at a high temperature (90 °C) for more than one day. All processes were carried out in a glovebox with nitrogen. All blend films had to be processed from hot solutions in order to prevent premature aggregation of the polymers. Polymer/PC71BM solutions were spin-coated onto MoO3 substrates, and then the films were left in a N2 atmosphere to dry. To deposit the electrodes, the samples were transferred into a vacuum chamber, and then LiF (0.6 nm)/Al (100 nm) were deposited. The current density–voltage (J–V) characteristics of the PSCs were measured using a source-measure unit (Keithley 4200) in the dark and under 100 mW cm−2 of AM 1.5 solar illumination in a N2-filled glove box. Light was generated with an Oriel 1 kW solar simulator referenced using a Reference Cell PVM 132 calibrated at the US National Renewable Energy Laboratory. A photomodulation spectroscopic set-up was used to measure the incident photon-to-current conversion efficiency as a function of the wavelength of the light.
Acknowledgements
This work was supported by a grant (Code No. 2011-0031628) from the Center for Advanced Soft Electronics under the Global Frontier Research Program of the Ministry of Science, ICT and Future Planning, Korea. The authors thank the Pohang Accelerator Laboratory for providing the synchrotron radiation sources at 3C, 9A beam lines used in this study.
References
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3–24 CrossRef CAS PubMed.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS.
- J.-D. Chen, C. Cui, Y.-Q. Li, L. Zhou, Q.-D. Ou, C. Li, Y. Li and J.-X. Tang, Adv. Mater., 2015, 27, 1035–1041 CrossRef CAS.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- S.-H. Liao, H.-J. Jhuo, P.-N. Yeh, Y.-S. Cheng, Y.-L. Li, Y.-H. Lee, S. Sharma and S.-A. Chen, Sci. Rep., 2014, 4, 6813 CrossRef CAS PubMed.
- T. L. Nguyen, H. Choi, S.-J. Ko, M. A. Uddin, B. Walker, S. Yum, J.-E. Jeong, M. H. Yun, T. J. Shin, S. Hwang, J. Y. Kim and H. Y. Woo, Energy Environ. Sci., 2014, 7, 3040–3051 CAS.
- S. B. Darling and F. You, RSC Adv., 2013, 3, 17633–17648 RSC.
- Y. Galagan, E. W. C. Coenen, B. Zimmermann, L. H. Slooff, W. J. H. Verhees, S. C. Veenstra, J. M. Kroon, M. Jørgensen, F. C. Krebs and R. Andriessen, Adv. Energy Mater., 2014, 4, 1300498 CrossRef.
- N. Espinosa, M. Hösel, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2014, 7, 855–866 CAS.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS.
- Y. Liang and L. Yu, Acc. Chem. Res., 2010, 43, 1227–1236 CrossRef CAS PubMed.
- D. Gendron and M. Leclerc, Energy Environ. Sci., 2011, 4, 1225–1237 CAS.
- I. Osaka, T. Kakara, N. Takemura, T. Koganezawa and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 8834–8837 CrossRef CAS.
- J. S. Kim, Z. Fei, S. Wood, D. T. James, M. Sim, K. Cho, K. M. J. Heeney and J.-S. Kim, Adv. Energy Mater., 2014, 4, 1400527 Search PubMed.
- P. Ding, C. Zhong, Y. Zou, C. Pan, H. Wu and Y. Cao, J. Phys. Chem. C, 2011, 115, 16211–16219 CAS.
- A. C. Stuart, J. R. Tumbleston, H. Zhou, W. Li, S. Liu, H. Ade and W. You, J. Am. Chem. Soc., 2013, 135, 1806–1815 CrossRef CAS.
- S. Subramaniyan, H. Xin, F. S. Kim, S. Shoaee, J. R. Durrant and S. A. Jenekhe, Adv. Energy Mater., 2011, 1, 854–860 CrossRef CAS.
- L. Huo, J. Hou, S. Zhang, H.-Y. Chen and Y. Yang, Angew. Chem., Int. Ed., 2010, 49, 1500–1503 CrossRef CAS PubMed.
- K. G. Jespersen, W. J. D. Beenken, Y. Zaushitsyn, A. Yartsev, M. Andersson, T. Pullerits and V. Sundström, J. Chem. Phys., 2004, 121, 12613–12617 CrossRef CAS.
- R. Steyrleuthner, M. Schubert, I. Howard, B. Klaumünzer, K. Schilling, Z. Chen, P. Saalfrank, F. Laquai, A. Facchetti and D. Neher, J. Am. Chem. Soc., 2012, 134, 18303–18317 CrossRef CAS.
- L. Huo, L. Ye, Y. Wu, Z. Li, X. Guo, M. Zhang, S. Zhang and J. Hou, Macromolecules, 2012, 45, 6923–6929 CrossRef CAS.
- I. C. Wu, C. H. Lai, D. Y. Chen, C. W. Shih, C. Y. Wei, B. T. Ko, C. Ting and P. T. Chou, J. Mater. Chem., 2008, 18, 4297–4303 RSC.
- T. Q. Nguyen, I. B. Martini, J. Liu and B. J. Schwartz, J. Phys. Chem. B, 2000, 104, 237–255 CrossRef CAS.
- J. C. Bijleveld, A. Zoombelt, S. G. J. Mathijssen, M. M. Wienk, M. Turbiez, D. M. Leeuw and R. A. J. Janssen, J. Am. Chem. Soc., 2009, 131, 16616–16617 CrossRef CAS PubMed.
- S. B. Darling and M. Sternberg, J. Phys. Chem. B, 2009, 113, 6215–6218 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- Y.-X. Xu, C.-C. Chueh, H.-L. Yip, F.-Z. Ding, Y.-X. Li, C.-Z. Li, X. Li, W.-C. Chen and A. K.-Y. Jen, Adv. Mater., 2012, 24, 6356–6361 CrossRef CAS.
- B. C. Schroeder, Z. Huang, R. S. Ashraf, J. Smith, P. D'Angelo, S. E. Watkins, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Adv. Funct. Mater., 2012, 22, 1663–1670 CrossRef CAS.
- J. S. Kim, J. H. Lee, J. H. Park, C. Shim, M. Sim and K. Cho, Adv. Funct. Mater., 2011, 21, 480–486 CrossRef CAS.
- K. H. Kim, S. Park, H. Yu, H. Kang, I. Song, J. H. Oh and B. J. Kim, Chem. Mater., 2014, 26, 6963–6970 CrossRef CAS.
- J. H. Park, J. S. Kim, J. H. Lee, W. H. Lee and K. Cho, J. Phys. Chem. C, 2009, 113, 17579–17584 CAS.
- W. Chen, M. P. Nikiforov and S. B. Darling, Energy Environ. Sci., 2012, 5, 8045–8074 CAS.
- F. Li, K. G. Yager, N. M. Dawson, Y.-B. Jiang, K. J. Malloy and Y. Qin, Chem. Mater., 2014, 26, 3747–3756 CrossRef CAS.
- F. Li, K. G. Yager, N. M. Dawson, Y.-B. Jiang, K. J. Malloy and Y. Qin, Polym. Chem., 2015, 6, 721–731 RSC.
- J. Rivnay, S. C. B. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012, 112, 5488–5519 CrossRef CAS PubMed.
- J.-H. Kim, J. H. Park, J. H. Lee, J. S. Kim, M. Sim, C. Shim and K. Cho, J. Mater. Chem., 2010, 20, 7398–7405 RSC.
- J. S. Kim, Y. Lee, J. H. Lee, J. H. Park, J. K. Kim and K. Cho, Adv. Mater., 2010, 22, 1355–1360 CrossRef CAS PubMed.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS PubMed.
- F. Liu, W. Zhao, J. R. Tumbleston, C. Wang, Y. Gu, D. Wang, A. L. Briseno, H. Ade and T. P. Russell, Adv. Energy Mater., 2014, 4, 1301377 Search PubMed.
- H.-C. Liao, C.-C. Ho, C.-Y. Chang, M.-H. Jao, S. B. Darling and W.-F. Su, Mater. Today, 2013, 16, 326–336 CrossRef CAS.
- M. Kim, J. H. Park, J.-H. Kim, J. H. Sung, S. B. Jo, M.-H. Jo and K. Cho, J. Phys. Chem. C, 2009, 113, 17579 Search PubMed.
- J. R. Tumbleston, B. A. Collins, L. Yang, A. C. Stuart, E. Gann, W. Ma, W. You and H. Ade, Nat. Photonics, 2014, 8, 385–391 CrossRef CAS.
- M. Kim, J. H. Park, J.-H. Kim, J. H. Sung, S. B. Jo, M.-H. Jo and K. Cho, Adv. Energy Mater., 2015, 5, 1401317 Search PubMed.
- C. Lin, Y. Lin, S. Li, C. Yu, C. Huang, S. Lee, C. Du, J. Lee, H. Chen and C. Chen, Energy Environ. Sci., 2011, 4, 2134–2139 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra21602h |
| ‡ Jisoo Shin and Min Kim contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.