
Competitive cation binding to phosphatidylinositol-4,5-bisphosphate domains revealed by X-ray fluorescence†
Z. T. Graber‡
a,
W. Wangb,
G. Singhc,
I. Kuzmenkod,
D. Vakninb and
E. E. Kooijman*e
aDepartment of Chemistry and Biochemistry, Kent State University, Kent, OH 44242, USA
bAmes Laboratory and Department of Physics and Astronomy, Iowa State University, Ames, IA 50011, USA
cDepartment of Physics, Kent State University, Kent, OH 44242, USA
dX-ray Science Division, Advanced Photon Source, Argonne National Laboratory, Lemont, IL 60439, USA
eDepartment of Biological Sciences, Kent State University, Kent, OH 44242, USA. E-mail: ekooijma@kent.edu
First published on 7th December 2015
Abstract
Phosphatidylinositol-4,5-bisphosphate (PIP2) is an important signaling phospholipid in the inner leaflet of the cell membrane. Due to the high negative charge of its headgroup, PIP2 strongly interacts with cellular cations. We have used synchrotron diffraction and fluorescence techniques to determine preferential cation binding to two-dimensional PIP2 templates. The natural, highly unsaturated PIP2 is manipulated as a Langmuir monolayer on a physiological buffer containing 100 mM KCl and varying amounts of Ca2+ and Mg2+. X-ray fluorescence shows an 800% surface enhancement in K+ concentration (bound to PIP2) compared to bulk concentration. Adding physiological levels of Ca2+( 1–100 μM) results in gradual replacement of K+ by Ca2+ ions, leading to a significant change in the organization of the PIP2 model membrane, while higher concentrations (100–1000 μM) lead to three orders of magnitude increase in surface [Ca2+]. Similar experiments with Mg2+ ions also show strong ion binding to PIP2 at physiological levels (1 mM) with a lesser structural effect. For mixed solutions of Mg2+ and Ca2+ we find that Ca2+ occupies the majority of binding sites. Remarkably we find that, with both 1 mM Mg2+ and 1 mM Ca2+ in the subphase there is still a 400% surface enrichment of K+ ions at the headgroup region.
1 Introduction
Although a minor phospholipid in cell membranes, phosphatidylinositol-4,5-bisphosphate (PIP2) is implicated in nearly all aspects of cell physiology.1 Its versatile functions stem from the highly charged and phosphorylated inositol headgroup which is involved in numerous enzyme regulation events. For example, PIP2 binds the tumor suppressor PTEN and activates it to prevent uncontrolled cell growth.2–4 Through its highly phosphorylated headgroup, PIP2 acts as a template for Ca2+ signaling and inwardly rectifying potassium channels.5,6 PIP2 is highly negatively charged at neutral pH and much of its signaling occurs via electrostatic interactions with various ions and charged moieties of proteins.7,8 Specific cations interact with PIP2 to mediate or reduce its interactions with signaling partners. For instance, it has been proposed that the highly cationic protein MARKS regulates PIP2 by engulfing it with cationic lysine residues, thus neutralizing PIP2 from binding to other targets until MARKS is removed via a calmodulin mediated mechanism.9,10 Other polyvalent cations, such as neomycin and spermine, have been found to inhibit PIP2 mediated activation of the ATP-sensitive potassium channel.11Ca2+ has been found to interact strongly with PIP2 and cause clustering that potentially enhances its signaling roles.12,13 Clustering has been observed in AFM imaging of Langmuir monolayers of PIP2 transferred to solid support and by condensation of PIP2 monolayers upon addition of Ca2+ to the physiologically prepared solution.13 In contrast, limited cluster formation is observed for monolayers of PIP2 that are spread on Mg2+ solutions.13 On mixed Ca2+/Mg2+ solutions, a higher intrinsic binding constant is found for Ca2+ than Mg2+, and Ca2+ replaces Mg2+.13 It is well established that Ca2+ flux is commonly exploited by the cell to trigger signaling events, potentially via PIP2.14
Here, we report on the competition between K+, Mg2+, and Ca2+ for PIP2 binding sites in a two-dimensional template formed by a PIP2 monolayer that is prepared at the vapor/solution interface as a model environment of the cytosol. The use of a lipid monolayer of natural, highly unsaturated, PIP2 that forms a uniform headgroup template allows direct evaluation of specific cation–PIP2 interactions without the complications of mixing with other lipids that may introduce non-specific ion binding. The pure PIP2 monolayers used in this study resemble the domain formation of PIP2 in model mixed-lipid membranes that may exist in living cells,12,13,15–17 especially in regions of active PIP2 synthesis. Here we employ a combination of X-ray fluorescence and reflectivity to directly characterize cation binding to PIP2. A previous study utilized X-ray reflectivity and grazing incidence X-ray diffraction to determine the influence of Ca2+ on a mixed DPPC/PIP2 monolayer with emphasis on the mechanism of synaptic vesicle fusion.18 Due to the mixed nature of the lipid-film no quantitative information on the specific interaction of Ca2+ with PIP2 is possible from those data. Our current study extends other work13,18–21 in a few ways, in particular, by providing a quantitative measure of the number of bound ions per molecule and by systematically and specifically monitoring competition among cytoplasmic ions.
2 Materials and methods
2.1 Materials
L-α-Phosphatidylinositol-4,5-bisphosphate (brain, porcine–triammonium salt) [brain PI(4,5)P2] was purchased from Avanti Polar Lipids (Birmingham, AL) in powder form and dissolved in a 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9:1 by volume mixture of chloroform, methanol, and water to form a lipid stock. Concentration was determined by carefully weighing the lipid powder on an analytical balance before dissolving. CaCl2, MgCl2, KCl, HCl, Tris, and EDTA of purity > 99% were purchased from Sigma Aldrich (St. Louis, MO), and the Ca/Mg/K salts were at least 99.99% pure based on trace metal basis. Buffer subphase contained 10 mM Tris and 100 mM KCl, along with varying amounts of CaCl2, MgCl2, and EDTA. Buffer pH was set to pH 7.2 ± 0.1 using high purity HCl. 100 mM KCl was used to replicate physiological salt levels. For buffers with no divalent cations, 0.1 mM EDTA was used to chelate any trace divalent cation impurities. MilliQ water was obtained from a MilliQ filter system and was determined to have 18.2 MΩ resistivity.
:9:1 by volume mixture of chloroform, methanol, and water to form a lipid stock. Concentration was determined by carefully weighing the lipid powder on an analytical balance before dissolving. CaCl2, MgCl2, KCl, HCl, Tris, and EDTA of purity > 99% were purchased from Sigma Aldrich (St. Louis, MO), and the Ca/Mg/K salts were at least 99.99% pure based on trace metal basis. Buffer subphase contained 10 mM Tris and 100 mM KCl, along with varying amounts of CaCl2, MgCl2, and EDTA. Buffer pH was set to pH 7.2 ± 0.1 using high purity HCl. 100 mM KCl was used to replicate physiological salt levels. For buffers with no divalent cations, 0.1 mM EDTA was used to chelate any trace divalent cation impurities. MilliQ water was obtained from a MilliQ filter system and was determined to have 18.2 MΩ resistivity.
2.2 Experimental set-up and data analysis
X-ray reflectivity (XR) and near-total-reflection fluorescence (XNTRF) measurements were conducted on the liquid surface spectrometer at beamline 9ID-C, Advanced Photon Source, Argonne National Laboratory (E = 8.0 keV; wavelength λ = 1.5497 Å). ESI Fig. S1† provides a sketch of the experimental setup. The monolayer is formed by drop-wise deposit (at a molecular area of 85–100 Å2 per molecule) of PIP2 stock solution onto the aqueous surface of the subphase solution that is contained in a Langmuir trough and maintained at constant temperature of 20 °C. The Langmuir trough is encapsulated in an enclosure with thin Kapton windows and purged with water-saturated helium gas to minimize background scattering from air and potential X-ray radiation damage to the samples. An oxygen sensor (S101, Qubit System Inc.) in the trough is used to monitor oxygen levels in the enclosure. The monolayer is compressed by a Teflon barrier to a desired surface pressure of 30 ± 2 mN m−1. Surface pressure is monitored by a microbalance made of a Wilhelmy microbalance and filter paper plate. The X-ray measurements are started after the oxygen–helium exchange reaches equilibrium, where the oxygen content inside the enclosure is reduced by a factor of at least 100 with respect to that of the ambient atmosphere. Radiation damage is monitored by systematically translating the trough laterally to probe fresh surfaces and examine measurement reproducibility.The specular reflection is collected by a Bicron scintillation detector at an X-ray exit angle αf = αi, αi being the incident angle. The angular-dependence of both XR and XNTRF are expressed as functions of Qz, the z-component (surface normal) of the scattering (wave vector transfer) vector Q. Qz is related to αi via Qz = (4π/λ)sinαi, and Qc represents the Qz that corresponds to the critical angle for total reflection, αc (= 0.154° at E = 8 keV).
The fluorescence signals are collected at a series of αi near the critical angle αc by a Vortex energy dispersive detector (EDD, silicon-drift, Vortex-90EX) with a collimator in the front end of the EDD probe, which accepts the X-ray fluorescence of emitted photons in the direction along the surface normal at ∼1° angular resolution.
X-ray reflectivity data, R(Qz), are normalized to the calculated Fresnel reflectivity (RF), for an ideally smooth, flat air–water interface.22,23 The R/RF data for a surface monolayer on an aqueous surface are accounted for in terms of a simplistic, two-box (i.e., two-layer) structural model, where one box contains the hydrophilic head group, and the other contains the hydrocarbon tail.22,24,25 Each box is characterized by a uniform electron density (ρ) and vertical height (l) relative to the interface. Given (ρH, lH) and (ρT, lT), in which subscripts “H” and “T” represent the headgroup and the hydrocarbon tail respectively, a step-like, discrete ED profile across the interfaces, ρ0(z), is constructed and its corresponding reflectivity R0(Qz) is calculated by the Parratt recursion method and smeared by a Gaussian22,25 (see ESI†). The structural refinements are carried out through a least squares optimization method.25 Surface fluorescence studies have been performed in the past26,27 and analytic routines to quantify the surface enrichment of ions have been established28,29 and are discussed in detail in the ESI.†
3 Results
3.1 Ca2+ reorients PIP2 in model lipid membranes
Fig. 1(a) shows normalized XR, R/RF, from natural PIP2 monolayers at physiologically relevant subphase conditions in the absence and presence of Ca2+. Addition of Ca2+ results in dramatic changes in the structure of PIP2. There is a systematic shift of the R/RF minimum to lower momentum transfers, Qz, and an overall rise in reflectivity as Ca2+ concentration increases. Qualitatively, these features indicate an increase in film thickness and a higher electron density in the film in the presence of Ca2+. Fig. 1(b) shows the corresponding electron (scattering) densities (ED) across the lipid layer calculated from the best fit parameters (see Table 1) to the reflectivity curves. The increase in ED is mainly in the headgroup region and the increase in thickness occurs in the acyl-chain region (see Table 1).24 The increased density in the headgroup region is due to Ca2+ binding and screening of PIP2 charge leading to a reduction of the molecular area due to reduced repulsion. As a result the hydrocarbon chains are able to pack more tightly (i.e. relative order increases, but they are still disordered as we observe no diffraction from the monolayer (data not shown)), and this results in a subsequent condensation of the PIP2 monolayer in agreement with the condensing effect of divalent cations on PIP2.13,19–21 The XR data allows for an estimation of the number of Ca2+ bound to the headgroup of PIP2, based on a space-filling model,25,28,29 and the estimated electron density of the Ca2+ ion (see Table 1 and ESI†). | ||
| Fig. 1 X-ray reflectivity of the PIP2 monolayer in the presence of varying [Ca2+]. (a) X-ray reflectivity measured from a PIP2 monolayer spread on a pH 7.2 ± 0.1 subphase containing 10 mM Tris, 100 mM KCl, and varying amounts of CaCl2 (from 0 to 1000 μM). (b) Electron density versus distance from the PIP2 headgroup/aqueous interface for modeling the corresponding reflectivities shown in (a). Electron density profiles are calculated from a 2-box refinement to the normalized reflectivity with the parameters that are listed in Table 1. | ||
| Subphase (CaCl2) | 0 mM | 1 μM | 10 μM | 100 μM | 1 mM |
|---|---|---|---|---|---|
| a Structural parameters derived from the reflectivity curves for a PIP2 monolayer spread on a pH 7.2 ± 0.1 subphase containing 10 mM Tris, 100 mM KCl, as well as 1 mM, 10 mM, 100 mM, or 1 mM CaCl2 respectively. lH (Å) describes the length of the headgroup region, ρH (e Å−3) is the electron density of the headgroup region, lT (Å) is the length of the acyl chain region, ρT (e Å−3) is the electron density of the acyl chain region, σ (Å) is the surface roughness, Amol (Å2) is the area per molecule in the monolayer calculated from the X-ray reflectivity, and Amol2 (Å2) is the area per molecule in the monolayer determined from the isotherms. Ca2+/PIP2 is the number of surface-bound calcium ions (Ca2+) per molecule (PIP2). | |||||
| lH (Å) | 10.3(7) | 9.2(9) | 7.6(4) | 7.2(4) | 8.2(6) |
| ρH (e Å−3) | 0.51(1) | 0.56(2) | 0.65(2) | 0.72(2) | 0.70(3) |
| lT (Å) | 13.4(3) | 14.7(4) | 16.2(2) | 18.1(2) | 18.4(3) |
| ρT (e Å−3) | 0.323(3) | 0.321(4) | 0.322(2) | 0.322(2) | 0.307(4) |
| lH + lT (Å) | 23.6(4) | 24.0(5) | 23.8(2) | 25.2(2) | 26.7(3) |
| σ (Å) | 4.2(1) | 4.3(2) | 4.6(1) | 4.7(1) | 4.5(1) |
| Amol (Å2) | 64–69 | 58–62 | 54–55 | 48–49 | 47–49 |
| Amol2 (Å2) | 70(5) | 57(6) | 48(10) | 45(10) | 42(10) |
| Ca2+/PIP2 | n/a | 0.3 ± 0.5 | 0.6 ± 0.3 | 0.9 ± 0.4 | 1.6 ± 0.9 |
3.2 Mg2+ affects the structure of PIP2 to a lesser degree
Fig. 2(a) shows X-ray reflectivity from PIP2 monolayers on buffer subphases containing 1 mM Mg2+ and various Ca2+ concentrations. It should be noted that the XR is not very sensitive to accumulation of the lighter Mg2+ ion, as its replacement with water molecules in the headgroup region does not affect the ED appreciably. While the addition of 10 μM Ca2+ (to the 1 mM Mg2+) is hardly noticeable in the XR data, increasing Ca2+ concentration has a more significant effect, indicating the exchange of Mg2+ by Ca2+ even at inferior Ca2+ concentrations, as may occur during Ca2+ signaling. More quantitatively, Fig. 2(b) shows the ED profiles that best fit the reflectivity (solid lines Fig. 2(a)) with the structural parameters listed in Table 2. The XR data allows for an estimation of the number of divalent cations (M2+) bound to the headgroup of PIP2, based on a space-filling model25,28,29 and the estimated electron density of the Mg2+ ion (see Table 2 and ESI†). The volume and electron density of the ion is dependent on the hydration state. If we assume a dehydrated Mg2+ ion we calculate a minimum of ∼2 Mg2+ ions per PIP2 headgroup. However, Slochower et al. have recently suggested that the Mg2+ ion remains hydrated while bound to PIP2.30 If we assume a hydration shell of six water molecules around each Mg2+ ion, we calculate a much lower Mg2+ density of ∼0.6 ions/PIP2.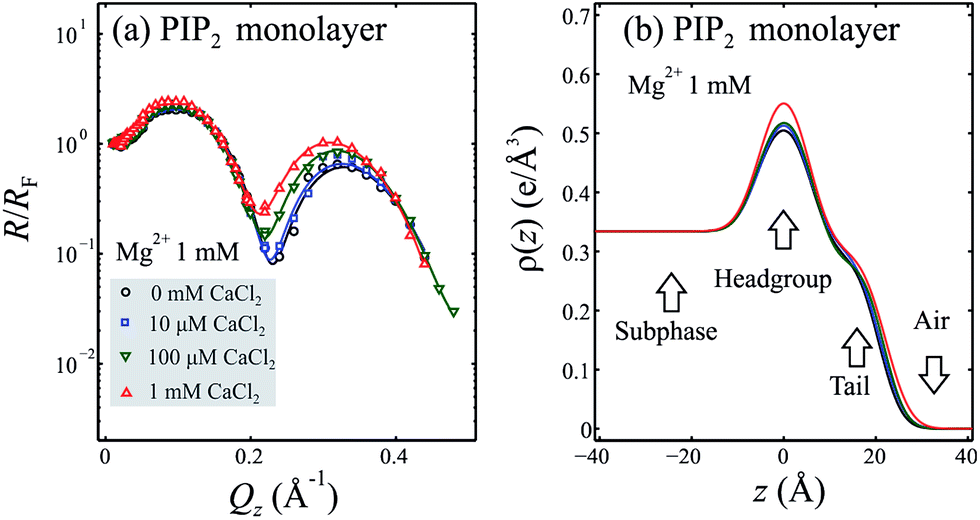 | ||
| Fig. 2 X-ray reflectivity of the PIP2 monolayer in the presence of 1 mM Mg2+ and varying [Ca2+]. (a) X-ray reflectivity measured from a PIP2 monolayer spread on a pH 7.2 ± 0.1 subphase containing 10 mM Tris, 100 mM KCl, 1 mM MgCl2, and varying amounts of CaCl2 (from 0 to 1000 μM). (b) Electron density versus distance from the PIP2 headgroup/aqueous interface for modeling the corresponding reflectivities shown in (a). Electron density profiles are calculated from a 2-box refinement to the normalized reflectivity with the parameters that are listed in Table 2. | ||
| Mg2+ 1 mM | 0 μM Ca2+ | 10 μM Ca2+ | 100 μM Ca2+ | 1 mM Ca2+ |
|---|---|---|---|---|
| a Structural parameters derived from the reflectivity curves for a PIP2 monolayer spread on a pH 7.2 ± 0.1 subphase containing 10 mM Tris, 100 mM KCl, 1 mM MgCl2, as well as 0 μM, 10 μM, 100 μM, or 1 mM CaCl2 respectively. Parameters are the same as those for Table 1. | ||||
| lH (Å) | 10(1) | 9(1) | 9(2) | 10.0(8) |
| ρH (e Å−3) | 0.57(4) | 0.60(4) | 0.63(5) | 0.63(3) |
| lT (Å) | 15.4(6) | 16.0(6) | 16.9(8) | 17.0(4) |
| ρT (e Å−3) | 0.303(7) | 0.308(8) | 0.30(1) | 0.300(5) |
| lH + lT (Å) | 25.4(6) | 25.3(6) | 25.8(9) | 27.0(4) |
| σ (Å) | 4.2(2) | 4.3(2) | 4.3(3) | 4.3(2) |
| Amol (Å2) | 55–64 | 53–57 | 50–55 | 51–58 |
| Amol2 (Å2) | 54(6) | 55(6) | 56(6) | 57(6) |
| Mg2+/PIP2 | 2.0 ± 1.8 | n/a | n/a | n/a |
3.3 X-ray fluorescence
A more direct way to determine cation binding to PIP2 model membranes is X-ray fluorescence. Fig. 3(a) shows X-ray fluorescence spectra (Qz < 0.0217 Å−1) for a pure 100 mM KCl buffer interface (no PIP2 present, black squares) compared to the same interface in the presence of PIP2 (red squares). The highly negative surface charge of PIP2 attracts K+ from the buffer solution as expected from the Poisson–Boltzman theory of the double layer. The K+ surface concentration is enriched by a factor of eight to 0.8 M when PIP2 is present as compared to 0.1 M in the bulk (surface concentration is based on the total volume calculated from the penetration depth of the X-ray (D ∼ 52 Åat Qz = 0.01 Å−1) and the molecular area of the PIP2 molecule (A ∼ 50 Å2, taken from isotherms and consistent with published results (Slochower et al.,21 see Fig. 1)). As we increase the [Ca2+] in the bulk, the surface [Ca2+] increases dramatically (green (1 μM), blue (0.1 mM), and orange (1 mM) data points) with a simultaneous reduction in [K+], as Ca2+ displaces K+ from the headgroup interface due to the higher binding constant for Ca2+ compared to that of K+. | ||
| Fig. 3 X-ray fluorescence reveals specific cation binding to the PIP2 monolayer. (a) X-ray fluorescence signal from surface K+ and Ca2+ ions in the absence (black) or with a PIP2 monolayer (red, labeled “pure subphase”) spread on a pH 7.2 ± 0.1 subphase containing 10 mM Tris, 100 mM KCl, and varying amounts of CaCl2 as indicated (from 0 to 1000 μM). The monolayer is held at a constant surface pressure of 30 ± 2 mN m−1. The fluorescence signal is integrated over Qz values from 0.010 to 0.021 Å−1, probing surface ions in the penetration depth of the evanescent beam (approximately 52 Å at Qz = 0.01 Å−1). (b) Same as (a) but the solution includes additional 1 mM Mg2+ (dark green). | ||
Fig. 3(b) compares the surface fluorescence (Qz < 0.0217 Å−1) for a PIP2 monolayer on a subphase with 1 mM Mg2+, 100 mM K+ and varying amounts of Ca2+ with that of the pure subphase with no monolayer (black squares). Data from a 100 mM K+ (labelled “pure subphase”) subphase covered with a PIP2 monolayer is shown as well (red squares). This and the bare subphase data are the same in Fig. 3(a and b). We note that the fluorescence signals from Mg2+ ions are too low in energy to be detected by the Vortex EDD, so we take advantage of the decrease in K+ signals to indirectly monitor Mg2+ exchange for K+. Indeed for 1 mM Mg2+ in the buffer solution we observe a significant decrease in the interfacial [K+] (dark green triangles) indicating Mg2+ migration to the interface ([K+] changes from 0.8 M to 0.5 M at the PIP2 headgroup). By increasing concentrations of Ca2+ to the Mg2+ and K+ containing solution, prominent Ca2+ signals emerge (light green diamonds and light blue triangles) in accordance with our reflectivity results. The Ca2+ fluorescence saturates at roughly 1 mM Ca2+ (cyan data vs. green data).
We now treat the fluorescence data more quantitatively to determine the ion concentration and the number of bound ions per PIP2 molecule for the various conditions described above (see ESI† for methods). Fig. 4(a) shows the ion density (K+ and Ca2+) at the PIP2 headgroup for the different subphase conditions investigated. Multiplying the ion density by the area per lipid molecule (determined during preparation of the PIP2 monolayer) we obtain the number of ions per lipid, Fig. 4(b). Fig. 4(a) and (b) thus summarize the observed ion density and ions/lipid with respect to Ca2+ concentration, with or without 1 mM Mg2+. In the absence of Ca2+ (or Mg2+) K+ density is high, with roughly 1.6 K+ per PIP2 molecule (Fig. 4(b), black data). As the [Ca2+] increases, the K+ density drops off as it is replaced by Ca2+. Even for 1 mM Ca2+ there are still ∼0.5 K+ per PIP2 molecule, compared to ∼1.6 Ca2+ per PIP2.
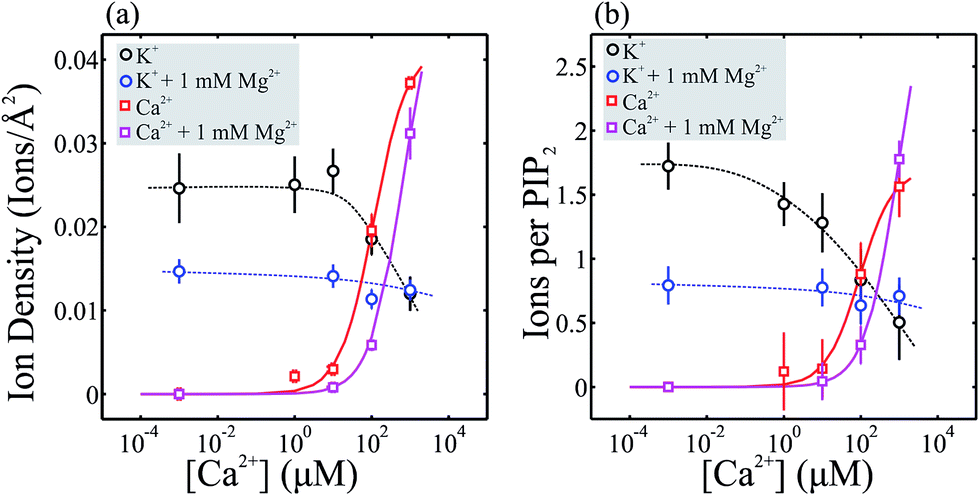 | ||
| Fig. 4 Calculated ion density for K+ and Ca2+ at the PIP2 monolayer. (a) Ion density plotted vs. [Ca2+] for K+ and Ca2+ ions at the surface of the PIP2 monolayer within a depth of ∼52 Å (at Qz = 0.01 Å−1) of the headgroup/solution interface. Ion density of K+ and Ca2+ is compared for samples in the presence (blue and magenta respectively) and absence (black and red) of 1 mM Mg2+. (b) Number of ions per lipid plotted vs. [Ca2+] for K+ and Ca2+. Measured points in the absence of Ca2+ and Mg2+ are included at 10−3 M. Lines are guides to the eye. | ||
As noted above, Mg2+ ion density could not be directly detected. However, the presence of 1 mM Mg2+ leads to a measurable reduction in K+ (blue data in Fig. 4(a and b)). In the absence of Ca2+, 1 mM Mg2+ reduces the K+ density to similar levels as observed for 1 mM Ca2+ indicating that the binding energies of both ions, although different, are of the same order of magnitude. We estimate ∼1 Mg2+ per PIP2 at this concentration based on the observed decrease in K+ signal and the increase in ED, as compared to 1.6 Ca2+ per PIP2.
Interestingly, a persistent K+ signal is observed in both cases (i.e. 1 mM Mg2+ alone and with increasing [Ca2+]), suggesting that some K+ ions reside in regions of PIP2 domains that even large amounts of Ca2+ ions cannot replace. The observation of remaining surface signal for K+, even with high concentrations of Ca2+ or Mg2+, suggests that K+ plays an important role in PIP2 signaling.
4 Discussion
The X-ray experiments provide a qualitative and quantitative picture of the interaction of PIP2 domains with mono- and di-valent cations. They clearly show a hierarchy of associated ions at the interface.4.1 K+ concentration increases 8-fold in the presence of PIP2
Based on electrostatics, there is a significant surface enhancement of K+ in the presence of PIP2 (approximately 8-fold, assuming a uniform K+ distribution within the X-ray penetration depth). At pH 7.2, PIP2 has a single remaining exchangeable proton and carries an equivalent of four electron charges.16 Our experiment shows that ∼1.6 K+ ions per PIP2 molecule associate with the headgroup. Based on a PIP2 charge of −4,16,31 this results in an effective charge of −2.4 for each PIP2. By effective charge we refer to the charge experienced by PIP2 targets within 1 nm from the charged monolayer. These remaining charges are compensated by ions in solution including the protons that are competing for space at the interface. The same is true with Mg2+ and Ca2+, i.e. the charges on PIP2 that are not compensated by these ions are compensated by protons, either as bound ions or as a distribution within 1–2 nm from the interface.32 The overall K+ ion concentration at the interface increases from 0.10 ± 0.01 to 0.8 ± 0.1 M, consistent with what Toner et al. have suggested based on zeta potential measurements (Toner et al. determined 0.7–1.5 K+ ions/PIP2 or 0.3–0.7 M (assuming a molecular area of PIP2 of 67 Å2 per molecule)), although our number, determined directly, is a bit higher.334.2 Ca2+ interacts strongly and partially replaces K+
For Ca2+ concentrations that are two to five orders of magnitude lower than the K+ concentration, we find that a significant number of Ca2+ ions bind and replace K+ ions at the interface. Based on the K/Ca exchange at the interface, we estimate that the binding constant for Ca2+ is at least 100 times greater than that of K+. With 1 mM Ca2+ and 100 mM KCl, 1.6 Ca2+ and 0.5 K+ ions bind per PIP2, reducing the effective charge of the PIP2 to −0.3. If we account for the increased deprotonation of PIP2 in the presence of divalent cations31 this charge increases to roughly −0.4. While the charge of PIP2 is always neutralized over a distance of 1 nm or so from the interface, the presence of these tightly bound Ca2+ ions may reduce the effect of the PIP2 charge and electrostatic interactions between PIP2 and protein targets. This is similar to the effect Ca2+ ions have on DMPA as reported recently.294.3 Mg2+ is replaced by Ca2+ but significant [K+] remain at the PIP2 headgroup
Mg2+, despite its similarities to Ca2+ in terms of charge, plays a distinctly different role in cells. While [Ca2+] ranges from nano to micro-molar and varies dramatically, [Mg2+] remains relatively constant at ∼1 mM free Mg2+.34 Although we could not directly detect Mg2+ ions, by monitoring K+ fluorescence signals at the interface we estimate the number of Mg2+ ions at ∼1 Mg2+ ion per PIP2 at the physiological concentration of 1 mM Mg2+. This is consistent with our rough estimate of 0.6 Mg2+ per PIP2 based on the increased electron density in the headgroup region observed in the XR data (see Fig. 1(b) and assuming a hydrated Mg2+ ion). At a subphase composition of 1 mM Mg2+ and 100 mM K+ we find ∼1 Mg2+ and 0.7 K+ ions bound per PIP2 resulting in an effective charge of −1.3, a noticeably higher charge as compared to the charge observed in the presence of 1 mM Ca2+. As we add Ca2+ to the 1 mM Mg2+ solution, we observe very little change in the interfacial K+ concentration (see Fig. 4(a) and (b)) consistent with the fact that there are K+ ions that are tucked in deeply in the PIP2 headgroup interface and are not replaced by either one or both divalent ions. These K+ ions may play a role in protein–PIP2 interactions, altering the local charge and shape of PIP2 as perceived by the protein's lipid-binding pocket. For a 1:1 Mg2+/Ca2+ ratio the Ca2+ almost completely replaces the Mg2+, while ∼0.5 K+ ions per PIP2 remain at the headgroup region.
4.4 Biological implications
The levels of ion binding determined in this study likely match those found in vivo for several reasons. First, our ionic strength matches that of the cytosol of mammalian cells, and while changes in ionic strength and exact ion concentrations in the cytosol will impact the exact number of bound ions, the 100 mM KCl concentration provides a realistic value. Second, our pure PIP2 monolayers match the in plane density that is found in 2D PIP2-rich domains in the membrane since the surface pressure of our experiments was chosen to match that determined for lipid bilayers. Additionally, we previously showed that the global concentration of PIP2 in model membranes has little impact on the charge of PIP2, likely due to nano-clustering of PIP2 resulting in PIP2 residing in regions of high local concentration.16 Binding of ions to PIP2 clusters induced by other lipid components (e.g. cholesterol and PI) is similarly not expected to lead to major deviations from the values we report here. Cholesterol and phosphatidylinositol (PI) were both shown to induce formation of macroscopic PIP2 rich clusters in model lipid membranes.15,31 However, in both cases a relatively small impact is observed on the charge of PIP2, suggesting that ion binding may also remain consistent with the values that we report here.31,35The varying Ca2+ concentration in the cell may have a significant effect on PIP2 mediated signaling events. Our results suggest that Ca2+ may have a dual effect on PIP2-mediated signalling: (1) Ca2+ may trigger clustering of PIP2 to bring PIP2 binding partners in close proximity allowing for increased binding (signaling) and (2), Ca2+ may neutralize the PIP2 charge leading to an effective sequestering effect similar to the MARCKS protein thus reducing the available pool of functional PIP2 in the membrane.36 This dual role of Ca2+ may be tightly regulated by the oscillating calcium concentration of the cell.14 In a resting cell, Ca2+ concentrations are quite low and only reach the micromolar range. According to our results, at these concentrations (1–10 μM) some Ca2+ binds to PIP2 at a ratio of roughly 0.1 ions/PIP2. With these reduced levels of Ca2+ we see an effect on the lipid packing in the presence of Ca2+ in the form of condensation of the PIP2 monolayer. This condensation is likely a result of clustering of PIP2 triggered by a screening effect of the calcium ions reducing the charge–charge repulsion between PIP2 headgroups and enabling increased hydrogen bond formation between PIP2 headgroups.16 Additionally, the calcium ions may play a ‘bridging role’ by linking adjacent headgroups. The increased interaction between PIP2 headgroups drives the reduced molecular area and results in the increased acyl chain length (see Tables 1 & 2). Ca2+-induced clustering of PIP2 at low [Ca2+] may lead to increased binding (signaling) of PIP2 binding proteins due to the increased local PIP2 concentration. Such binding can be stabilized by hydrogen bond interactions as summarized in the electrostatic-hydrogen bond switch model previously proposed for phosphatidic acid.37 Previous studies have also suggested that 1 μM Ca2+ is enough to cause formation of small PIP2 clusters.12,13 However, these studies have not always accounted for the large number of competing cations (such as K+ and Mg2+, etc.) within the cytosol, which may reduce the impact of 1 μM Ca2+ and prevent the formation of Ca2+ induced PIP2 clustering. Indeed, we find a reduced Ca2+ induced condensation in the presence of 1 mM Mg2+. During Ca2+ signaling events, large amounts of Ca2+ are released via calcium channels, and Ca2+ concentrations reach 10–100 μM.14 It is possible that near calcium channels local concentrations may even reach millimolar values. This can lead to large amounts of Ca2+ binding to PIP2 to a peak of around 1.6 Ca2+ ions per lipid. At these high Ca2+ concentrations, Ca2+ will start to neutralize the PIP2 charge and may play more of a sequestering role, while Ca2+ induced clustering will also be promoted. Many proteins that bind to PIP2 rely on electrostatic interactions to dock to PIP2, and the presence of Ca2+ ions may reduce or eliminate binding between a protein and PIP2. Alternatively, the additional Ca2+ ions might be shared between two PIP2 molecules, acting as a bridge to promote stronger PIP2 clustering. Thus, we suggest that Ca2+ may facilitate PIP2-mediated signalling events in different ways depending on its local concentration. At moderate [Ca2+] PIP2 signaling might be facilitated due to clustering and enhanced binding, while at higher [Ca2+] PIP2 signaling may be inhibited due to charge neutralization.
K+ concentration in the cytosol is between 100 and 150 mM, and that of Na+ one or two orders of magnitude lower (the reverse is found on the outside of a typical mammalian cell). Coupled with the intracellular location of PIP2, the observation that the PIP2 binding constant for K+ is ∼100 fold lower than the Ca2+ binding constant, and the fact that the fluorescence signals for Na+ are too weak to be detected by the Vortex EDD is the reason we focused on K+ in this work. The PIP2 binding affinity of Na+ is expected to be similar to that of K+. Our results show that as many as 1.6 K+ ions per PIP2 are present in PIP2 rich domains. The cytosol also commonly contains roughly 1 mM free Mg2+. At this concentration roughly one Mg2+ ion binds to each PIP2, replacing approximately one K+ ion, but maintaining a significant amount (∼0.5 ions) of K+ bound to the PIP2 headgroup.
5 Conclusions
Employing surface sensitive (synchrotron) X-ray diffraction and spectroscopic techniques we determine the structure of natural PIP2 in the form of a monolayer on physiological solutions with various relevant ions to quantify ion adsorption and binding to PIP2 domains in plasma membranes. We find that in buffer solutions with 100 mM KCl approximately 1.6 K+ ions are associated with each headgroup of PIP2 increasing by eight-fold the concentration at the surface compared to that in the bulk. Upon addition of Ca2+ or Mg2+ our results show a few effects: first, the divalent ions replace the K+, and second, binding leads to increased packing of the PIP2 monolayer, consistent with recent observations.13,19,21,31 Remarkably, we find that at least 0.5 K+/PIP2 remain at the PIP2 interface even at physiological levels of Ca2+ or Mg2+. K+ ions tightly bound to PIP2 effectively change the binding partner for PIP2 binding proteins from just PIP2 to PIP2–K+. This should be taken into account when considering protein–PIP2 interactions, e.g. in MD simulations.For mixed Mg2+/Ca2+ we demonstrate that Ca2+ has a stronger affinity to binding than Mg2+ as it replaces most of the bound Mg2+ ions. Our results also show that the divalent ions have measurable effect on the structure of PIP2 in domains in particular on the molecular area that can decrease by almost 20% in the presence of 1 mM Ca2+ or Mg2+ in physiological solutions.21 Such an increase in in-plane density may be related to clustering of PIP2 in cells and PIP2 clusters may form important platforms for PIP2 signaling,38–41 for example by increasing protein binding affinity as was recently demonstrated for PIP2 clusters induced by cholesterol.15
Acknowledgements
ZTG and EEK gratefully acknowledge financial support from NSF through grant CHE 1216827. The work at the Ames Laboratory was supported by the Office of Basic Energy Sciences, U.S. Department of Energy under Contract No. DE-AC02-07CH11358. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.Notes and references
- G. Di Paolo and P. de Camilli, Nature, 2006, 443, 651–657 CrossRef CAS PubMed.
- N. R. Leslie, I. H. Batty, H. Maccario, L. Davidson and C. P. Downes, Oncogene, 2008, 27, 5464–5476 CrossRef CAS PubMed.
- R. E. Redfern, D. Redfern, M. L. Furgason, M. Munson, A. H. Ross and A. Gericke, Biochemistry, 2008, 47, 2162–2171 CrossRef CAS PubMed.
- A. H. Ross and A. Gericke, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1297–1298 CrossRef CAS PubMed.
- B. Hille, E. J. Dickson, M. Kruse, O. Vivas and B. C. Suh, Biochim. Biophys. Acta, 2015, 1851, 844–856 CrossRef CAS PubMed.
- M. Soom, R. Schonherr, Y. Kubo, C. Kirsch, R. Klinger and S. H. Heinemann, FEBS Lett., 2001, 490, 49–53 CrossRef CAS PubMed.
- M. Langner, D. Cafiso, S. Marcelja and S. McLaughlin, Biophys. J., 1990, 57, 335–349 CrossRef CAS PubMed.
- R. V. Stahelin, J. L. Scott and C. T. Frick, Chem. Phys. Lipids, 2014, 182, 3–18 CrossRef CAS PubMed.
- A. Arbuzova, J. Wang, D. Murray, J. Jacob, D. S. Cafiso and S. McLaughlin, J. Biol. Chem., 1997, 272, 27167–27177 CrossRef CAS PubMed.
- S. McLaughlin, G. Hangyas-Mihalyne, I. Zaitseva and U. Golebiewska, Biochem. Soc. Symp., 2005, 189–198 CrossRef CAS.
- Z. Fan and J. C. Makielski, J. Biol. Chem., 1997, 272, 5388–5395 CrossRef CAS PubMed.
- M. J. Sarmento, A. Coutinho, A. Fedorov, M. Prieto and F. Fernandes, Biochim. Biophys. Acta, 2013, 1838, 822–830 CrossRef PubMed.
- Y. H. Wang, A. Collins, L. Guo, K. B. Smith-Dupont, F. Gai, T. Svitkina and P. A. Janmey, J. Am. Chem. Soc., 2012, 134, 3387–3395 CrossRef CAS PubMed.
- M. D. Bootman, P. Lipp and M. J. Berridge, J. Cell Sci., 2001, 114, 2213–2222 CAS.
- Z. Jiang, R. E. Redfern, Y. Isler, A. H. Ross and A. Gericke, Chem. Phys. Lipids, 2014, 182, 52–61 CrossRef CAS PubMed.
- E. E. Kooijman, K. E. King, M. Gangoda and A. Gericke, Biochemistry, 2009, 48, 9360–9371 CrossRef CAS PubMed.
- I. L. Salvemini, D. M. Gau, J. Reid, L. A. Bagatolli, A. Macmillan and P. D. Moens, Chem. Phys. Lipids, 2014, 177, 51–63 CrossRef CAS PubMed.
- S. K. Ghosh, S. Castorph, O. Konovalov, T. Salditt, R. Jahn and M. Holt, Biophys. J., 2012, 102, 1394–1402 CrossRef CAS PubMed.
- I. Levental, A. Cebers and P. A. Janmey, J. Am. Chem. Soc., 2008, 130, 9025–9030 CrossRef CAS PubMed.
- I. Levental, D. A. Christian, Y. H. Wang, J. J. Madara, D. E. Discher and P. A. Janmey, Biochemistry, 2009, 48, 8241–8248 CrossRef CAS.
- D. R. Slochower, Y. H. Wang, R. Radhakrishnan and P. A. Janmey, Phys. Chem. Chem. Phys., 2015, 17, 12608–12615 RSC.
- J. Als-Nielsen and D. McMorrow, Elements of modern X-ray physics, Wiley, Hoboken, 2nd edn, 2011 Search PubMed.
- P. S. Pershan and M. L. Schlossman, Liquid Surfaces and Interfaces: Synchrotron X-ray methods, Cambridge University Press, New York, 2012 Search PubMed.
- K. Kjaer, Phys. B, 1994, 198, 100–109 CrossRef CAS.
- D. Vaknin, in Characterization of Materials, ed. E. N. Kaufmann, John Wiley & Sons, New York, 2012, vol. 2, pp. 1393–1423 Search PubMed.
- J. Daillant, L. Bosio, J. J. Benattar and C. Blot, Langmuir, 1991, 7, 611–614 CrossRef CAS.
- W. B. Yun and J. M. Bloch, J. Appl. Phys., 1990, 68, 1421–1428 CrossRef CAS.
- W. Bu, K. Flores, J. Pleasants and D. Vaknin, Langmuir, 2009, 25, 1068–1073 CrossRef CAS PubMed.
- W. Wang, N. A. Anderson, A. Travesset and D. Vaknin, J. Phys. Chem. B, 2012, 116, 7213–7220 CrossRef CAS PubMed.
- D. R. Slochower, P. J. Huwe, R. Radhakrishnan and P. A. Janmey, J. Phys. Chem. B, 2013, 117, 8322–8329 CrossRef CAS PubMed.
- Z. T. Graber, A. Gericke and E. E. Kooijman, Chem. Phys. Lipids, 2014, 182, 62–72 CrossRef CAS PubMed.
- W. Bu, D. Vaknin and A. Travesset, Langmuir, 2006, 22, 5673–5681 CrossRef CAS PubMed.
- M. Toner, G. Vaio, A. McLaughlin and S. McLaughlin, Biochemistry, 1988, 27, 7435–7443 CrossRef CAS PubMed.
- B. Alberts, Molecular Biology of the Cell: Reference edition, Garland Science, 2008 Search PubMed.
- Z. T. Graber, Z. Jiang, A. Gericke and E. E. Kooijman, Chem. Phys. Lipids, 2012, 165, 696–704 CrossRef CAS PubMed.
- J. Y. Wang, S. McLaughlin and D. Murray, Biophys. J., 2003, 84, 461a Search PubMed.
- E. E. Kooijman, D. P. Tieleman, C. Testerink, T. Munnik, D. T. Rijkers, K. N. Burger and B. de Kruijff, J. Biol. Chem., 2007, 282, 11356–11364 CrossRef CAS PubMed.
- X. Gao, P. R. Lowry, X. Zhou, C. Depry, Z. Wei, G. W. Wong and J. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14509–14514 CrossRef CAS.
- A. Honigmann, G. van den Bogaart, E. Iraheta, H. J. Risselada, D. Milovanovic, V. Mueller, S. Mullar, U. Diederichsen, D. Fasshauer, H. Grubmuller, S. W. Hell, C. Eggeling, K. Kuhnel and R. Jahn, Nat. Struct. Mol. Biol., 2013, 20, 679–686 CAS.
- K. Kwiatkowska, Cell. Mol. Life Sci., 2010, 67, 3927–3946 CrossRef CAS PubMed.
- L. Picas, J. Viaud, K. Schauer, S. Vanni, K. Hnia, V. Fraisier, A. Roux, P. Bassereau, F. Gaits-Iacovoni, B. Payrastre, J. Laporte, J. B. Manneville and B. Goud, Nat. Commun., 2014, 5, 5647 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Calculation of molecular area from reflectivity data, and calculation of # of divalent ions bound to PIP2. See DOI: 10.1039/c5ra19023a |
| ‡ Currently at: Department of Chemistry, 231 S 34th street, University of Pennsylvania, Philadelphia, PA 19104. |
| This journal is © The Royal Society of Chemistry 2015 |