
A theoretical and experimental study of the crystal structure of H2V3O8†
Yoann Mettan*a,
Riccarda Caputo*b and
Tapan Chatterjic
aBelenos Clean Power Holding Ltd, c/o Renata AG, CH-4452 Itigen, Switzerland. E-mail: yoann.mettan@belenoscleanpower.com
bETH Zürich, Swiss Federal Institute of Technology, Department of Chemistry and Applied Biosciences, Institute of Inorganic Chemistry, Vladimir-Prelog-Weg 1, 8093 Zürich, Switzerland. E-mail: riccarda.caputo@inorg.chem.ethz.ch
cInstitute Lau-Langevin, 71 Avenue des Martyrs, CS 20156 – 38042, Grenoble, Cedex 9, France
First published on 7th December 2015
Abstract
The present study aims at clarifying the positions of hydrogen atoms in H2V3O8. Starting with the structure provided by Oka (J. Solid State Chem., 1990, 89, 372) a model was established using first-principles total energy calculations. Optimized structure data were obtained for two possible configurations within the orthorhombic symmetry representation of the V–O frame. The hydrogen atoms were located close to the oxygen atom labelled O(6) by Oka, in a water-like H–O–H moiety. The two different configurations are distinguished by either parallel or perpendicular orientation of the H–O–H plane with regards to the crystallographic b-axis. In addition, neutron diffraction measurements at 4 K and ambient temperature were performed. The hydrogen atom positions were found in proximity of O(6), in a similar arrangement as predicted theoretically. In particular, the structure obtained from the analysis of the 4 K data matches the model with H–O–H plane parallel to the b-axis.
1 Introduction
The first reference to H2V3O8 in literature dates back to the late 1960s. By treating hydrated VO2 in aqueous hydrogen peroxide at 453 K Théobald and Bernard discovered a green product (which they named “phase-F”) with a vanadium average oxidation state between 4.60 and 4.84. In a consecutive publication, they derived an orthorhombic structural model based on the X-ray diffraction (XRD) pattern of the pure so-called F-phase. The chemical composition was established as V3O7·H2O.1 In 1990, Oka2 presented a hydrothermally-synthesized V3O7·H2O as H2V3O8 with orthorhombic Pnma (IT No 62) space group symmetry. The limitations of the X-ray powder diffraction analysis did not make possible the localization of hydrogen atoms. Until today a detailed structural investigation of the positions of hydrogen atoms is missing. Oka2 suggested that the oxygen atoms having the longest V–O bond should be those bound to hydrogen atoms. Without any further evidence, the position of hydrogen atoms in the V3O8 framework remained an assumption. Therefore, to our best knowledge, the problem of the location of the hydrogen atoms in the V3O8 framework is still an open question. Locating protons in a mixed-valence transition metal oxide may also provide further insights into other physico-chemical properties of the material. A distinction between structural water, free protons and/or hydroxyl group is crucial to understand the electronic structure of such a transition metal compound. Hydrogen atoms can be accommodated in transition metal oxides by forming hydrogen metal bronzes with general formula HxMemOn, where Me stands for the transition metal. For example, in manganese(III–IV) oxide, two types of protons have been characterized, which are referred to as Rütschi and Coleman protons, after the name of their discoverers3 (and reference therein). Rütschi protons refer to substitution protons, which occupy the transition metal sites and Coleman protons are interstitial sites protons. The water moiety and especially the protons play an important role in the stabilization of the structure and influence the electrochemical behavior. In recent years, several groups reported about the electrochemical behavior of H2V3O8 in lithium batteries. H2V3O8 was found to be able to intercalate reversibly more than four equivalents of Li ions and is therefore an interesting candidate as positive electrode material.4–9 Overall, up to date the published reports focused either on lithium intercalation or on synthesis. In the present work, we aimed to clarify the positions of the hydrogen atoms in the V3O8 framework and characterize the nature of the oxygen–proton bonding, by combining crystal structure prediction, via first-principles calculations, and targeted experiments.2 Methodology
2.1 Synthesis
H2V3O8 was synthesized by using the known hydrothermal method reported by Oka.2 Vanadyl sulfate was prepared in-house since commercial products are hydrates of unknown stoichiometry and often contain impurities. In a typical synthesis, 2.2 g V2O5 (Alfa Aesar), 0.65 ml H2SO4 (97–98% by Sigma Aldrich), 22 ml H2SO4 (4–5% by Sigma Aldrich) and 380 ml deionized water were mixed in a 500 ml three-neck (condenser with nitrogen in, nitrogen out and thermometer) round bottom flask equipped with a condenser for reflux. The apparatus was kept under nitrogen during the reaction. The suspension was heated to 353 K. The reaction took two hours to complete. The colour of the reaction mixture changed quickly from orange-yellow to green. Once all vanadium(5+) was reduced to vanadium(4+), a clear deep blue solution of vanadyl sulfate (typically 0.06 M) was obtained. The chemical reaction is known as the contact process:| V2O5 + H2SO4 + H2SO3 ⇀ 2VOSO4 + 2H2O | (1) |
After the hydrothermal step, the as-synthesized green H2V3O8 compound was collected by filtration, rinse twice with deionized water and once with isopropanol. The solid was dried in air stream at room temperature for 3 hours and at 393 K in air overnight. The phase purity and the structure of the product was probed by performing different analyses, SEM, XRD, DTA-TG and TG-MS.
2.2 Spectroscopic measurements
The presence of unpaired electrons of vanadium ions was probed by using a continuous-wave EPR spectrometer operating at X-band (9.5 GHz) (Bruker EMX). The spectra were recorded at 123 K, and the temperature was monitored by a Eurotherm controller.The 1H-MAS NMR experiment was performed at an operating frequency of 272.1 MHz, corresponding to a field strength of 16.4 T, on a Bruker Avance ITM 700 MHz Ultrashield. The spectra were acquired using a Bruker double resonance MAS probe head equipped with a 2.5 mm ZrO2 rotor that was spun at a frequency of 21 kHz. The probe was tuned to 1H (700.14 MHz) and the spectrum was referenced to distilled water by setting it to 0 ppm.
The IR spectra were recorded on a Brucker FTIR model Vertex 70 and the measurements carried out in air. The resolution was ±4 cm−1.
2.3 Thermal analysis
The DTA was performed with a Netzsch STA 409 in corundum crucibles with heating ramp of 10 K min−1 from room temperature to 973 K. We used nitrogen as carrier gas.2.4 Neutron diffraction
Neutron diffraction measurements on H2V3O8 were carried out on the high resolution powder diffractometer D2B at the Institute Laue-Langevin, Grenoble and also on the high resolution powder diffractometer SPODI at the FRM II reactor, Garching. The structure refinement was based on the neutron powder diffraction experiments by using the Rietveld method implemented in the program GSAS,10 part of the EXPGUI package.11 Graphical representations of the structure and the nucleus densities were created with VESTA crystal structure visualization program.122.5 Computational settings
The set of modelled structures were optimized as the atomic positions and the lattice parameters, without any symmetry constraints, by using the DFT-based code, CASTEP, implemented in Materials Studio 6.0.13 We used norm-conserving pseudopotentials and reciprocal space representation for all atoms, and for V and O atoms, the valence shells contain respectively the electrons in the s–p orbitals, the 2s22p4 and 4s23d3. The Brillouin zone was sampled by using a fine mesh commensurate to the specific lattice dimensions, but with the actual spacing below 0.03 Å−1. The energy threshold, the maximum atomic displacement, the maximum atomic force and the lattice stress were set to 0.005 meV per atom, 0.001 Å, 0.001 eV Å−1 and 0.002 GPa, respectively. We used the generalized gradient form (GGA) of the exchange-correlation functional and in particular the Perdew–Burke–Ernzerhof96 (PBE) one.14 The dispersion term being one of the important long range contribution to the total energy, non-covalent forces, and in particular van der Waals interactions, were introduced by using damped atom-pairwise dispersion corrections of the form C6R−6, proposed by Grimme,15,16 as implemented in CASTEP.173 Results and discussion
3.1 Experimental results
The stoichiometry V3O8 given by Theobald and Oka in their respective works1,2,18 on V3O8 was also confirmed by our diffraction experiments. Nevertheless, the hydrogen stoichiometry could deviate from 2. V3O8 is known to be an intercalation compound and therefore the structure may be stable for the hydrogen content in the range 2 ± δ. For this reason, we review some of the experimental results obtained by techniques detecting either hydrogen or unpaired electrons in H2V3O8. Electron paramagnetic resonance spectroscopy (EPR) was used to probe unpaired electrons. Since H2V3O8 is green, vanadium is expected to be found in the two oxidation states: 4+ and 5+. Each vanadium(IV) ion contains one unpaired (3d1) electron which strongly interacts through exchange forces with the surrounding atoms. The EPR signal originating from the unpaired electron, d1 in V4+, is shown in Fig. 1.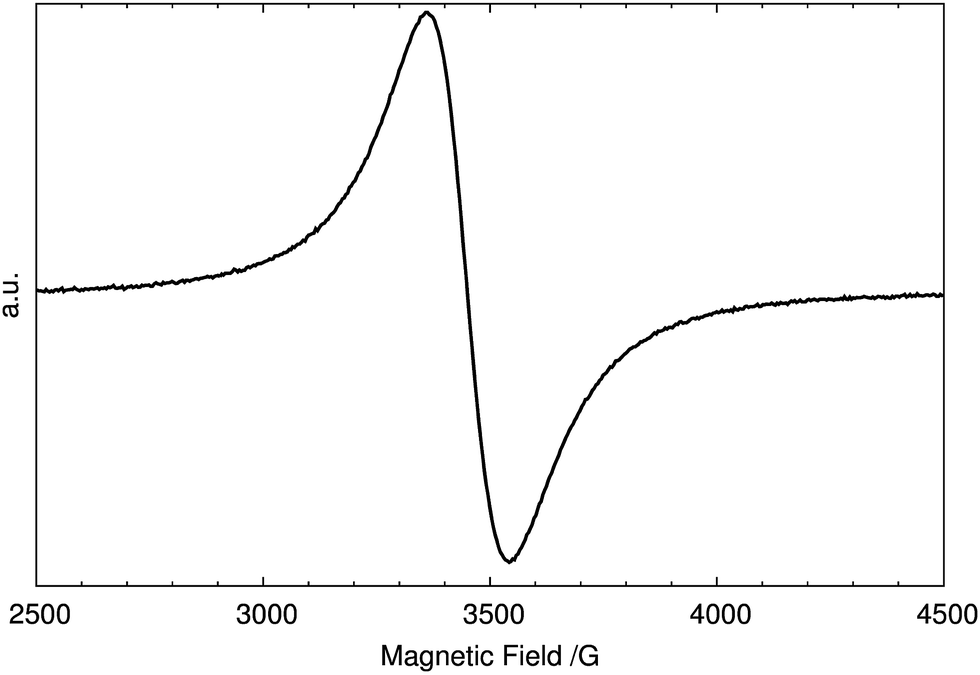 | ||
| Fig. 1 The EPR spectrum of the as-synthesized H2V3O8. | ||
In a second spectroscopic investigation, 1H-MAS-NMR was used to probe the protons in pristine H2V3O8. The spectrum in Fig. 2 displays one signal at 41 ppm with asymmetric spinning sidebands, i.e. all protons were found magnetically equivalent within the chosen experimental parameters. The spinning sideband pattern reflects the anisotropy of the coupling interaction between the protons and the paramagnetic centers, which can be described by the appropriate tensor. The small peaks around 5 ppm are due to background subtraction.
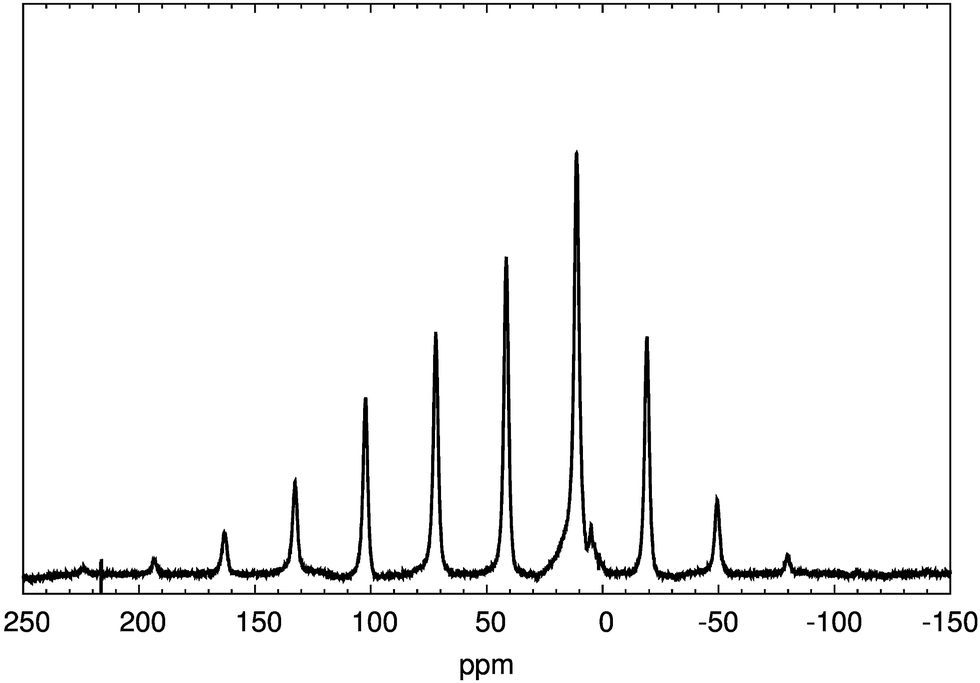 | ||
| Fig. 2 Experimental NMR spectrum of H2V3O8. | ||
Using the combined information delivered by EPR and H-NMR, we can state that H2V3O8 contains protons and unpaired electrons. At this point, the composition H2±δV3O8 is confirmed, although no quantification of the protons and unpaired electrons can be made based on that information alone. The IR spectra were recorded in order to gain more information about the nature of hydrogen–oxygen bonding. The experimental IR spectrum in Fig. 3 showed four main bands, in agreement with the literature.7,19–21 They are: the bridging oxygen–vanadium–oxygen bond at around 780 cm−1; the stretching of vanadium(IV)–oxygen bond and vanadium(V)–oxygen bonds at 969 cm−1 and 1009 cm−1, respectively; and the bending vibration of water at 1658 cm−1. The broad and very low intense peak in the range of 3500 cm−1 can be attributed to the O–H stretching modes. Their intensity is reduced because of the constrained environment of the O–H moieties. For comparison, in the vibrational spectra of liquid water, the bending mode, ν2, is reported in the range of 1632–1643 cm−1, while for example, the bending mode of water molecules in zeolites22 falls in the range 1500–1700 cm−1. The presence of the bending vibration of water-like moiety indicates that at least some of the protons are bound to oxygen as in molecular water and confirms the presence of H–O–H configuration.
 | ||
| Fig. 3 Experimental IR spectrum of H2V3O8. | ||
The TG-DTA measurements were conducted in order to get quantitative information on the exact content of hydrogen. The thermal gravimetric measurements of the as-synthesized H2V3O8, qualitatively in agreement with the literature data,7,8,19–21,23,24 showed the weight loss of 6.31%, which corresponds to one equivalent of molecular water per formula unit. A molecular ion with m/z = 18 was, in fact, detected in a TG-MS measurement.25 The thermogravimetric analysis as a function of the temperature is shown in Fig. 4. In particular, the DTA curve in Fig. 4 shows an endothermic peak related to a completed water loss at 620 K, immediately followed by an exothermic peak at 695 K. For temperature above no further weight loss was observed, indicating that the thermal decomposition is complete and that any subsequent process does not change the chemical composition but may involve a phase change. In fact, the exothermic process, which has been recently reported21 to occur at 650 K, was attributed by the authors to the formation of V3O7 phase. The molecular water is a structural building unit in H2V3O8, whose orthorhombic lattice collapses upon H2O removal and recrystallizes about 690 K in one or multi-component vanadium oxide phases (see the ESI for details†). Although H2V3O8 may occur as a proton-deficient or a proton-rich compound, our results agree with the stoichiometry H2V3O8 so far reported in literature.
 | ||
| Fig. 4 The TG- and DTA-curves of the as-synthesized H2V3O8. | ||
Two neutron powder diffraction experiments, one at ambient temperature and one at 4 K are compared in Fig. 5.
 | ||
| Fig. 5 Rietveld refinements of H2V3O8 based on neutron diffraction data obtained at ambient temperature (a) and 4 K (b). The observed (red), calculated (green) and difference (purple) patterns are shown, reflection positions are marked in black. | ||
The starting model structure was that provided by Oka.2 Since the bound coherent neutron scattering length for vanadium is very small (−0.3824(12) fm) compared to the ones of oxygen (5.803(4) fm) and hydrogen (−3.7390(11) fm), the positions of the vanadium atoms were taken from a Rietveld refinement based on synchrotron powder diffraction data of a partially deuterated sample and excluded from the subsequent refinements. To further reduce the number of free parameters, the displacement parameters of all atoms were constrained to be equal. For the ambient temperature data, the hydrogen atom positions H(1) and H(2) in Fig. 6 were located in the difference Fourier map and their positional parameters were refined freely, whereas the occupancy factors were set to 0.5 each. A free refinement of the occupancy factors of the disordered positions converges at a total occupancy of about 2/3 with almost equal distribution on both sites, which corresponds to a sum formula of H1.3V3O8. However, it is obvious that the smeared nucleus density resulting from the disorder cannot be described properly by simple isotropic displacement parameters of the hydrogen atoms. Therefore, in principle, it might be possible that the missing 1/3 of a hydrogen atom is contained in the residual nucleus density, not covered by the displacement parameters of H(1) and H(2), as shown in Fig. 6, panel (a).
 | ||
| Fig. 6 (a and b) The coordination spheres of V(2) and the partially occupied hydrogen atom positions H(1) and H(2) bonded to O(6), shown together with the isosurfaces of the nucleus density (−0.78 fm in blue and 0.78 fm in yellow). (c and d) Observed Fourier map of a mean plane through the partially occupied hydrogen atom positions. H(1), H(2) and their symmetry equivalent positions, isovalues in steps of 0.2 fm from −3.74 to 3.74, blue for negative, H(1) and H(2), and red for positive, O(6), values, respectively. Results of (a) and (b) are derived from data obtained at ambient temperature and 4 K, respectively. The V(2)–O(6) bond length is larger (2.346 Å, in the structure 1 and 2.371 Å in the structure 2) that the other five V–O bonds, for that it is not considered in the coordination of V(2) atoms. A 3-D view of the modelled structures: structure 2 (e) and structure 1 (f). In (e) the two non-equivalent H atoms are coloured differently (light blue and white, which in the highlighted V–O group becomes light blue and dark blue) and in (f) the equivalent H atoms are coloured in white (which turn in blue in the highlighted V–O group). The V atoms are in grey and the O atoms in red, which in the highlighted V–O group becomes dark-green and brown, respectively. | ||
For the 4 K data, only one hydrogen atom position was found close to O(6) corresponding to the H(2) position of the ambient temperature data, and refined with free positional parameters. A refinement of the occupancy factor converges at about 0.6, very similar to the value found at ambient temperature. It is remarkable, that in both refinements, the total occupancy of the hydrogen atoms converge only at around 0.6, instead close to a full occupancy, as it would be the case for a water molecule. However, it has to be emphasized, that the resolution of the neutron diffraction pattern does not allow unambiguously to prove a partial occupancy of the hydrogen atom positions. Therefore the occupancies for both, ambient temperature and 4 K, were set to 1 for the final refinement. Crystallographic data and structure refinement at room temperature and 4 K are provided in Table 1. It seems that the disorder of H(1) and H(2) observed for ambient temperature data is dynamic and the two non-symmetry equivalent positions merge to just one symmetry-equivalent hydrogen position at 4 K, shown in Fig. 6, panel (c). No further significant residual nucleus density was identified (largest residual peaks with about ±0.1 fm). In Fig. 6, panels (e) and (d) the two model structures found via first-principles modelling are reported. They are described in the following section.
| a The standard uncertainties determined from powder data are based on counting statistics only and neglect all systematic errors. In fact, more realistic Rp values are about ten times larger. | ||
|---|---|---|
| Empirical formula | H2V3O8 | H2V3O8 |
| Temperature | 293(2) K | 4 K |
| Crystal system | Orthorhombic | Orthorhombic |
| Space group | Pnma – D162h | Pnma – D162h |
| Unit cell dimensionsa | a = 16.8593(7) Å | a = 16.7510(6) Å |
| b = 3.63401(13) Å | b = 3.64051(10) Å | |
| c = 9.3260(4) Å | c = 9.25217(29) Å | |
| Volume, Z | 571.37(4) Å3, 4 | 4564.219(31) Å3, 4 |
| Density | 3.280 g cm−3 | 3.321 g cm−3 |
| Measurement | Neutron beamline D2B | Beamline SPODI |
| Institute Laue-Langevin, | Forschungs-Neutronenquelle | |
| Grenoble, France | Hans-Maier-Leibnitz (FRM II) | |
| TU München | ||
| Wavelength | λ = 1.594 Å | λ = 1.54815(2) Å |
| No. variables | 36 | 33 |
| Final R values | wRp = 0.0181 | wRp = 0.0238 |
| Rp = 0.0137 | Rp = 0.0179 | |
| χ2 = 2.663 | χ2 = 10.52 | |
| R(F2) = 0.0804 | R(F2) = 0.1174 | |
3.2 Theoretical localization of hydrogen atoms
From the structural modelling standpoint, the crystal structure problem can be handled in two ways. One could consider the structure of vanadium heptaoxide and add in the stoichiometric amount of H2O to form H2V3O8. Alternatively, one can start from the molecular skeleton of V3O8 (ref. 2) and add two hydrogen atoms per formula unit. H2V3O8 and V3O7·H2O are reported in the literature indicating the same compound because of a lack of knowledge about the exact location of the hydrogen atoms. Clearly, the orthorhombic lattice of H2V3O8 cannot be derived from the reaction V3O7 + H2O. As a consequence, the formulation H2V3O8 and the position of vanadium and oxygen atoms, suggested by Oka2 appears to us as the convenient piece of information to start with. In the following we report the results obtained by using the V3O8 framework as the starting model structure. From the structural modelling standpoint, the problem of the localization of the hydrogen atoms in V3O8 framework can be reduced to a search for symmetry-allowed sites, which occupancy preserve the orthorhombic symmetry representation of the lattice. Monte Carlo calculations, and in particular bias Monte Carlo method implemented in Materials Studio 6.0, provided the most likely sites where hydrogen atoms can reside. This was done by sequential addition of hydrogen atoms to the host lattice formed by the V3O8 network. A canonical Monte Carlo sampling through a number of temperature cycles of the simulated annealing algorithm was used for that purpose. The lowest energy sites for the hydrogen atoms in the framework of V3O8 were identified by repeatedly searching the configurational space of the V3O8–H system. Fig. 7 shows a top-view of the V3O8 framework with hydrogen atom locations suggested by Monte Carlo simulations. | ||
| Fig. 7 A top-view along the b-direction of the double cell of H2V3O8, in which the hydrogen atoms are localized with their motion sphere as suggested by the Monte Carlo simulations. The radius of the motion spheres is for visualization purpose. Representing colours: vanadium, gray; oxygen, red; hydrogen, white. | ||
By symmetry the general site (x, 0.25, z) in the orthorhombic lattice, with x, z chosen to be close to the [V4+O] moiety, can be occupied by hydrogen atoms up to reach the stoichiometry of H2V3O8. That means two sets of sites (x1, 0.25, z1) and (x2, 0.25, z2). In addition, the general position (x, 0.5, z) has multiplicity of two with the hydrogen atoms lying along the b-direction. The optimized structure data of the two possible configurations with the orthorhombic symmetry representation of the V–O frame, are reported in Table 2.
| Species | x | y | z | Site |
|---|---|---|---|---|
| a a = 16.6597 Å, b = 3.6313 Å, c = 9.2647 Å. Vc = 560.4807 Å3, Z = 4.b a = 17.2885 Å, b = 3.5919 Å, c = 9.5270 Å, Vc = 591.613 Å3, Z = 4. | ||||
| Structure 1a | ||||
| H(1) | 0.2652 | 0.5350 | 0.1735 | 8d |
| V(1) | 0.4550 | 1/4 | 0.1287 | 4c |
| V(2) | 0.1543 | 1/4 | 0.4286 | 4c |
| V(3) | 0.0562 | 1/4 | 0.0873 | 4c |
| O(1) | 0.0391 | 1/4 | 0.5741 | 4c |
| O(2) | 0.4054 | 1/4 | 0.2810 | 4c |
| O(3) | 0.8676 | 1/4 | 0.5246 | 4c |
| O(4) | 0.0623 | 1/4 | 0.2885 | 4c |
| O(5) | 0.2256 | 1/4 | 0.3036 | 4c |
| O(6) | 0.2168 | 1/4 | 0.6169 | 4c |
| O(7) | 0.4796 | 1/4 | 0.5782 | 4c |
| O(8) | 0.1485 | 1/4 | 0.0319 | 4c |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Structure 2b | ||||
| H(1) | 0.2719 | 1/4 | 0.5855 | 4c |
| H(2) | 0.2026 | 1/4 | 0.6941 | 4c |
| V(1) | 0.4541 | 1/4 | 0.1279 | 4c |
| V(2) | 0.1442 | 1/4 | 0.4187 | 4c |
| V(3) | 0.0530 | 1/4 | 0.0887 | 4c |
| O(1) | 0.0379 | 1/4 | 0.5760 | 4c |
| O(2) | 0.4013 | 1/4 | 0.2710 | 4c |
| O(3) | 0.8781 | 1/4 | 0.5338 | 4c |
| O(4) | 0.0572 | 1/4 | 0.2857 | 4c |
| O(5) | 0.2168 | 1/4 | 0.3097 | 4c |
| O(6) | 0.2160 | 1/4 | 0.5953 | 4c |
| O(7) | 0.4829 | 1/4 | 0.5797 | 4c |
| O(8) | 0.1437 | 1/4 | 0.0409 | 4c |
A 3D view of the optimized structures is reported in Fig. 8. The polyhedra show the coordination of vanadium atoms. The V–O bonds are cut through up to a distance of 2.35 Å.
 | ||
| Fig. 8 A top-view of the unit cell of the orthorhombic structure of H2V3O8. The location of hydrogen atoms are (a) perpendicular to b-direction in the structure 1 and (b) in-plane, ac-plane, in structure 2. Representing colours: hydrogen, white; vanadium, green; oxygen, red. | ||
The two structures differ by 17.75 kJ mol−1 being lower in energy the one with the hydrogen atoms aligned along the b-direction (structure 1). The two possible locations of hydrogen atoms can be thought as two possible states easily accessible by thermal activation from one state to the other. In addition, the energy difference between the two structures is of the same order of the free rotational energy of molecular H2O. Therefore, the vibrational and rotational modes are sufficient to mimic a sort of de-localized proton between two adjacent V3O8 units. Interestingly, in the higher energy structure (structure 2) there are two non-equivalent hydrogen atoms by symmetry, while in the lowest energy structure all hydrogen atoms are equivalent. Our NMR calculations confirmed that in the lowest energy structure all protons have the same chemical shift, while in the higher energy structure the two hydrogen atoms are not magnetically equivalent, which is reflected into the different values of the corresponding chemical shifts. Therefore, the lattice parameters of the room-temperature structure can be considered as the mean values of the corresponding lattice parameters of the two structures found computationally. A comparison of the low temperature (4 K) neutron diffraction pattern with the simulated patterns of the two predicted positions of hydrogen atoms in H2V3O8, orthorhombic symmetry space group, is reported in Fig. 9. The two alignments of the hydrogen atoms in the V3O8 framework induce a relative shift of the diffraction peaks of the planes (2 0 0) and (1 0 1), which are the planes mainly affected by the O–H interactions between H–O–H moieties. Differences in the diffraction peaks mirror the dynamics of the hydrogen atoms into the lattice. Deviations from the experimental patterns indirectly highlight a sort of (de)localization of hydrogen atoms, which might be not correctly accounted in the particular functionals (exchange-correlation and dispersion contribution) used. Clearly, the exact identification of the sites most visited by the hydrogen atoms in the lattice is an important piece of information, because their localization affects the lattice parameters, the length of the V–O bonds and the oxidation state of V atoms.
 | ||
| Fig. 9 (a) The simulated neutron diffraction pattern of the lowest energy structure of H2V3O8 is overlapped with the experimental neutron data at T = 4 K. (b) A detailed view in the region 10–11 degrees of 2θ shows the relative shift of the diffraction peak of the (1 0 1) plane due to the lattice relaxation and the dynamics of hydrogen atoms into the lattice. | ||
4 Conclusions
In our DFT-optimized crystal structure model of H2V3O8, the hydrogen atoms reside close to the O(6) oxygen atoms in two possible configurations differing by 17.75 kJ mol−1, with H–O–H plane either perpendicular or parallel to the crystallographic b-axis. The two possible locations of hydrogen atoms can be thought of as two possible states easily accessible by thermal activation. Neutron diffraction were performed at room temperature and 4 K. For the ambient temperature data, two non-equivalent hydrogen atom positions have been found. For the 4 K data, only one hydrogen atom position was found close to O(6) corresponding to the H(2) position of the ambient temperature data. It is remarkable, that in both refinements, the total occupancy of the hydrogen atoms converged only at around 0.6, instead to a full occupancy, 1.0, as it would be the case for a water molecule. On the other side, it has to be emphasized that the refinement of the neutron diffraction data does not allow to prove unambiguously the site occupancy by hydrogen atoms. The chemical nature of the structural H2O moiety in H2V3O8 is definitely more complex than that of simply crystal-trapped molecular water. Water cannot be removed from H2V3O8 without destroying the original structure and even if the V(2)–O(6) distance is longer than the typical V–O single bond distance in vanadates, the O(6) resides in the first coordination shell of vanadium. Furthermore, the combination of the long-range hydrogen bond interactions between H–O–H moieties and the thermally-activated motion of the O–H bonds may cause randomness of the H atoms in the room-temperature structure and consequently induce changes in the oxidation state of vanadium atoms in close proximity to the oxygen of H–O–H moiety. This ought to be a typical problem of the (de)localization and dynamics of hydrogen atoms in transition metal oxides.Acknowledgements
The authors thank Christian Mensing, Michael Wörle, Santhosh Matam, Reinhard Kissner and René Verel for the DFT-TG, XRD, IR, EPR and NMR measurements. Special thank to Michael Wörle for the analysis of the neutron diffraction data and to Reinhard Nesper for his valuable comments. The computer facilities at ETH, Zürich are highly appreciated. The authors acknowledge supports of the neutron scattering centers of Institut Laue-Langevin, Grenoble and MLZ, FRM II. and thank E. Suard and A. Senyshyn for their helps in neutron diffraction data collection.References
- F. Theobald and J. Bernard, C. R. Seances Acad. Sci., Ser. C, 1970, 270, 2138 CAS.
- Y. Oka, T. Yao and N. Yamamoto, J. Solid State Chem., 1990, 89, 372 CrossRef CAS.
- C. Pitteloud, M. Nagao, K. Itoh and R. Kanno, J. Solid State Chem., 2008, 181, 467 CrossRef CAS.
- K. Waltersson, B. Forslund, K. Wilhelmi, S. Andersson and J. Galy, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 2644 CrossRef.
- G.-C. Li, S.-P. Pang, Z.-B. Wang, H.-R. Peng and Z. Zhi-kun, Eur. J. Inorg. Chem., 2005, 11, 2060 CrossRef.
- S.-I. Mho, C. Reddy and Y. Kim, J. Korean Phys. Soc., 2009, 54, 2420 CrossRef CAS.
- V. L. Volkov and G. S. Zakharova, Russ. J. Inorg. Chem., 2009, 54, 1704 CrossRef.
- G. S. Zakharova, V. L. Volkov, C. Taeschner, I. Hellmann, A. Leonhardt, R. Klingeler and B. Buechner, Solid State Commun., 2009, 149, 814 CrossRef CAS.
- J. Prado-Gonjal, B. Molero-Sánchez, D. Ávila-Brande, E. Morán, J. Pérez-Flores, A. Kuhn and F. Gárcia-Alvarado, J. Power Sources, 2013, 232, 173 CrossRef CAS.
- A. Larson and R. von Dreele, General Structure Analysis System (GSAS), Los Alamos National Laboratory, Report LAUR 86–748, 1994 Search PubMed.
- B. Toby, J. Appl. Crystallogr., 2001, 34, 210 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
- S. Clark, M. Segall, C. Pickard, P. Haspin, M. Probert, K. Refson and M. Payne, Z. Kristallogr., 2005, 220, 567 CAS.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- E. R. McNellis, J. Meyer and K. Reuter, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 205414 CrossRef.
- F. Theobald and J. Bernard, C. R. Seances Acad. Sci., Ser. C, 1969, 268, 60 CAS.
- H. Li, T. Zhai, P. He, Y. Wang, E. Hosonoa and H. Zhou, J. Mater. Chem., 2011, 21, 1780 RSC.
- V. Channu Reddy, R. Holze and B. Rambabu, Curr. Appl. Phys., 2013, 13, 237 CrossRef.
- Q. An, J. Sheng, X. Xu, Q. Wei, Y. Zhu, C. Han, C. Niu and L. Mai, New J. Chem., 2014, 38, 2075 RSC.
- C. Geiger, Eur. J. Mineral., 2012, 24, 439 CrossRef CAS.
- S. Gao, Z. Chena, M. Wei, K. Wei and H. Zhou, Electrochim. Acta, 2009, 54, 1115 CrossRef CAS.
- Y. Zhang, M. Zhou, M. Fan, C. Huang, C. Chen, Y. Cao, H. Li and X. Liu, Curr. Appl. Phys., 2011, 11, 1159 CrossRef.
- Y. Mettan, PhD thesis, ETH Zürich, Diss. Nr 19382, 2011.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra19665e |
| This journal is © The Royal Society of Chemistry 2015 |