
Microwave assisted rapid conversion of fructose into 5-HMF over solid acid catalysts†
Abstract
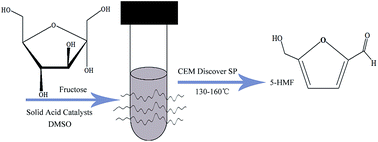
In this work, we investigated the dehydration of fructose into 5-hydroxymethylfurfural using solid catalysts with microwave assistance in dimethyl sulfoxide. Five kinds of solid catalyst ZrO2, WOx/ZrO2, MoOx/ZrO2, SO42−/WOx–ZrO2 and SO42−/MoOx–ZrO2 were prepared and characterized by XRD, UV-DRS, FTIR, XPS, TEM, TPD, BET, and Raman and the surface acid amount was obtained by combining pyridine adsorption and UV spectrometry. The SO42−/WOx–ZrO2 catalyst was found be the most active catalyst, and a fructose conversion of 95.80% with 83.90% 5-HMF yield was obtained at 150 °C in a relatively short reaction time of 5 min. The value of the activation energy was comparable with previous values reported in the literature, implying that SO42−/WOx–ZrO2 was efficient for the dehydration of fructose to 5-HMF and that microwave radiation heating had a remarkable accelerating effect on the fructose conversion.
 Please wait while we load your content...
Please wait while we load your content...

