
Synthesis and characterization of organotriphosphonate-functionalized TM-containing polyoxotungstates†
Yu Huo,
Dandan Li,
Rong Wan,
Pengtao Ma,
Dongdi Zhang,
Jingyang Niu* and
Jingping Wang*
Henan Key Laboratory of Polyoxometalate Chemistry, Institute of Molecular and Crystal Engineering, College of Chemistry and Chemical Engineering, Henan University, Kaifeng, Henan 475004, P. R. China. E-mail: jyniu@henu.edu.cn; jpwang@henu.edu.cn; Fax: +86-371-23886876
First published on 1st December 2015
Abstract
A new family of organotriphosphonate-functionalized TM-containing polyoxotungstates, K4H6[H4{(AsW9O33)TM(H2O)W5O11(N(CH2PO3)3)}2(μ2-O)2]·24H2O [TM = Ni (1), Co (2), Mn (3)], have been synthesized from the preformed POM precursor [As2W19O67(H2O)]14−. They were thoroughly characterized by single crystal X-ray diffraction, IR and UV spectra, thermogravimetric (TG), X-ray powder diffraction (XRPD), X-ray photoelectron spectroscopy (XPS) and elemental analyses. The structures are comprised of two Wells–Dawson building blocks in opposite directions that are connected by two μ2-oxo groups acting as a “hinge”, with the two ATMP ligands serving as a stanchion. The electrochemical behavior of these compounds has also been investigated.
Introduction
Polyoxometalates (POMs), especially incorporated with organic groups, have received considerable interest in recent years owing to their remarkable structural varieties and applications in catalysis, medicine, electrochemistry and magnetism.1 Preparation of organophosphonate-based POMs has been attracting increasing attention, especially because some rare earth metals and some transition metals (TMs) have been introduced into POM-based organophosphonate derivatives and potentially generate new properties.2 Over the past few decades, a number of organophosphonate derivatives have been simply grafted onto the surface of polyoxotungstates (POTs).3 Generally, Keggin, and Lindqvist-type polyoxoanions are used as the inorganic building blocks. Despite Mialane, Dolbecq, and co-workers having reported a series of TM-substituted organodiphosphonate-based POTs, [{(B-α-PW9O34)TM3(OH)(H2O)2(O3PC(O)(C3H6NH3)PO3)}2TM]14− (TM = Co, Ni), TM-containing organotriphosphonate-functionalized POTs are still largely unexplored.4To the best of our knowledge, POTs incorporated with 3d metals have been widely studied for their catalytic, magnetic, and remarkable electrochemical properties.5 Thus, we mainly focussed to explore the interaction of organophosphonic acids and polyoxotungstates with embedding 3d metals, which are expected to possess good catalytic or biological activities. In this context, we chose the system of [As2W19O67(H2O)]14−, organophosphonate and TMs due to the following considerings. (i) Compared to the Keggin and Lindqvist tungstate anions, the dilacunaryarsenotungstate [As2W19O67(H2O)]14− can partially decompose to provide the necessary fragment to get the new structural architectures.6 (ii) Organophosphonate is a multidentate and good oxygen-containing organic ligand, which is efficient in terms of charge compensation. (iii) Organophosphonic acid can be considered as structure-directing components owing to their flexibility in altering the organic groups. In addition, the incorporation of organophosphonate into POMs offers an opportunity to fine-tune the properties.7 Based on aforementioned points, we attempt to utilize the [As2W19O67(H2O)]14− as precursor to construct intriguing and functionalized structures by the incorporation of TM salts and the multidentate and flexible amino trimethylenephosphonic acid (ATMP).
In this paper, we report a novel series of organotriphosphonate-functionalized TM-containing POTs, K4H6[H4{(AsW9O33)TM-(H2O)W5O11(N(CH2PO3)3)}2(μ2-O)2]·24H2O [TM = Ni (1), Co(2), Mn (3)], which were synthesized in water by treating [As2W19O67(H2O)]14− precursor, ATMP and transition metals. In addition to structural details we also present the electrochemical properties.
Results and discussion
Synthesis
The compounds 1–3 were conventionally synthesized in a one-pot reaction of the precursor [As2W19O67(H2O)]14−, trisphosphonate ligand ATMP and chloride salts of the transition metals Ni, Co and Mn in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:4 in 80 °C aqueous solution under stirring for 2 h. Parallel experiments show that the final products appear to depend on three key factors: (i) the presence or absence of additional potassium cations. The extra potassium cations are necessary to obtain pure bulk samples of 1–3, and the title compounds can not be obtained with the countercations replaced by Li+/Na+/Cs+ cations. The results confirm that countercations can exert significant effects on the synthesis, improve the solubility of the POM reaction intermediates, for the further operation of superstructures.8 (ii) the introduction of ATMP ligand. Its presence during the course of the reaction is crucial because replacement of N-(phosphonomethyl) iminodiacetic acid or 1-hydroxyethylidene-1,1-diphosphonic acid does not produce the desired results. As we know, ATMP as a multidentate organic ligand is flexible in space morphology and stable over a wide range of temperatures and pH values, which mainly encouraging us to investigate the synthesis of organophosphonate-based POMs. (iii) The suitable pH of the reaction. Parallel experiments confirm that the optimal pH for the crystals of compounds 1–3 ranges from 2.0 to 3.0, no title compounds produced when the pH less than 1.5, while a tri-nuclear sandwiched tungstoarsenate will be produced in the pH values of 3.5 or more high. Notably, the identical structures and the similar reaction conditions for 1–3 indicate that the positions substituted by TM atoms are highly active, which provides us the possibility of forming novel POMs by introducing lanthanide ions or organic ligands.
:4:4 in 80 °C aqueous solution under stirring for 2 h. Parallel experiments show that the final products appear to depend on three key factors: (i) the presence or absence of additional potassium cations. The extra potassium cations are necessary to obtain pure bulk samples of 1–3, and the title compounds can not be obtained with the countercations replaced by Li+/Na+/Cs+ cations. The results confirm that countercations can exert significant effects on the synthesis, improve the solubility of the POM reaction intermediates, for the further operation of superstructures.8 (ii) the introduction of ATMP ligand. Its presence during the course of the reaction is crucial because replacement of N-(phosphonomethyl) iminodiacetic acid or 1-hydroxyethylidene-1,1-diphosphonic acid does not produce the desired results. As we know, ATMP as a multidentate organic ligand is flexible in space morphology and stable over a wide range of temperatures and pH values, which mainly encouraging us to investigate the synthesis of organophosphonate-based POMs. (iii) The suitable pH of the reaction. Parallel experiments confirm that the optimal pH for the crystals of compounds 1–3 ranges from 2.0 to 3.0, no title compounds produced when the pH less than 1.5, while a tri-nuclear sandwiched tungstoarsenate will be produced in the pH values of 3.5 or more high. Notably, the identical structures and the similar reaction conditions for 1–3 indicate that the positions substituted by TM atoms are highly active, which provides us the possibility of forming novel POMs by introducing lanthanide ions or organic ligands.
Structural descriptions
The crystallographic data and structural refinements for three compounds 1–3 were given in Table 1 and their structural representations are available in Fig. 1a. X-ray crystal structure analyses reveal that compounds 1–3 crystallize in the monoclinic space group C2/c and the general features are identical for the compounds, the only difference is the TM atom substituted the equatorial position. The phase purity of 1–3 was confirmed using X-ray powder diffraction (Fig. S2†).| 1 | 2 | 3 | |
|---|---|---|---|
| Formula | H74As2C6K4N2Ni2O134P6W28 | H74As2C6K4N2Co2O134P6W28 | H74As2C6K4N2Mn2O134P6W28 |
| Mr (g mol−1) | 8075.57 | 8076.05 | 8068.06 |
| T (K) | 296(2) | 296(2) | 296(2) |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | C2/c | C2/c | C2/c |
| A (Å) | 29.917(2) | 29.880(1) | 29.848(4) |
| B (Å) | 27.671(2) | 27.639(1) | 27.794(4) |
| C (Å) | 20.667(1) | 20.626(8) | 20.700(3) |
| α (deg) | 90 | 90 | 90 |
| β (deg) | 122.318(1) | 122.184(1) | 121.555(2) |
| Γ (deg) | 90 | 90 | 90 |
| V (Å3) | 14458.8(2) |
14416.8(1) |
14634(4) |
| Z | 4 | 4 | 4 |
| Dc (g cm−3) | 3.703 | 3.714 | 3.657 |
| μ (mm−1) | 23.170 | 23.207 | 22.809 |
| Limiting indices | −35 ≤ h ≤ 27 | −35 ≤ h ≤ 35 | −35 ≤ h ≤ 29 |
| −31 ≤ k ≤ 33 | −32 ≤ k ≤ 26 | −29 ≤ k ≤ 33 | |
| −12 ≤ l ≤ 24 | −24 ≤ l ≤ 21 | −24 ≤ l ≤ 24 | |
| Params | 789 | 791 | 785 |
| Reflns collected | 37338 |
37039 |
37013 |
| GOF | 0.997 | 1.022 | 0.956 |
| R1, wR2 [I > 2σ(I)] | R1 = 0.0438, wR2 = 0.1094 | R1 = 0.0381, wR2 = 0.0969 | R1 = 0.0691, wR2 = 0.1578 |
| R1, wR2 [all data] | R1 = 0.0669, wR2 = 0.1182 | R1 = 0.0594, wR2 = 0.1038 | R1 = 0.1457, wR2 = 0.1847 |
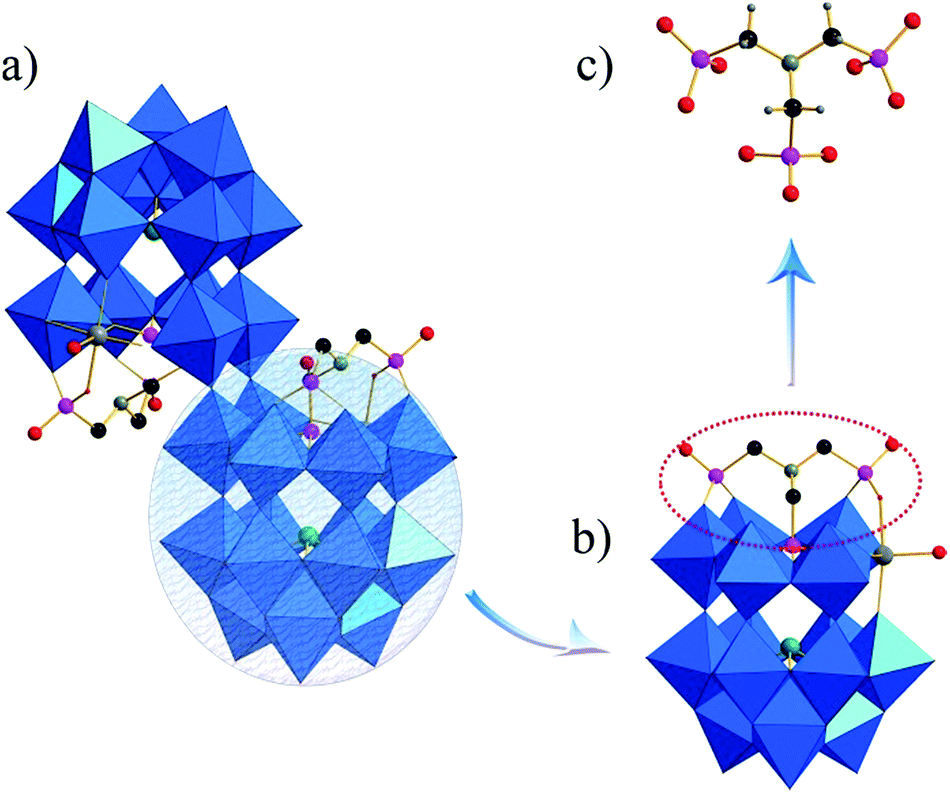 | ||
| Fig. 1 (a) Polyhedral/ball-and-stick representation of the polyanion. (b) Combined polyhedral/ball-and-stick representation of the subunit. (c) Coordination mode of the ATMP ligand. Color code: As, green; C, black; N, dark yellow; P, pink; WO6 octahedral, blue. | ||
These structures are composed of two Wells–Dawson building blocks in opposite direction linked by two μ2-oxo atoms. The building block arises from the {As2W19O67(H2O)} precursor fragmenting to afford {(AsW9O33)} unit and free tungstate, which is favored to rearrange to a new unit. An intriguing structural feature is the subunit contains two different heteroatoms (As1, P1) with the distance in the range of 3.71–3.73 Å. The central P atom in the subunit was provided by the ATMP ligand with the other two {PO3C} groups of ATMP fixing to the surface of the complex as peripheral linkage units (Fig. 1b). Thus, ATMP acts as a stanchion to support the structure. One equatorial position of the Wells–Dawson structure is substituted by the TM atom. The TMII cation achieves an octahedral coordination environment through coordination to a single terminal aqua ligand with TM–O(W) bonds range between 2.00 and 2.15 Å in length, which has two TM–O–P linkages with the angles in the range 128.4(9)–130.5(6)° for the three compounds. The edge-sharing linkages TM–O–W are all approximate 111°, while the corner-sharing linkages have TM–O–W angles of 146.0(5)–162.2(6)°.
The band valence sum (BVS) calculations confirm that the oxidation states of the As, P, W and TM atoms are +3, +3, +6 and +2, respectively, and the results of P and W are further confirmed by XPS spectra (Fig. S3†). In the polyanions, the P–O bond lengths are in the range of 1.482(1)–1.601(2) Å, while the W–O bonds range between 1.590(2) and 2.449(9) Å in length, with BVS calculations indicating one proton associated with the terminal oxygen atoms (O54, O55). The O–W–O bond angles are between 70.5(2)° and 169.8(3)°. According to the charge-balance consideration, six protons are needed to compensate the negative charges of [{(AsW9O33)TM(H2O)W5O11(N(CH2PO3)3)}2(μ2-O)2]14− except the four K+ cations. Additionally, the polyanions are connected to each other by the coordination of four additional K+ ions to form a two-dimensional layerwork structure (Fig. S1†).
IR, UV spectra,and TG analysis
The IR spectra of compounds 1–3 exhibit very similar characteristic vibrations in the region between 400 and 4000 cm−1 (Fig. S4†), indicating the polyanions in 1–3 are isostructural, which are in good agreement with the results of single-crystal X-ray structural analysis. The characteristic bands at 963 cm−1 can be assigned to the terminal W–Ot vibrations, peaks at 872 and 812 cm−1 are attributed to the two types of W–O–W stretching vibrations and the peak at 722 cm−1 can be assigned to the W–O(–As) stretch.9 The P–O vibration bands in 1–3 appear in 1191, 1122 and 1090 cm−1. The bands located in the low wavenumber region from 723 to 561 cm−1 are dominated by the P–C stretching vibrations. A sharp band of moderate intensity at 1026 cm−1 is likely due to an As–O stretch of the trigonal-pyramid coordination geometry of the AsIII atoms.10 Additionally, broad peaks at 3436 cm−1 and 1618 cm−1 are mainly associated with the stretching and bending modes of lattice and coordinated water molecules.The UV spectra of 1–3 exhibit a characteristic absorption band at 279 nm for 1, 278 nm for 2, and 263 nm for 3, respectively, (Fig. S5†), which is assigned to the pπ–dπ charge-transfer transitions of the Ob,c → W bonds.11 In the visible region of 2 (Fig. S6†), an absorption band appears at 532 nm, which is assigned to the Co-centered d–d transitions.12 Whereas, no absorption band is observed in the visible region of 1 and 3 due to the rather weak absorbance corresponding to the Ni/Mn-centered d–d transitions. Thus, the characteristic of the Co band features distinct fingerprint of the three POMs. To the best of our knowledge, the POMs are sensitive to the pH value of the studied media. This leads us to study the influences of the pH value on the stability of compounds. For this purpose, the UV spectra of 3 were measured (Fig. 2). The pH values were lowered by using dilute HCl solution and raised by using dilute NaOH solution. The initial pH value of 3 in water was 3.4. The exact energies and shapes vary slightly with the pH ranging from 1.0 to 5.2. Upon increasing the pH to 5.8 or higher, the absorbance band at 265 nm gradually disappears, which suggests the decomposition of the POM skeleton of 3. The results indicate that compound 3 could not exist in the solution whose pH is higher than 5.2.
 | ||
| Fig. 2 The influence of the pH values on the stability of 3 in aqueous solution. | ||
Thermal gravimetric analyses (TGA) of compounds 1–3 were investigated under a N2 atmosphere from 25 °C to 800 °C (Fig. S7†). All the TG curves of 1–3 show two slow steps of weight loss, the first weight loss of 6.91% for 1 (calcd 6.91%), 6.41% for 2 (calcd 6.91%), and 7.14% for 3 (calcd 6.92%) in the temperature of 25–400 °C, respectively, which arise from the loss of twenty-seven crystal water molecules, two coordinated water molecules and five structure water molecules. The second weight loss between 400–800 °C of 3.11% (calcd 3.84%), 3.71% (calcd 3.84%), and 3.41% (calcd 3.84%) for 1–3, respectively, are attributed to two {–N(CH2)3} groups and the sublimation of As2O3 molecules.13
Electrochemistry
In order to explore the electrochemical behaviors of compounds 1–3, the electrochemistry of 1–3 was carried out at different scan rates in a pH 3 sulfate medium (0.2 M Na2SO4 + H2SO4). As shown in Fig. 3, the cyclic voltammograms of 1–3 are constituted by three reversible waves in the potential range from −1.0 to 0.3 V. Their main reduction peak potentials located at E1/2 = (Epa + Epc)/2 at −0.41, −0.59 and −0.85 V for 1, −0.40, −0.57 and −0.83 V for 2,−0.40, −0.56 and −0.82V for 3 (vs. SCE), respectively, which can be ascribed to the reduction processes of W in the polyoxoanion framework.14 The cyclic voltammograms of the three complexes are very similar in shape. The peak potentials change gradually following the scan rates from 100 to 200 mV s−1: the anodic peak potentials are varied with increasing scan rates and the corresponding cathodic current values shift toward the negative direction. The results indicate that the redox ability of 1–3 can be maintained. As the scan rate varied from 100 to 200 mVs−1, the peak currents related to the tungsten redox processes were proportional to the square root of the scan rate (Fig. 3, inset), which suggests that redox processes are diffusion-controlled.15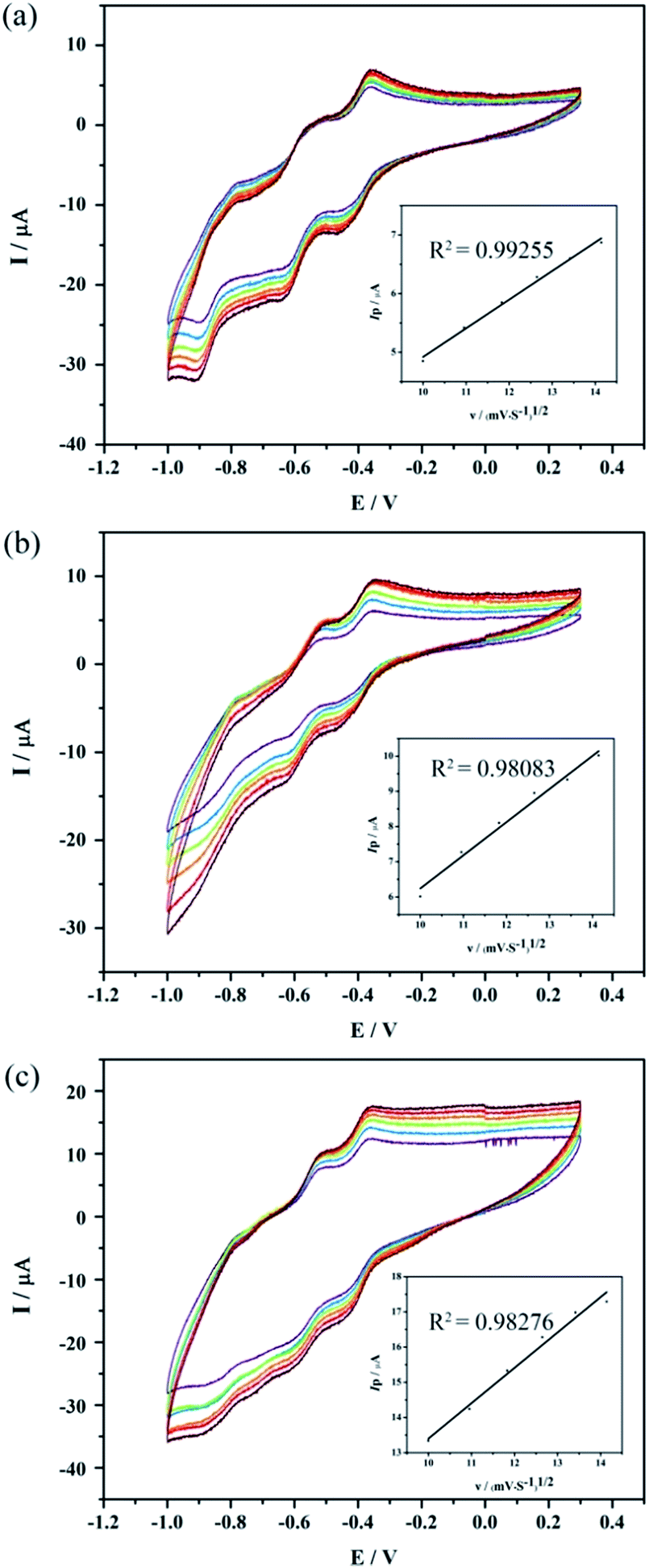 | ||
| Fig. 3 Cyclic voltammograms of (a) 1, (b) 2 and (c) 3 at different scan rates (from inner to outer: 100, 120, 140, 160, 180, 200 mV s−1). Insert: Variation of the peak current intensity for the first WVI wave as a function of the square root of the potential scan rate. Working electrode, glassy carbon; reference electrode, SCE. | ||
As a complement to compound 3, a quasi-reversible and an irreversible oxidation peak observed with the E1/2 peak potentials to be located at +0.946 and +1.484 V respectively (Fig. 4), which are assigned to the main characteristics of the oxidation of the MnII centers of 3.16 The characteristic sharpness of the oxidation wave indicates the presence of a surface-active species.17
 | ||
| Fig. 4 Cyclic voltammograms of 3. Scans are restricted to the redox processes attributed to MnII centers at scan rates (from inner to outer) of 30 to 70 mV s−1; reference electrode SCE. | ||
Experimental
Materials and methods
The POM precursor K14[As2W19O67(H2O)] was synthesized as described in the literature.18 Other chemical reagents were purchased and used without further purification. Elemental analyses (C, H) were conducted on a Perkin-Elmer 2400-II CHNS/O analyzer. ICP analyses were performed on a Perkin-Elmer Optima 2000 ICP-OES spectrometer. Infrared spectra (using KBr in pellets) were recorded on a Bruker VERTEX 70 IR spectrometer (4000–400 cm−1). The following abbreviations were used to assign the peak intensities: w = weak, m = medium, and s = strong. UV absorption spectra were obtained with a U-4100 spectrometer at room temperature. Thermogravimetric analyses (TGA) were performed under N2 atmosphere on a Mettler-Toledo TGA/SDTA851e instrument with the heating rate of 10 °C min−1 from 25 to 800 °C. XRPD was performed on a Bruker AXS D8 Advance diffractometer using Cu Kα radiation in the range 2θ = 5–40° at 293 K. XPS were recorded by an Axis Ultra (Kratos, U.K.) photoelectron spectroscope with Al Kα (1486.7 eV) irradiation. The electrochemical studies were measured on the CorrTest CS-150 analyzer (Wuhan) using saturated calomel electrode (SCE) as reference electrode.X-ray crystallography
The crystallographic data for compounds 1–3 were given in Table 1. Intensity data were collected at 296 K on a Bruker APEX-II CCD diffractometer using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Routine Lorentz and polarization corrections were applied, and an absorption correction was performed using the SADABS program. The structures were solved by direct methods and refined by the full-matrix least-squares method on F2 using the SHELXTL-14 program package. In the crystal structure, there are a number of short connections between Ow(H2O) and O(POM) in the range 2.74–2.84 Å, which indicate extensive H-bonding interactions among them. Non-hydrogen atoms were refined anisotropically and all H atoms on solvent water molecules were directly included into the final molecular formula. Hydrogen atoms of organic ligands were fixed in calculated positions. Futhermore, coordination and lattice H2O and the counter cations stated in the formula could not be distinguished based on electron densities, which were determined based on elemental analysis, thermogravimetric analysis and the charge balance consideration. CCDC 987931 (1), CCDC 987933 (2), CCDC 987934 (3) contain the supplementary crystallographic data for this paper.†Conclusions
In conclusion, a novel series of TM-substituted organotriphosphonate-based polyoxotungstate derivatives has been successfully synthesized under similar reaction condition. To our knowledge, the compounds represent the first series of TM-containing organotriphosphonate-functionalized POTs. The identical structures show that the position substituted by TM atoms is highly active, providing the great potentialities of constructing novel functionalized and multidimensional POMs. Further research will be focused on the synthesis of new aggregates based on [As2W19O67(H2O)]14− by introducing lanthanide ions and organic ligands.Acknowledgements
We gratefully acknowledge financial support from the Natural Science Foundation of China, Foundation of Education Department of Henan Province, and Natural Science Foundation of Henan Province for financial support.Notes and references
- (a) A. Proust, R. Thouvenot and P. Gouzerh, Chem. Commun., 2008, 16, 1837 RSC; (b) Y. Zhang, R. Cao, R. T. Guo, H. Robinson, S. Papapoulos, A. H. Wang, T. Kubo and E. Oldfield, J. Am. Chem. Soc., 2009, 131, 5153 CrossRef CAS; (c) A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009 CrossRef CAS PubMed; (d) P. C. Yin, D. Li and T. B. Liu, Chem. Soc. Rev., 2012, 41, 7368 RSC; (e) A. Banerjee, B. S. Bassil, G. V. Roschenthaler and U. Kortz, Chem. Soc. Rev., 2012, 41, 7590 RSC.
- (a) E. Burkholder, V. Golub, C. J. O'Connor and J. Zubieta, Inorg. Chem., 2004, 43, 7014 CrossRef CAS PubMed; (b) N. G. Armatas, W. Ouellette, K. Whitenack, J. Pelcher, H. X. Liu, E. Romaine, C. J. O'Connor and J. Zubieta, Inorg. Chem., 2009, 48, 8897 CrossRef CAS PubMed; (c) J. Y. Niu, X. Q. Zhang, D. H. Yang, J. W. Zhao, P. T. Ma, U. Kortz and J. P. Wang, Chem.–Eur. J., 2012, 18, 6759 CrossRef CAS PubMed.
- (a) C. R. Mayer, P. Herson and R. Thouvenot, Inorg. Chem., 1999, 38, 6152 CrossRef CAS PubMed; (b) U. Kortz, C. Marquer, R. Thouvenot and M. Nierlich, Inorg. Chem., 2003, 42, 1158 CrossRef CAS PubMed; (c) C. R. Mayer, M. Hervé, H. Lavanant, J. C. Blais and F. Sécheresse, Eur. J. Inorg. Chem., 2004, 5, 973 CrossRef; (d) H. Driss, K. Boubekeur, M. Debbabi and R. Thouvenot, Eur. J. Inorg. Chem., 2008, 23, 3678 CrossRef.
- (a) H. E. Moll, A. Dolbecq, J. Marrot, G. Rousseau, M. Haouas, F. Taulelle, G. Rogez, W. Wernsdorfer, B. Keita and P. Mialane, Chem.–Eur. J., 2012, 18, 3845 CrossRef PubMed; (b) H. E. Moll, G. Rousseau, A. Dolbecq, O. Oms, J. Marrot, M. Haouas, F. Taulelle, E. Rivière, W. Wernsdorfer, D. Lachkar, E. Lacôte, B. Keita and P. Mialane, Chem.–Eur. J., 2013, 19, 6753 CrossRef.
- (a) Z. M. Zhang, Y. G. Li, S. Yao and E. B. Wang, Dalton Trans., 2011, 40, 6475 RSC; (b) S. T. Zheng and G. Y. Yang, Chem. Soc. Rev., 2012, 41, 7623 RSC; (c) J. M. Clemente-Juan, E. Coronado and A. Gaita-Ariño, Chem. Soc. Rev., 2012, 41, 7464 RSC; (d) Y. C. Liu, C. H. Fu, S. T. Zheng, J. W. Zhao and G. Y. Yang, Dalton Trans., 2013, 42, 16676 RSC.
- (a) C. Ritchie, E. G. Moore, M. Speldrich, P. Kögerler and C. Boskovic, Angew. Chem., Int. Ed., 2010, 49, 7702 CrossRef CAS PubMed; (b) C. Ritchie, V. Baslon, E. G. Moore, C. Reber and C. Boskovic, Inorg. Chem., 2012, 51, 1142 CrossRef CAS PubMed; (c) X. J. Feng, H. Y. Han, Y. H. Wang, L. L. Li, Y. G. Li and E. B. Wang, CrystEngComm, 2013, 15, 7267 RSC.
- (a) D. Compain, P. Mialane, J. Marrot, F. Sécheresse, W. Zhu, E. Oldfield and A. Dolbecq, Chem.–Eur. J., 2010, 16, 13748 CrossRef PubMed; (b) A. Banerjee, F. S. Raad, N. Vankova, B. S. Bassil, T. Heine and U. Kortz, Inorg. Chem., 2011, 50, 11667 CrossRef CAS PubMed; (c) H. E. Moll, W. Zhu, E. Oldfield, L. M. Rodriguez-Albelo, P. Mialane, J. Marrot, N. Vila, I. M. Mbomekallé, E. Rivière, C. Duboc and A. Dolbecq, Inorg. Chem., 2012, 51, 7921 CrossRef PubMed.
- (a) J. D. Compain, P. Deniard, R. Dessapt, A. Dolbecq, O. Oms, F. Secheresse, J. Marrot and P. Mialane, Chem. Commun., 2010, 46, 7733 RSC; (b) T. B. Gasa, C. Valente and J. F. Stoddart, Chem. Soc. Rev., 2011, 40, 57 RSC; (c) J. M. Wang, S. G. Li, P. J. Yang, X. J. Huang, X. J. Yang and B. Wu, CrystEngComm, 2013, 15, 4540 RSC.
- (a) L. G. Detusheva, L. I. Kuznetsova, L. S. Dovlitova and V. A. Likholobov, Russ. Chem. Bull., 2003, 52, 370 CrossRef CAS; (b) C. Craciun, D. Rusu, L. Pop-Fanea, M. Hossu, M. Rusu and L. J. David, J. Radioanal. Nucl. Chem., 2005, 264, 589 CrossRef CAS; (c) Y. Wang, X. P. Sun, S. Z. Li, P. T. Ma, J. P. Wang and J. Y. Niu, Dalton Trans., 2015, 44, 733 RSC.
- (a) I. M. Muller and W. S. Sheldrick, Eur. J. Inorg. Chem., 1998, 12, 1999 CrossRef; (b) C. Ritchie, M. Speldrich, R. W. Gable, L. Sorace, P. Kögerler and C. Boskovic, Inorg. Chem., 2011, 50, 7004 CrossRef CAS PubMed.
- (a) J. Y. Niu, K. H. Wang, H. N. Chen, J. W. Zhao, P. T. Ma, J. P. Wang, M. L. Li, Y. Bai and D. B. Dang, Cryst. Growth Des., 2009, 9, 4362 CrossRef CAS; (b) L. J. Chen, D. Y. Shi, J. W. Zhao, Y. L. Wang, P. T. Ma, J. P. Wang and J. Y. Niu, Cryst. Growth Des., 2011, 11, 1913 CrossRef CAS; (c) Q. Tang, S. X. Liu, Y. W. Liu, S. J. Li, F. J. Ma, J. X. Li, S. T. Wang and C. Z. Liu, Dalton Trans., 2013, 42, 8512 RSC.
- (a) M. D. Ritorto, T. M. Anderson, W. A. Neiwert and C. L. Hill, Inorg. Chem., 2004, 43, 44 CrossRef CAS PubMed; (b) L. Lisnard, P. Mialane, A. Dolbecq, J. Marrot, J. M. Clemente-Juan, E. Coronado, B. Keita, P. de Oliveira, L. Nadjo and F. Secheresse, Chem.–Eur. J., 2007, 13, 3525 CrossRef CAS PubMed.
- (a) C. Y. Sun, Y. G. Li, E. B. Wang, D. R. Xiao, H. Y. An and L. Xu, Inorg. Chem., 2007, 46, 1563 CrossRef CAS; (b) J. Y. Niu, J. A. Hua, X. Ma and J. P. Wang, CrystEngComm, 2012, 14, 4060 RSC; (c) X. L. He, Y. P. Liu, K. N. Gong, Z. G. Han and X. L. Zhai, Inorg. Chem., 2015, 54, 1215 CrossRef CAS PubMed.
- (a) U. Kortz, I. M. Mbomekalle, B. Keita, L. Nadjo and P. Berthet, Inorg. Chem., 2002, 41, 6412 CrossRef CAS PubMed; (b) I. M. Mbomekalle, B. Keita, M. Nierlich, U. Kortz, P. Berthet and L. Nadjo, Inorg. Chem., 2003, 42, 5143 CrossRef CAS PubMed; (c) I. M. Mbomekalle, B. Keita, L. Nadjo, P. Berthet, W. A. Neiwert, C. L. Hill, M. D. Ritorto and T. M. Anderson, Dalton Trans., 2003, 13, 2646 RSC.
- (a) B. S. Bassil, M. Ibrahim, S. S. Mal, A. Suchopar, R. N. Biboum, B. Keita, L. Nadjo, S. Nellutla, J. V. Tol, N. S. Dalal and U. Kortz, Inorg. Chem., 2010, 49, 4949 CrossRef CAS PubMed; (b) J. I. Nambu, T. Ueda, S. X. Guo, J. F. Boasc and A. M. Bond, Dalton Trans., 2010, 39, 7364 RSC; (c) D. Gabb, C. P. Pradeep, H. N. Miras, S. G. Mitchell, D. L. Long and L. Cronin, Dalton Trans., 2012, 41, 10000 RSC.
- (a) I. M. Mbomekalle, B. Keita, L. Nadjo, P. Berthet, W. A. Neiwert, C. L. Hill, M. D. Ritorto and T. M. Anderson, Dalton Trans., 2003, 13, 2646 RSC; (b) I. M. Mbomekalle, B. Keita, M. Nierlich, U. Kortz, P. Berthet and L. Nadjo, Inorg. Chem., 2003, 42, 5143 CrossRef CAS.
- (a) J. Liw, F. Ortega, P. Sethuraman, D. E. Katsoulis, C. E. Costello and M. T. Pope, J. Chem. Soc., Dalton Trans., 1992, 12, 1901 Search PubMed; (b) S. G. Mitchell, T. Boyd, H. N. Miras, D. L. Long and L. Cronin, Inorg. Chem., 2011, 50, 136 CrossRef CAS PubMed.
- U. Kortz, M. G. Savelieff, B. S. Bassil and M. H. Dickman, Angew. Chem., Int. Ed., 2001, 40, 33 CrossRef.
Footnote |
| † CCDC 987931 (1), 987933 (2), 987934 (3). Packing modes for polyanions (Fig. S1); XRPD of 1–3 (Fig. S2); XPS spectra of 1–3 (Fig. S3); IR and UV spectra of 1–3 (Fig. S4 and S5); visible spectra of 1–3 (Fig. S6); TG curves of 1–3 (Fig. S7); selected bond distances of 1–3 (Table S1); BVS results of As, P and the protonated oxygen atoms (Tables S2 and S3). For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra20806h |
| This journal is © The Royal Society of Chemistry 2015 |