
Magnetic nano-organocatalysts: impact of surface functionalization on catalytic activity†
E. Nehlig,
L. Motte and
E. Guénin*
UFR SMBH, Université Paris 13, Sorbonne Paris Cité, 74 Rue Marcel Cachin, 93017 Bobigny Cedex, France. E-mail: guenin@univ-paris13.fr
First published on 26th November 2015
Abstract
Iron oxide nanoparticles (γ-Fe2O3) have been synthesized using soft chemistry in aqueous media. The particles were then stabilized on a surface using bifunctional coating agents bearing terminal functional groups which enable post functionalization with the desired catalyst (proline derivatives, peptides). The hybrid nanomaterials were characterized with various techniques in order to determine their properties. The catalysts’ activities were evaluated using aldolization and 1,4-Michael addition as model reactions. For both reactions the crucial impact of the nanocatalyst surface functionalisation on the catalytic properties is demonstrated. For the Michael addition, good selectivity was achieved using a small amount of nano-catalyst.
1. Introduction
Nowadays, interest in new catalytic systems has increased exponentially thanks to various industrial applications in both fine and pharmaceutical chemistry. Numerous catalysts have been developed for a perpetually increasing number of organic reactions. Lots of homogeneous catalysts are expensive and/or contain noble metals, and are difficult to adapt for industrial processes, due to poor extraction, poor recycling and many reuse problems. During the last decade, more efficient and greener new catalysts have been developed in organometallic, photocatalytic and organocatalytic areas. Recently immobilization of organocatalysts onto magnetic nanoparticles (MNPs) has appeared as a really efficient method with a low impact on the environment.1–4 Organocatalysts present several advantages like low toxicity, affordability, availability, and robustness.5–8 Since a report by O’Dálaigh et al.9 in 2007, many organocatalysts have been grafted onto MNPs, usually with a sturdy link and with or without a silica shell. But developing new eco-friendly processes is still required. Moreover, precise control of the functionalization and an efficient determination of the average number of catalysts per MNP are a necessity when there are catalytic aims. Another important parameter to control is the possible interactions of the nanoparticle surface during the catalysis, as it cannot be considered totally inert as firstly described by Gleeson et al.10 As a part of our research on magnetic nanocatalysts,11,12 we proposed to synthesize, stabilize and functionalize iron oxide NPs in water and anchor different organocatalysts on their surface by simple and efficient click chemistry methodologies13–17 applicable to nanosurfaces.18–22 Using grafting to or grafting from methodologies we prepared several nanocatalysts to be evaluated in aldolization and 1,4-Michael addition reactions.2. Results and discussion
MNPs were synthesized in water using a direct micellar process, according to a previously established procedure.23 Briefly, ferrous dodecyl sulfate was mixed with dimethylamine in water at 28 °C for 2 hours, and after several washing steps, the bare MNPs were obtained. The as-synthesized MNPs were spherical, with an inorganic core size of 10.3 ± 0.25 nm in diameter, determined using transition electron microscopy (TEM). The magnetic properties determined using vibrating sample magnetometer (VSM) measurements confirmed their superparamagnetic behavior24 with a magnetization saturation of 55.2 ± 3 A m2 kg−1 (Fig. 1).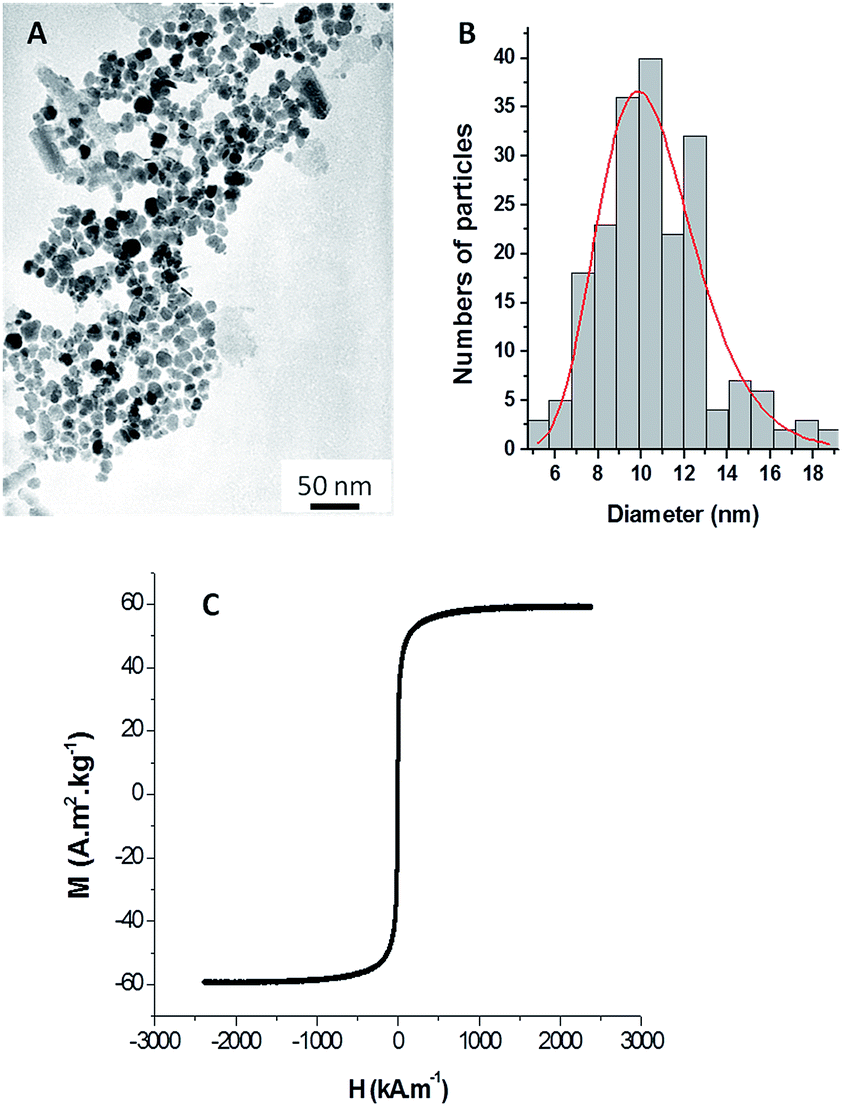 | ||
| Fig. 1 TEM image (A), size distribution (B) and magnetization curve (C) of the bare MNPs. | ||
The bare nanoparticles were then coated in water with small bifunctional molecules from the bisphosphonic acid family. The bisphosphonic function ensured anchoring to the MNP surface while the lateral chain enabled post functionalization with organocatalysts. Two synthetic molecules of the bisphosphonic acid family were used, alendronate-proline 1 for the “grafting to” methodology, and an already described bisphosphonic acid bearing a terminal alkyne functionality ((1-hydroxy-1-phosphonohept-6-ynyl)phosphonic acid or BPheptyne) which enables chemoselective functionalization of the MNP surface.25 Bisphosphonate 1 was obtained by reacting alendronate and (Boc)-L-Pro-OSu in a water/DMF (1/1) mixture in the presence of di-isopropylethylamine (DIPEA) for 4 days at room temperature. After a classic acidic deprotection step using TFA/CH2Cl2 (1/1) for 30 min and precipitation, 1 was obtained with good yield and characterized using NMR and HR-MS (see ESI†). The MNPs were then functionalized with 1 using an aqueous grafting process for 2 h at 90 °C (Scheme 1).
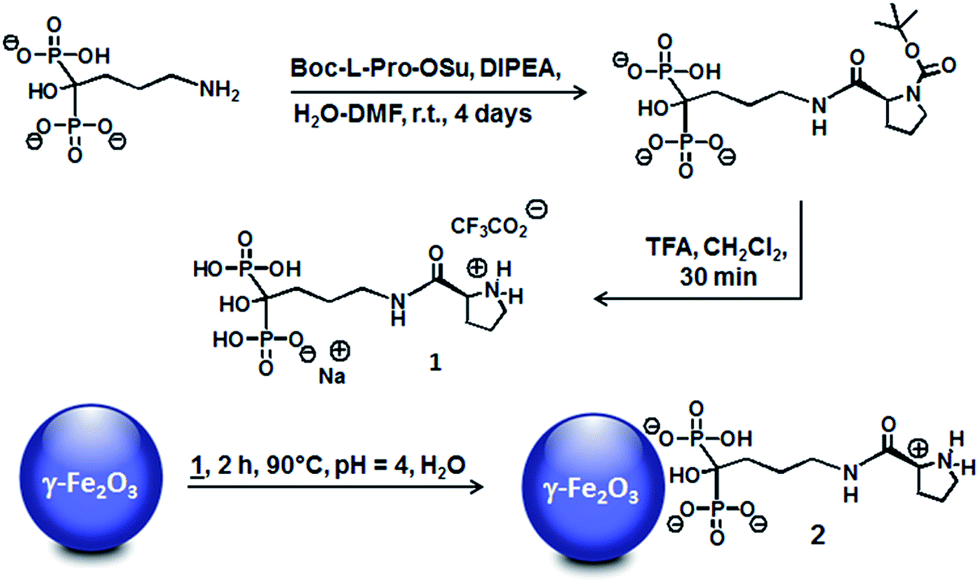 | ||
| Scheme 1 Synthesis of 1 and preparation of MNP@2. | ||
The functionalized MNP@2 was stable in a wide range of pH (5 < pH < 10) (see ESI†). At pH 7 the hydrodynamic diameter (Dh) and zeta potential (ζ) were respectively 25 ± 5 nm and −36 ± 5 mV. Energy dispersive X-ray spectroscopy (EDX) measurements enabled us to evaluate the number of molecules of bisphosphonic acid 1 as 400 ± 50 per nanoparticle. This result corresponds to a surface area of 26.5 Å2 occupied by 1. This result is in accordance with a previous observation for such chelating molecules.26
For the “grafting from” methodology, anchoring of BPheptyne 3 was realized according to a previously described procedure.25 Two click methodologies were used for the functionalization of MNP@3 with organocatalysts. A prolinamide derivative bearing azide functionality 4, and a Boc-deprotected commercial azido-proline 5 (see ESI†) were grafted using a copper azide alkyne cycloaddition (CuAAC).27,28 Thiol–yne click coupling25,27 was used in the case of a commercial peptide 6 which derivates from Professor H. Wennemers’ peptide sequence29–31 (Scheme 2). Thermogravimetric analysis (TGA) was performed for MNP@3-4 and MNP@3-5 (see ESI†), indicating the grafting of 150 ± 50 molecules of catalyst 4 for MNP@3-4 and 250 ± 50 molecules of catalyst 5 for MNP@3-5 (see ESI†). At pH 7, the Dh and ζ potential are 28 ± 5 nm/−37 ± 5 mV and 26 ± 5 nm/−45 ± 5 mV respectively for MNP@3-4 and MNP@3-5. Considering the thiol–yne methodology, by EDX 150 ± 50 molecules of catalyst 6 per nanoparticle were found for MNP@3-6 (see ESI†). The Dh and ζ potential were 25 ± 5 nm and −44 ± 5 mV in water at pH 7.
 | ||
| Scheme 2 Preparation of MNP@3-4, MNP@3-5 and MNP@3-6. | ||
The grafting of the molecules to the surface was further demonstrated by Fourier transform infra-red (FTIR) analysis. This analysis confirmed the presence of 1 at the MNP surface, as shown in Fig. 2A, as a vibration band corresponding to the bisphosphonic functional groups could be found at around 1100 cm−1. Moreover the shift observed for the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and P–O vibration bands confirmed the anchorage via the phosphonate moieties on the MNPs’ surface.32 The strong vibration band at 579 cm−1 is characteristic of Fe–O vibrations typical of maghemite structures. For both MNP@3-4 and MNP@3-5, the appearance of vibrations bands in the FTIR spectra between 1400 and 1800 cm−1 is characteristic of triazole ring formation.28 Furthermore, the appearance of an amide vibration band near 1640 cm−1 is observable for MNP@3-4 in the spectrum. For MNP@3-5, the characteristic carboxylic acid vibration bands between 1690 and 1760 cm−1 appear after functionalization. All these data confirm the presence of 4 and 5 on the MNPs’ surface. The large vibration band between 1665 and 1760 cm−1 in the FTIR spectrum confirms that 6 is grafted onto the nanoparticles’ surface.
O and P–O vibration bands confirmed the anchorage via the phosphonate moieties on the MNPs’ surface.32 The strong vibration band at 579 cm−1 is characteristic of Fe–O vibrations typical of maghemite structures. For both MNP@3-4 and MNP@3-5, the appearance of vibrations bands in the FTIR spectra between 1400 and 1800 cm−1 is characteristic of triazole ring formation.28 Furthermore, the appearance of an amide vibration band near 1640 cm−1 is observable for MNP@3-4 in the spectrum. For MNP@3-5, the characteristic carboxylic acid vibration bands between 1690 and 1760 cm−1 appear after functionalization. All these data confirm the presence of 4 and 5 on the MNPs’ surface. The large vibration band between 1665 and 1760 cm−1 in the FTIR spectrum confirms that 6 is grafted onto the nanoparticles’ surface.
 | ||
| Fig. 2 FTIR spectra of 1 (dash) and 2 (A); MNP@3 (black), MNP@3-4 (red) and MNP@3-5 (blue) (B); MNP@3 (dash) and MNP@3-6 (C). | ||
First we evaluated the catalytic activity of 2 in an aldolization reaction between 4-p-nitrobenzaldehyde and cyclohexanone in isopropanol (iPrOH) at room temperature33–35 (Table 1). The reaction conditions were previously optimized with L-proline as a model catalyst, and 10 mol% of the catalyst appeared to be the best condition with full conversion in less than a day (entry 1, other data not shown). Unfortunately, after 7 days using nano-catalyst 2 under the same conditions, no conversion was observed (entry 2). In order to understand the influence of the MNPs and/or bisphosphonic acid, bare nanoparticles and 3 were used separately as additives for L-proline (entries 3 and 4). Both additions did not impair the reactivity, so neither the MNPs nor the bisphosphonate have an effect on L-proline’s activity. In fact it appeared that it was compound 1 itself which was completely unreactive: using 10 mol% of 1, even after 7 days, no conversion was detected (entry 5), even if N-methyl morpholine (NMM) was added as an additive (entry 6). Our hypothesis was that the amine group in compound 1 may interact with bisphosphonic functional groups in the free molecule as well as on the MNPs disabling its catalytic efficiency (Scheme 3).

 | ||
| Scheme 3 The addition of calcium may prevent catalyst deactivation. | ||
To validate this hypothesis we took advantage of the high affinity of bisphosphonic functional groups for calcium ions.36 We hoped that the presence of calcium ions would decrease the undesired interactions thus allowing the catalytic activity. So the reaction was performed with 1 in the presence of 10 mol% of anhydrous CaCl2 (entry 7). Conversion of some of the aldehyde was obtained but only 40% after 10 days. The conversion rate was still slow but tended to confirm our hypothesis. Besides, when CaCl2 was added to L-proline (entry 9), the conversion decreased to 60% after 7 days indicating that although having no catalytic efficiency (entry 8), CaCl2 greatly decreases the kinetic rate of the reaction (entry 9). To further ascertain our findings we performed catalysis with nano-catalyst 2 in presence of 10 mol% of anhydrous CaCl2 (entry 10). Thus, a good conversion (up to 60%) was achieved after only 3 days. The kinetic rate was still low compared to proline alone but nano-catalyst 2 was more efficient than catalyst 1 when CaCl2 was used. The same phenomenon of complete un-efficiency of the catalyst without the presence of calcium ions was found when MNP@3-4 and MNP@3-5 were used (data not shown). Again, no activity was observed even though the link between the bisphosphonate and proline was stiffened by the presence of the triazole ring. One must note that several reports have documented such interactions between a capping agent and nanomaterial core which modify the catalytic performance.37 The results being disappointing with the aldol reaction, we decided to move to another reaction also catalyzed by proline derivatives: the conjugate addition reaction of aldehydes to nitro-olefins.38,39 This reaction moreover presents another advantage in that it requires a lower percentage of catalyst thus allowing the use of a smaller amount of MNPs.
We first used the three parent molecules 4, 5 and 6 to evaluate their catalytic activities for conjugate addition reactions of aldehydes to nitro-olefins and to serve as reference. The reaction consisted of condensation of n-butyraldehyde with trans-β-nitrostyrene.31,38,39 The reaction conditions were optimized (data not shown) and the ideal conditions were found to be reaction in a CHCl3/iPrOH (9/1) medium in the presence of 30 mol% of NMM using 3 mol% of catalyst (Table 2). After less than a day, 4, 5 and 6 led to full conversion (entries 1, 2 and 3). Both 4 and 5 led to full conversion in 18 h with good diastereoselectivity ratios of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and reasonable to good enantioselectivities for 4 (ee = 60%) and 5 (ee = 75%). The results were more disappointing for peptide 6, which gave a lower diastereoselectivity ratio of 7:1 and a lower enantioselectivity (ee = 55%). It appeared that compared to the parent peptide described by Wennemer’s group the adjunction of the extra amino-acids at the N-terminus to allow the thiol–yne coupling greatly impaired the catalytic activity. When MNP@3-4, MNP@3-5 and MNP@3-6 were evaluated, an important decrease in the reaction kinetics and lower conversion yields in general were observed. For MNP@3-4, a good conversion of 70% was still achieved (entry 4) but after 6 days. For MNP@3-5 complete conversion was obtained after 4 days (entry 5) and for MNP@3-6 a maximum conversion of 45% was reached after 6 days. Though the kinetic rates were lower the diastereoselectivity as well as the ee were globally conserved for the three catalysts after anchoring them to the MNPs, indicating that their stereoselective abilities were not lost in the grafting process. One hypothesis to explain the slowing down of the kinetic rates could be that in the organic media the dispersion of the nanoparticles is not optimal, inducing some aggregation and decreasing the catalyst accessibility. Differences between MNP@3-4 and MNP@3-5 can be found in the variation of the linker rigidity, which in the case of MNP@3-5 would allow for better stability and/or orientation of the catalyst. Thus it could also have a lesser hypothetical deactivation as observed in the case of MNP@2. Recyclability experiments were conducted with MNP@3-5. The MNPs were recovered after the first run owing to their magnetic properties, washed several times and reused. For the second run, the results were unchanged after 96 h, and the same yield, enantioselectivity and diastereoselectivity were found. However, after the third run, though the stereoselectivities were unmodified, the conversion was lower reaching only 80%. This decrease cannot be attributed to leaching of the catalyst as TGA and EDX measurements didn’t show any blatant modification (data not shown). When the reaction time was longer, the conversion improved showing that this result could be explained by partial deactivation or loss of the nanocatalyst during the washing steps. No further experiments were run as the kinetic rate of the reaction was already low and we preferred to try to optimize the catalyst.
:1 and reasonable to good enantioselectivities for 4 (ee = 60%) and 5 (ee = 75%). The results were more disappointing for peptide 6, which gave a lower diastereoselectivity ratio of 7:1 and a lower enantioselectivity (ee = 55%). It appeared that compared to the parent peptide described by Wennemer’s group the adjunction of the extra amino-acids at the N-terminus to allow the thiol–yne coupling greatly impaired the catalytic activity. When MNP@3-4, MNP@3-5 and MNP@3-6 were evaluated, an important decrease in the reaction kinetics and lower conversion yields in general were observed. For MNP@3-4, a good conversion of 70% was still achieved (entry 4) but after 6 days. For MNP@3-5 complete conversion was obtained after 4 days (entry 5) and for MNP@3-6 a maximum conversion of 45% was reached after 6 days. Though the kinetic rates were lower the diastereoselectivity as well as the ee were globally conserved for the three catalysts after anchoring them to the MNPs, indicating that their stereoselective abilities were not lost in the grafting process. One hypothesis to explain the slowing down of the kinetic rates could be that in the organic media the dispersion of the nanoparticles is not optimal, inducing some aggregation and decreasing the catalyst accessibility. Differences between MNP@3-4 and MNP@3-5 can be found in the variation of the linker rigidity, which in the case of MNP@3-5 would allow for better stability and/or orientation of the catalyst. Thus it could also have a lesser hypothetical deactivation as observed in the case of MNP@2. Recyclability experiments were conducted with MNP@3-5. The MNPs were recovered after the first run owing to their magnetic properties, washed several times and reused. For the second run, the results were unchanged after 96 h, and the same yield, enantioselectivity and diastereoselectivity were found. However, after the third run, though the stereoselectivities were unmodified, the conversion was lower reaching only 80%. This decrease cannot be attributed to leaching of the catalyst as TGA and EDX measurements didn’t show any blatant modification (data not shown). When the reaction time was longer, the conversion improved showing that this result could be explained by partial deactivation or loss of the nanocatalyst during the washing steps. No further experiments were run as the kinetic rate of the reaction was already low and we preferred to try to optimize the catalyst.

In order to improve the catalytic efficiency we decided to evaluate the effects of modification of the lipophilicity of the nanoparticle surface on the catalysis. To prepare a more lipophilic catalyst we took advantage of the possibility to multi-functionalize the surface of our MNPs. Indeed we previously described the possibility to chemoselectively di-functionalize the MNP@3 nanoplatform using two sequential click chemistry reactions: CuAAC and thiol–yne. Herein, we applied this double click functionalization to further graft two thioalkanes differing by their terminal group onto the surface of MNP@3-5. Dodecanthiol 7 and 1-mercapto-dodecanoic acid 8 were thus added to MNP@3-5 in a second functionalization step using a thiol–yne reaction not impairing the catalytic sites (Scheme 4). Both molecules have long alkyl chains which could create a promiscuous environment near the catalyst enhancing accessibility of the reactant and thus improving the kinetic rate of the reaction. 7 and 8 were grafted using the protocol previously described and the number of grafted molecules was determined using EDX measurements (see ESI†). 170 ± 25 molecules of 7 and 250 ± 25 molecules of 8 were respectively found at the surface of MNP@3-5-7 and MNP@3-5-8. It should be noted that the adjunction of 7 modified the zeta potential from −45 mV to −14 mV (see ESI†) on the resulting nanoparticles, whereas the addition of 8 only slightly modified it to −39 mV.
 | ||
| Scheme 4 Double click methodology for MNP@3-5-7 and MNP@3-5-8. | ||
The two modified catalysts were then evaluated under the same conditions (Table 2). The results were contrasting, as for MNP@3-5-7 full conversion was obtained (entry 7) but with an increased length of time (6 days of reaction instead of 4 days). Once again the kinetic rate was modified but without impairing the stereoselective ability, as a comparable diastereoselectivity and enantioselectivity were found. On the other hand, when MNP@3-5-8 was evaluated this time full conversion was achieved in 3 days instead of the 4 needed previously. The diastereoselectivity was unchanged and a negligible decrease of the ee was observed. To explain these results, it could be hypothesized that whilst improving the lipophilicity of the nanocatalyst the addition of 7 decreased the electrostatic repulsion between the nanoparticles as it shields surface charges. This phenomenon would thus impair the catalyst accessibility by increasing particle interactions through possible chain inter-digitation in the organic media. This is consistent with the fact that when adding 8 the presence of the terminal carboxylate still ensures electrostatic repulsion between the nanoparticles, thus preventing alkyl chain interactions and improving the catalyst’s access to the reactants. Finally, this double controlled click functionalization permitted us to achieve good conversion using a small amount of catalyst (3 mol%) in 3 days.
3. Experimental
3.1. General
Boc-Pro-Osu (≥98%), 1-mercapto-dodecanoic acid (96%), 1-dodecanthiol (≥98%), dimethylamine (40 wt% in H2O), and tris(trimethyl-silyl)phosphite were purchased from Sigma-Aldrich and were used without further purification. 4-p-Nitrobenzaldehyde (99%), cyclohexanone (99%), n-butyraldehyde (99%), trans-β-nitrostyrene, TFA (99%) and DMF (99%) were purchased from Alfa Aesar and were used without further purification. Peptide 6 (H-L-Pro-L-Pro-Glu-AHX-Cys-NH2) (HPLC > 95%) was purchased from Eurogentec. Solvents: methanol (RS HPLC), isopropanol (HPLC), dichloromethane (RE amylene stabilized), ethyl acetate, hexane and diethylether (RE stabilized) were purchased from Carlo Erba SDS. All the other reagents were obtained from current commercial suppliers and were used without purification. Water was purified with a Millipore system (resistivity 18.2 MΩ cm). 1H NMR spectra (400 MHz) and proton-decoupled 13C NMR spectra (100.63 MHz) were recorded on a Bruker Advance III 400 spectrometer. Chemical shifts are reported in parts per million (ppm) on the δ scale. The residual solvent peaks were used as internal references (1H NMR: CHCl3 7.26 ppm, H2O 4.79 ppm; 13C NMR: CDCl3 77.2 ppm). Data are reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, qt = quintuplet, m = multiplet, and coupling constant(s) are given in Hz. FTIR spectra were recorded as KBr pellets or between NaCl plates (for liquid products) on a Thermo Scientific Nicolet 380 FTIR spectrophotometer and are reported in wavenumbers (cm−1). TEM images were obtained using a FEI CM10 microscope (Philips). HPLC was performed with an Agilent 1200 Infinity device, using a Daicel AD-H chiral column, fitted with a guard column. The aldolization and Michael adducts are previously known and their spectroscopic data were in accordance with the literature.29–31,38,393.2. MNPs synthesis
For the MNPs synthesis refer to a publication by Lalatonne et al.23 and the ESI.†3.3. Synthesis of 1
Sodium alendronate (500 mg, 1.85 mmol) was dissolved in 50 mL of pure water and the pH was adjusted to 7 with NaOH (1 M) (10 mL). (Boc)-L-Pro-OSu (1.20 mg, 3.7 mmol) previously dissolved in 10 mL of DMF and DIPEA (700 μL, 4.1 mmol) was added to the water solution. The resulting mixture was stirred for 4 days and then washed twice with 15 mL of Et2O. The white product was obtained with 75% yield after precipitation in MeOH/water mixture [9:1]. A classical deprotection was performed using 6 mL of a CH2Cl2/TFA (1/1) mixture for 30 min at room temperature. Then the solvents were removed under reduced pressure. The product was washed twice with 10 mL of Et2O. After crystallization, a white powder was obtained with 75% yield. The product (1) was obtained as a TFA salt. 1H (D2O, 25 °C): δ = 4.20 (dd, 1H); 3.35–3.10 (m, 4H); 2.34–2.2 (m, 2H); 2–1.62 (m, 6H) ppm. 13C{1H} (D2O, 25 °C): δ = 169.33; 73.35 (t, JC,P = 138.3 Hz); 59.78; 46.34; 40.02; 30.79; 29.64; 23.50; 23.16 ppm. 31P{1H} (D2O, 25 °C): δ = 18.26 ppm. IR (KBr): 3400; 3294; 3103; 2983; 2786; 2394; 1685; 1681; 1573; 1441; 1435; 1385; 1206; 1138; 1068; 930; 840; 801; 722; 667; 580; 538; 461 cm−1. HRMS: (ESI-Q Tof) C9H21N2O8P2 m/z (M + H)+: 347.08; calc: 347.08. [α]25D (589 nm, acidic pH, H2O) = −91.3°.
3.4. Synthesis of the MNPs 2
To 4 mL of an aqueous solution of 1 (43 mg, 83 μmol, pH = 4), an aqueous solution of the bare MNPs (4 mL, [Fe] = 0.16 M, pH = 4) was added. The resulting mixture was vigorously stirred for 2 h at 90 °C. The nanoparticles 2 were then washed 5 times with filtration using 30 kDa Amicon filters, in order to remove the excess unbound 1. The MNPs 2 were dispersed in pure water and the pH of the solution was adjusted to 7 using NaOH solution (0.1 M).3.5. General procedure for the CuAAC
For the BPheptyne synthesis refer to a publication by Demay-Drouhard et al.25 The CuAAC procedure was performed according to ref. 27 and 28. Briefly, 5 equivalents of azido-proline (4 or 5), copper sulfate hexahydrate (5%) and sodium ascorbate (20%) were added to 2.5 mL of an aqueous solution of MNP@3 ([Fe] = 0.1–0.2 M, [3] = 2–5 mM) and reacted in a sealed vial under microwave irradiation for 8 min (Tmax (°C) = 100 °C). The as-synthesized nanoparticles were washed 5 times using magnetic separation with acidic pure water (pH = 2) and then dissolved in water at pH 7. The same protocol was used for the double functionalization, see the ESI.†3.6. General procedure for the thiol–yne reaction
The thiol–yne reaction procedure was performed according to a publication by Demay-Drouard et al.25 An aqueous solution of MNP@3 ([Fe] = 0.1–0.15 M, [3] = 2.5–3.8 mM, pH = 7, V = 3 mL) and 3 mL of DMF were mixed with a radical initiator, 1-hydroxycyclohexylphenylketone (10%). The thiol molecules were added (2 equivalents to 3, 10 for the double functionalization) and the mixture was stirred for one and a half hours under UV irradiation (360 nm). The as-functionalized MNPs were then washed 5 times with ethanol and 5 times with HCl 0.01 M using magnetic separation (for 6, only two ethanol washings were performed). The MNPs were dispersed in water and the pH was adjusted to 7.3.7. General procedure for the aldolization
The catalyst (10 mol%, 0.04 mmol) was dissolved in 420 μL of iPrOH. To this mixture, 60.4 mg of 4-p-nitrobenzaldehyde (0.4 mmol) and 210 μL of cyclohexanone (2 mmol) were added. The resulting mixture was mechanically mixed (600 rpm) at room temperature. The reaction was monitored using 1H NMR. After the reaction, the crude mixture was extracted three times with Et2O. The organic phase was then washed with water, dried with MgSO4 and the solvent was removed under reduced pressure. The product, an orange powder, was purified on a silica column using a hexane/AcOEt gradient. When using the MNPs as a catalyst: catalyst (2 μmol), 4-p-nitrobenzaldehyde (0.02 mmol), cyclohexanone (0.1 mmol), and iPrOH (21 μL) were used. The water was removed from the MNPs by magnetic separation.3.8. General procedure for the Michael addition
The catalyst (18.8 μmol) was first dissolved in 1 mL of CHCl3/iPrOH (9/1) and NMM (0.188 mmol, 23 μL), and mechanically stirred (600 rpm) for 5 min at room temperature. Then trans-β-nitrostyrene (0.7 mmol, 105 mg) and n-butyraldehyde (2.1 mmol, 191 μL) were added. The reaction was monitored using 1H NMR. After the reaction, the crude mixture was extracted three times with CH2Cl2, and three times with water. The organic phase was then dried with MgSO4 and the solvent and excess aldehyde were removed under reduced pressure. The yellow-orange product was purified on a silica column (hexane/AcOEt). With the MNPs: catalyst (1 μmol), trans-β-nitrostyrene (32.3 μmol, 4.8 mg), n-butyraldehyde (97 μmol, 8.8 μL), CHCl3/iPrOH (9/1) (50 μL), and NMM (30 mol%) were used. The water was removed from the MNPs by centrifugation.4. Conclusions
In conclusion, new nano-catalysts were developed using two methodologies. A “grafting to” methodology, on one hand, that permits anchoring of numerous catalytic moieties on the MNP’s surface. On the other hand, a “grafting from” click methodology that led to easily functionalized nano-catalysts and allowed for controlled multi-functionalization of the nanoparticle surface. Evaluating the obtained catalysts, we showed that the nature of the surface has a crucial impact on the catalysis efficiency. Use of 2 in aldolization made evident a deactivation mechanism of the catalyst, probably due to strong interactions with the anionic charges present at the surface. In 1,4-Michael addition reactions, we observed a decrease of the kinetic rate of the reaction when grafting the catalyst onto the nanoparticle surface but with a maintaining of their diastereoselectivity and enantioselectivity. A double controlled click methodology permitted us to improve the kinetic rate and emphasized the fact that once again the activity was closely related to the tailoring of the nanoparticle surface. Finally, nano-catalyst MNP@3-5-8 was shown to be efficient in the 1,4-Michael addition reaction with complete conversion after 72 h and good diastereoselectivity and enantioselectivity, using a small amount of catalyst (3 mol%).Acknowledgements
This work was supported by Région Ile de France. We are grateful to N. Liévre (University of Paris 13) for TEM observations, J. Perard (Paris 5 University) for specific optical rotation measurements and J. Hardouin (Rouen University) for mass spectrometry measurements.References
- R. B. N. Baig and R. S. Varma, Chem. Commun., 2013, 49, 752 RSC.
- M. B. Gawande, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371 RSC.
- R. Mrowczynski, A. Nan and J. Liebscher, RSC Adv., 2014, 4, 5927 RSC.
- R. Varma, Sustainable Chem. Processes, 2014, 2, 11 CrossRef.
- K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243 CrossRef CAS.
- P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS.
- B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef CAS.
- P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138 CrossRef CAS PubMed.
- C. O'Dálaigh, S. A. Corr, Y. Gun’ko and S. J. Connon, Angew. Chem., Int. Ed., 2007, 46, 4329 CrossRef PubMed.
- O. Gleeson, G.-L. Davies, A. Peschiulli, R. Tekoriute, Y. K. Gun’ko and S. J. Connon, Org. Biomol. Chem., 2011, 9, 7929 CAS.
- E. Nehlig, L. Motte and E. Guénin, Catal. Today, 2013, 208, 90–96 CrossRef CAS.
- E. Nehlig, B. Waggeh, N. Millot, Y. Lalatonne, L. Motte and E. Guenin, Dalton Trans., 2015, 44, 501 RSC.
- A. Dondoni, Angew. Chem., Int. Ed., 2008, 47, 8995 CrossRef CAS PubMed.
- A. Dondoni and A. Marra, Eur. J. Org. Chem., 2014, 2014, 3955 CrossRef CAS.
- B. D. Fairbanks, T. F. Scott, C. J. Kloxin, K. S. Anseth and C. N. Bowman, Macromolecules, 2008, 42, 211 CrossRef PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed.
- H. He and C. Gao, Curr. Org. Chem., 2011, 15, 3667 CrossRef CAS.
- N. Li and W. H. Binder, J. Mater. Chem., 2011, 21, 16717 RSC.
- T. P. Lodge, Macromolecules, 2009, 42, 3827 CrossRef CAS.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745 RSC.
- A. K. Tucker-Schwartz, R. A. Farrell and R. L. Garrell, J. Am. Chem. Soc., 2011, 133, 11026 CrossRef CAS PubMed.
- Y. Lalatonne, C. Paris, J. M. Serfaty, P. Weinmann, M. Lecouvey and L. Motte, Chem. Commun., 2008, 2553 RSC.
- C. de Montferrand, Y. Lalatonne, D. Bonnin, N. Lièvre, M. Lecouvey, P. Monod, V. Russier and L. Motte, Small, 2012, 8, 1945 CrossRef CAS.
- P. Demay-Drouhard, E. Nehlig, J. Hardouin, L. Motte and E. Guénin, Chem.–Eur. J., 2013, 19, 8388 CrossRef CAS PubMed.
- E. Guénin, Y. Lalatonne, J. Bolley, I. Milosevic, C. Platas-Iglesias and L. Motte, J. Nanopart. Res., 2014, 16, 1 CrossRef.
- J. Bolley, E. Guenin, N. Lievre, M. Lecouvey, M. Soussan, Y. Lalatonne and L. Motte, Langmuir, 2013, 29, 14639 CrossRef CAS PubMed.
- E. Guénin, J. Hardouin, Y. Lalatonne and L. Motte, J. Nanopart. Res., 2012, 14, 1 CrossRef.
- Y. Arakawa and H. Wennemers, ChemSusChem, 2012, 6, 242 CrossRef PubMed.
- Y. Arakawa, M. Wiesner and H. Wennemers, Adv. Synth. Catal., 2011, 353, 1201 CrossRef CAS.
- M. Wiesner, G. G. Upert, G. Angelici and H. Wennemers, J. Am. Chem. Soc., 2009, 132, 6 CrossRef PubMed.
- F. Benyettou, I. Chebbi, L. Motte and O. Seksek, J. Mater. Chem., 2011, 21, 4813 RSC.
- F. Giacalone, M. Gruttadauria, P. Agrigento, P. Lo Meo and R. Noto, Eur. J. Org. Chem., 2010, 2010, 5696 CrossRef.
- N. Mase, Y. Nakai, N. Ohara, H. Yoda, K. Takabe, F. Tanaka and C. F. Barbas, J. Am. Chem. Soc., 2005, 128, 734 CrossRef PubMed.
- J. Mlynarski and S. Bas, Chem. Soc. Rev., 2014, 43, 577 RSC.
- J. Galezowska and E. Gumienna-Kontecka, Coord. Chem. Rev., 2012, 256, 105 CrossRef CAS.
- N. Yan, Y. Yuan and P. J. Dyson, Dalton Trans., 2013, 42, 13294 RSC.
- M. Wiesner, M. Neuburger and H. Wennemers, Chem.–Eur. J., 2009, 15, 10103 CrossRef CAS PubMed.
- M. Wiesner, J. D. Revell, S. Tonazzi and H. Wennemers, J. Am. Chem. Soc., 2008, 130, 5610 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra20644h |
| This journal is © The Royal Society of Chemistry 2015 |