
Cation ordering induced semiconductor to half metal transition in La2NiCrO6
Jing Wanga,
Xiangfeng Haoa,
Yuanhui Xua,
Zhiping Lia,
Ningning Zub,
Zhijian Wub and
Faming Gao*a
aKey Laboratory of Applied Chemistry, Yanshan University, Qinhuangdao 066004, P. R. China. E-mail: fmgao@ysu.edu.cn
bState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China
First published on 21st May 2015
Abstract
La2NiCrO6, previously proposed to be a candidate of half metallic antiferromagnetism, is revisited using the first-principles calculation. Electron correlation is considered and cation ordering effects are studied by arranging Ni and Cr atoms along [111] and [001]. For the [111] case, which corresponds to an ordered double perovskite, a monoclinic structure is predicted to be the most stable. In contrast to the previous study, it is insulating from the calculation of electron structure. Attractively, the magnetic coupling of Ni and Cr is sensitive to electron correlation, i.e., it is antiferromagnetic in the GGA calculation, whereas the ferromagnetic state is favoured when electron correlation (U) is turned on. For the [001] case, it is ferromagnetic whether U is included or not. Interestingly, a semiconductor to half metal transition is expected according to the GGA + U method, and the half metallic character could be preserved under both compressive and tensile strain.
Introduction
Double perovskites (A2BB′O6, e.g., La2NiMnO6 and La2CoMnO6), a class of ferromagnetic (FM) insulator that is rarely found in the nature, have gained considerable attention in the last few years.1–14 They are attractive because of multifunctional properties such as magnetocapacitance,1 magnetodielectronic effect,1 and magnetoresistance up to relatively high temperature,2 which provide intriguing prospects in industrial applications. On the other hand, understanding the underlying mechanism behind the FM insulator is also impressive for fundamental physics. In double perovskite, the coordination number of each transition metal is six, i.e., the transition metal and the neighboring oxygens form an octahedron (Fig. 1). The five degenerate orbitals then split into two groups, i.e., the lower t2g (threefold) and higher eg (twofold) states. In the Ni/Mn ordered case, the insulating nature is easy to understand, in which the 3d orbitals of Ni2+ (3d8, t2g6eg2↑) and Mn4+ (3d3, t2g3↑)3,4 are half filled (eg2 for Ni, t2g3 for Mn). However, it is not so obvious for La2CoMnO6, in which the 3d orbitals of Co2+ (3d7, t2g3↑t2g2↓eg2↑) are partially filled. Indeed, previous density functional calculations give a half metallic (HM) solution even when electron correlation (U) is included for both Co and Mn.5 Until recently, an alternative explanation gives the origin as the Coulomb-assisted spin–orbit coupling (SOC).6 To date, many aspects are still under debate, and cation ordering is proposed as the main contributor. For instance, the Curie temperature (Tc) is about 240 K for the ordered La2CoMnO6 and it is reduced to ∼150 K for randomly ordered case.7 The saturation magnetization is 5.7 μB per f.u. for the ordered state,8 close to 6.0 μB per f.u. for high spin (HS) states of Co2+ (3d7, t2g3↑t2g2↓eg2↑) and Mn4+ (3d3, t2g3↑), and it falls to 3.5 μB per f.u. in the disordered sample.9,10 Disorder also leads to a structure transition from monoclinic to orthorhombic symmetry. Similarly, in La2NiMnO6, both the total magnetic moment (from 4.8 μB per f.u. to 3.7 μB per f.u.) and Tc (from ∼270 K to ∼138 K) are found to be decreased by disordering.11–13 These are generally due to the different ionic configurations in the disordered case, i.e., Co3+/Mn3+ and Ni3+/Mn3+. Recently, by designing a Ni/Mn(Co/Mn) ordering along the [001] and [110] direction, HM electronic structure is obtained in La2NiMnO6 and La2CoMnO6, which is useful for modern functional materials.14 In contrast to the disordered case, the valence states are expected to be between 3+ and 4+ for Mn and between 3+ and 2+ for Ni and Co. | ||
| Fig. 1 Schematic structure of La2NiCrO6 with Ni/Cr along the [111] and [001] directions. The green and purple octahedra are NiO6 and CrO6, respectively, with Ni and Cr in the center. Small red spheres are oxygen and big grey spheres are La atoms. | ||
La2NiCrO6, the counterpart of La2NiMnO6 and La2CoMnO6, was initially studied by Pickett with the aim to search for half metallic antiferromagnetism (HM-AFM).15 Both Ni and Cr were supposed to be 3+. In the ideal cubic structure, the crystal field splitting is small; thus, the d7 ion (Ni) will be in HS state (3d7, t2g3↑t2g2↓eg2↑) and it will balance the d3 moment of Cr (3d3, t2g3↓). However, the FM coupled Ni and Cr is found to be lower by 0.15 eV. This is similar to the magnetic configuration of La2NiMnO6 and La2CoMnO6. It is noticed that although structural relaxation is expected to change the magnetic state, no further studies are available. Thus, it is worthwhile to investigate whether the HM-AFM character could survive under structure relaxation. Inspired by these considerations, La2NiCrO6 is revisited in this study by the first principles calculation based on the density functional theory (DFT). Electron correlation, which is important for 3d electrons but not considered in the previous study,15 is included. Importantly, the cation disordering effect is simulated by changing the arrangement of Ni/Cr arranged along [111] (corresponding to ordered double perovskite) to [001] directions. In reality, this can be implemented by layer-by-layer growth techniques in the form of a thin film.
Computational method
The initial structures were predicted by the SPUDS program.16 This program has been widely employed to predict the structures of un-synthesized compounds. According to the Glazer tiling notation,17,18 seven symmetries are predicted for double perovskites, namely, Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (no. 225, a0a0a0), Pn (no. 201, a+a+a+), R (no. 148, a−a−a−), P4/mnc (no. 128, a0a0c+), I4/m (no. 87, a0a0c−), P21/n (no. 14, a−b+a−) and I2/m (no. 12, a0b−b−). All these structures are then optimized by the Vienna Ab Initio Simulation Package (VASP),19–22 based on the projector augmented-wave (PAW) method.23,24 The calculations were also performed using the WIEN2K program,25,26 which is based on the full-potential linearized augmented plane wave (FPLAPW) method with the dual basis set. The reliability of the calculations in the two basis sets has been cross-checked.
m (no. 225, a0a0a0), Pn (no. 201, a+a+a+), R (no. 148, a−a−a−), P4/mnc (no. 128, a0a0c+), I4/m (no. 87, a0a0c−), P21/n (no. 14, a−b+a−) and I2/m (no. 12, a0b−b−). All these structures are then optimized by the Vienna Ab Initio Simulation Package (VASP),19–22 based on the projector augmented-wave (PAW) method.23,24 The calculations were also performed using the WIEN2K program,25,26 which is based on the full-potential linearized augmented plane wave (FPLAPW) method with the dual basis set. The reliability of the calculations in the two basis sets has been cross-checked.
For the self-consistent calculation with the projector augmented-wave, the plane wave cut-off energy was chosen to be 500 eV. The meshes of k-points (including the magnitude of the cell and number of atoms) adopted are listed in Table 1. For the self-consistent field iterations, the convergence tolerances were selected as the difference in total energy and the maximum force being within 1.0 × 10−5 eV and 1.0 × 10−2 eV Å−1, respectively. The plane wave expansion cut-offs were 7.0 for expanding the wave function (RKMAX) in the FPLAPW method. In the complete Brillouin zone, 1000 k-points were used. The values of the atomic sphere radii (RMT) were chosen as 2.3, 1.9, 1.9 and 1.5 a.u. for La, Ni, Cr and O, respectively.
| Magnitude of cell | Number of atoms | Meshes of k-points | ||
|---|---|---|---|---|
| Optimization | Energy calculation | |||
| Fmm |
4 | 40 | 5 × 5 × 5 | 7 × 7 × 7 |
| Pn |
4 | 40 | 5 × 5 × 5 | 7 × 7 × 7 |
| R |
1 | 10 | 5 × 5 × 5 | 7 × 7 × 7 |
| P4/mnc | 2 | 20 | 5 × 5 × 3 | 7 × 7 × 5 |
| I4/m | 2 | 20 | 5 × 5 × 3 | 7 × 7 × 5 |
| P21/n | 2 | 20 | 5 × 5 × 3 | 7 × 7 × 5 |
| I2/m | 2 | 20 | 5 × 5 × 3 | 7 × 7 × 5 |
For both the methods, the exchange–correlation energy was treated by the generalized gradient approximation (GGA-PBE).27 Electron–electron Coulomb repulsion interactions or electron correlation (U) for the transition elements in combination with the double-counting correction were considered in the rotationally invariant form (GGA + U) with a single effective Hubbard parameter, Ueff = U − J.28 A series of U values were selected for transition metals, i.e., 0.5–6.0 eV for Cr and 0.5–8.0 eV for Ni, (with UNi > UCr as Ni follows Cr in the periodic table). Our calculations indicate that for the selected U values, the main conclusions are consistent. Therefore, unless specified, we choose the results from UNi = 6.0 eV and UCr = 4.0 eV for discussion.
Results and discussion
Ni/Cr arranged along [111] direction
When Ni/Cr is arranged along the [111] direction, it corresponds to an ordered double perovskite, i.e., there are six Ni–O–Cr linkages for each Ni(Cr). To determine the ground state (including both the crystal symmetry and magnetic configurations), structure relaxations were carried out for all the seven crystal structures predicted from the SPUDS program with both magnetic coupling (i.e., Ni/Cr FM and AFM coupled) and electron correlations considered. The energy difference is listed in Table 2.| FM | AFM | |||
|---|---|---|---|---|
| GGA | GGA + U | GGA | GGA + U | |
| Fmm |
1109.7 | 2032.4 | 1345.9 | 2447.5 |
| Pn |
566.9 | 1185.8 | 655.7 | 899.2 |
| R |
93.1 | 205.2 | 115.9 | 296.8 |
| P4/mnc | 520.4 | 566.5 | 60.9 | 1433.2 |
| I4/m | 328.2 | 955.7 | 312.7 | 955.7 |
| P21/n | 0 | 0 | −54.2 | 109.8 |
| I2/m | 100.4 | 106.2 | 172.8 | 245.8 |
In the GGA calculation, the AFM coupled Ni and Cr in monoclinic symmetry is favoured, which is in agreement with the expectation that structure relaxation will change magnetic state. However, when electron correlation is included (even with a very small value, i.e., UNi = UCr = 0.5 eV), the FM state is the most stable. This means that the ground state is sensitive to electron correlation. A similar phenomenon is observed in other double perovskite transition metal oxides. Taking La2CrFeO6 as an example,29 it is ferrimagnetic in the GGA calculation, which is in contradiction with the experimentally reported ferromagnetic state, whereas the inclusion of electron correlation will make the ferromagnetic coupling more stable.
Regardless of the magnetic state, monoclinic P21/n is the most stable, indicating the presence of structure distortions. Moreover, both the correlation and magnetic coupling have little effect on the structural parameters (less than 0.3%). Thus, only the structural parameters from GGA (FM) are given in Table 3. It is found that, due to the crystal distortion, the six Ni–O(Cr–O) bonds are divided into three groups. This will make the degenerated d states split. However, all the bond lengths are close to each other. Therefore, the sub-orbits are expected to be close in energy. Given that the radii of Ni3+ (0.70 Å for low spin (LS) and 0.74 Å for HS) are smaller than that of Cr3+ (0.76 Å), the larger Ni–O bond lengths suggests a Ni2+ (0.83 Å)/Cr4+ (0.69 Å) configuration, in contrast to the previous study.15 The Ni–O–Cr bond angles (Table 3) substantially deviate from the ideal 180° angle, which weakens the electron hopping interactions in this bridge.
| [111] | [001] | |||
|---|---|---|---|---|
| Space group | P21/n | P21/b | ||
| a | 5.517 Å | 5.493 Å | ||
| b | 5.577 Å | 5.499 Å | ||
| c | 7.802 Å | 7.839 Å | ||
| α/β | 89.99° | 90.01° | ||
| O1 | x | 0.7216 | 0.7168 | |
| y | 0.2961 | 0.2836 | ||
| z | 0.0408 | 0.0400 | ||
| O2 | x | 0.7981 | 0.7168 | |
| y | 0.7798 | 0.2833 | ||
| z | 0.0425 | 0.4644 | ||
| O3 | x | 0.0781 | 0.0722 | |
| y | 0.4807 | 0.4883 | ||
| z | 0.2418 | 0.2422 | ||
| Ni–O1 | 2.078 Å × 2 | |||
| Ni–O2 | 2.079 Å × 2 | 1.980 Å × 2, 1.979 Å × 2 | ||
| Ni–O3 | 2.063 Å × 2 | 2.060 Å × 2 | ||
| Cr–O1 | 1.937 Å × 2 | 1.987 Å × 2, 1.984 Å × 2 | ||
| Cr–O2 | 1.946 Å × 2 | |||
| Cr–O3 | 1.937 Å × 2 | 1.941 Å × 2 | ||
| Bond angles | In-plane | 155.1°(Ni–O1–Cr) | 157.9°(Ni–O2–Ni) | |
| 154.1°(Ni–O2–Cr) | 156.3°(Cr–O1–Cr) | |||
| Out-of-plane | 154.3°(Ni–O3–Cr) | 156.8°(Ni–O3–Cr) | ||
It is interesting to note that for either the Ni3+(S = 3/2)/Cr3+(S = 3/2) or Ni2+(S = 1)/Cr4+(S = 1) configuration, once the spin is antiparallel, the magnetization is zero. The calculated spin magnetic moment is 1.66 μB for Cr and −1.48 μB for Ni (Table 4), close to the theoretical value of the Ni2+(S = 1)/Cr4+(S = 1) configuration. Including this in the interstitial sites, the net magnetic moment is zero, consistent with the abovementioned analysis. Because it is AFM coupling in the GGA method, the next concern is whether it is half metallic to obtain HM-AFM. In the density of states (DOS) (Fig. 2, left, GGA), it is observed that there is a gap as large as 1.2 eV in the spin-down channel, which is formed between the filled Ni 3d states and unoccupied Cr 3d states. In the spin-up channel, it is the hybrid Ni 3d-O 2p-Cr 3d electrons in the neighborhood of the Fermi level. From the band structure (inset of Fig. 2), it is clear that a small gap of ∼0.1 eV is formed in the spin-up channel, making the [111] case a semiconductor. This is similar for La2NiMnO6 in which GGA gives an insulating nature. However, it is different from La2CoMnO6, in which the insulating nature is driven by the Coulomb-enhanced SOC.
| Ni | Cr | Total | ||
|---|---|---|---|---|
| [111] | GGA | −1.48 | 1.66 | 0.00 |
| GGA + U | 1.76 | 1.95 | 4.00 | |
| [001] | GGA | 1.17 | 2.19 | 4.00 |
| GGA + U | 1.55 | 2.31 | 4.00 | |
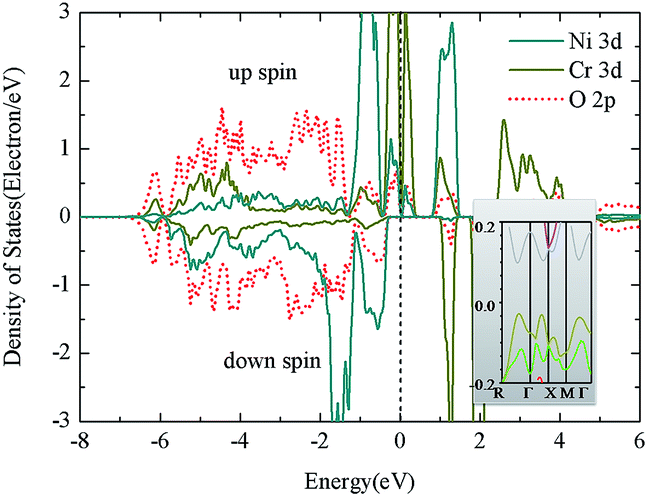 | ||
| Fig. 2 Density of states for Ni/Cr along [111] arrangement from the GGA method. The inset is the band structure of the spin-up channel near the Fermi level. | ||
Turning to the FM state from the GGA + U calculations, it is also insulating (Fig. 3). The band gap in the vicinity of the Fermi level becomes larger with increasing U values (not shown). Fig. 3 shows the DOS and band structure from UNi = 6.0 eV and UCr = 4.0 eV, which are the typical values for these two elements.30–32 The small gap in the spin-up channel is 0.90 eV and the larger one in the spin-down channel is 2.8 eV. In the spin-up channel, the Ni 3d states are all occupied, and from the partial DOS (Fig. 4, left), it is seen that three orbitals, namely, dyz, dxz and dx2y2, are occupied (the three orbitals correspond to conventional t2g states if the crystallographic xy plane is rotated by 45° from the basal plane). This indicates that Ni is in the 2+ state (3d8, t2g6eg2↑, S = 1), similar to that in La2NiMnO6 (ref. 4). The calculated orbital occupancy for each orbital (Table 4) is also consistent with this configuration. For Cr 3d states (Fig. 4, right), which mainly occupy the spin-up channel, they are split and this is the reason for the insulating behavior. Concerning the orbital occupancy, it might not be so obvious because of the hybridization, for instance, it is only 0.76 for the fully populated dyz (Table 5).
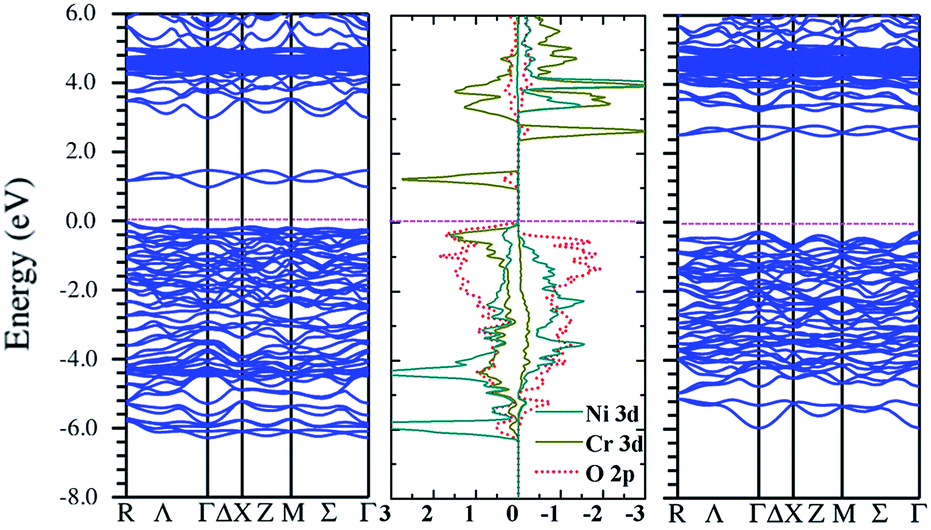 | ||
| Fig. 3 Density of states and band structure for Ni/Cr along [111] arrangement from the GGA + U (UNi = 6.0 eV and UCr = 4.0 eV) method. | ||
 | ||
| Fig. 4 Partial density of states of Ni/Cr along the [111] arrangement of Ni/Cr from GGA + U (UNi = 6.0 eV and UCr = 4.0 eV) method. | ||
| dz2 | dx2y2 | dxy | dxz | dyz | |||
|---|---|---|---|---|---|---|---|
| [111] | Ni | up | 0.94 | 0.93 | 0.95 | 0.92 | 0.91 |
| dn | 0.10 | 0.85 | 0.16 | 0.89 | 0.90 | ||
| Cr | up | 0.36 | 0.57 | 0.37 | 0.45 | 0.76 | |
| dn | 0.18 | 0.08 | 0.18 | 0.08 | 0.06 | ||
| [001] | Ni | up | 0.94 | 0.92 | 0.84 | 0.91 | 0.92 |
| dn | 0.18 | 0.90 | 0.18 | 0.83 | 0.88 | ||
| Cr | up | 0.27 | 0.83 | 0.26 | 0.53 | 0.83 | |
| dn | 0.15 | 0.05 | 0.13 | 0.06 | 0.05 | ||
Compared with the GGA calculation, the spin magnetic moment is increased to 1.74 μB for Ni and 1.95 μB for Cr (Table 4) in GGA + U calculations. This is understandable because the application of U localizes the electrons.
Ni/Cr arranged along [001] direction
Perovskite oxide films and superlattices are recent hot topics owing to the development of film-grown technologies such as pulsed laser deposition (PLD) and molecular beam epitaxy (MBE). It is feasible to grow cations with different stacking arrangements, which provides an opportunity to better understand the influence of disordering. In the following study, Ni/Cr was arranged along the [001] direction. Different from the [111] case, for each Ni(Cr) there are four Ni–O–Ni(Cr–O–Cr) linkages (in-plane) and two Ni–O–Cr linkages (out-of-plane). This internal environment variation leads to the change in the crystal structure into P21/b, which is still monoclinic. According to the structure parameters mentioned in Table 3, it is seen that the in-plane parameters (a and b) are smaller but c is slightly larger compared with the [111] case. It is also seen that the in-plane Ni–O bond distances are considerably smaller than the out-of-plane ones, making the NiO6 octahedra elongated. In contrast, the CrO6 octahedra are compressed. The bond angles are also far from the ideal 180° due to distortion.In contrast to the [111] case, the FM state is favored with and without U for the [001] case. In the GGA calculation, the energy difference is small, i.e., 4.5 meV, and it increases with increasing U. Fig. 5 plots the DOS for the [001] case. It is on the end-point of half-metal to metal transition in the GGA method because the Ni spin-down conduction band is on the edge of the Fermi level. Obviously, a small U value will push the unoccupied state to higher energy region, forming a gap in the spin-down channel. In the DOS from the GGA + U calculation with UNi = 6.0 eV and UCr = 4.0 eV (Fig. 5), a large gap (∼3.6 eV) in the spin-down channel is formed. Moreover, in the spin-up channel, most Ni 3d states are pushed down to lower energy region and Cr 3d states are almost split. However, a small amount of electrons are preserved in the neighborhood of the Fermi level (inset of Fig. 5) making this case half metallic.
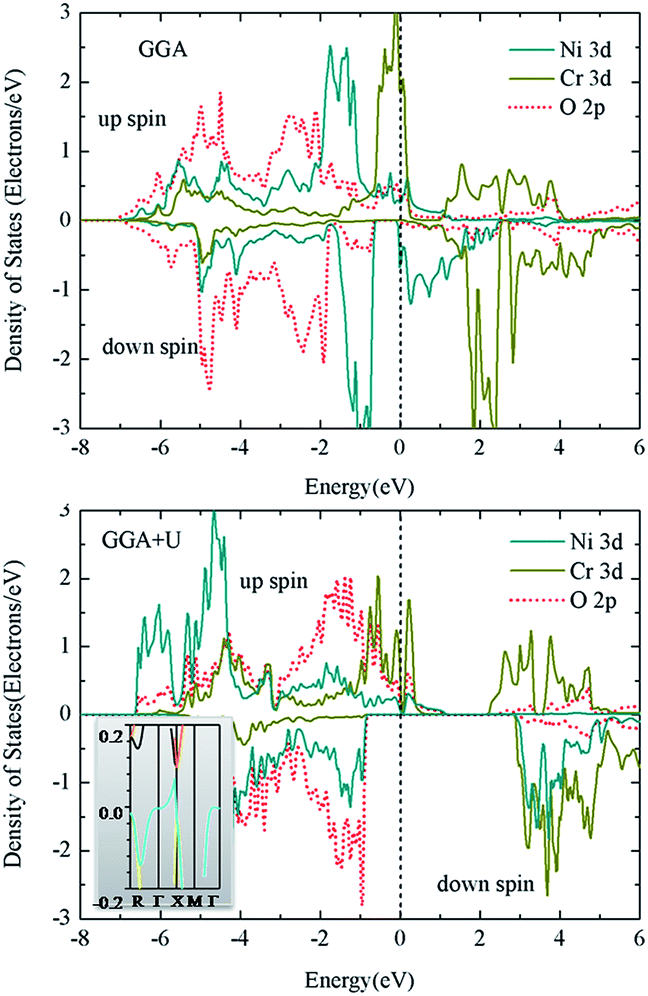 | ||
| Fig. 5 Density of states for Ni/Cr along [001] arrangement from the GGA and GGA + U (UNi = 6.0 eV and UCr = 4.0 eV) methods. The inset is the band structure near the Fermi level of the spin-up channel. | ||
For 3d metals, electron correlation is important. For instance, besides the electronic structure change, valence states of transition metals would also be different when U is considered in the un-synthesized NiCrO3 (ref. 32). For the present cases, small U values will introduce an interesting feature, i.e., a semiconductor to half-metal transition. A similar transition is reported for La2NiMnO6 and La2CoMnO6 (ref. 14). For these two cases, the origin is explained by the extension of eg states as a result of additional Mn–O–Mn, Ni–O–Ni or Co–O–Co linkages. This holds true for the Ni–O–Ni bridges of the title compound. As seen in the PDOS (Fig. 6), the Ni dxy electrons run over the Fermi level, which is fully filled in the [111] case. Thus, the number of electrons reduces by 0.11 (from 0.95 to 0.84) (Table 5). To balance this, the Cr 3d electrons in the conduction band move downwards to provide additional electrons; moreover, they play the role of conduction electrons. Obviously, the reduction of Ni 3d states makes the valence state of Ni enhanced, whereas the situation for Cr is the opposite. Therefore, similar to La2NiMnO6 and La2CoMnO6 (ref. 14), mixed valence states of Ni2+/Cr4+ and Ni3+/Cr3+ are assumed.
 | ||
| Fig. 6 Partial density of states of Ni/Cr along [001] arrangement from GGA + U (UNi = 6.0 eV and UCr = 4.0 eV) method. | ||
From Table 4, it is seen that the magnetic moment of Ni is reduced. Hence, Ni3+ should be in the LS state (3d7, t2g6eg1↑, S = 1/2), disagreement with previous speculation.15 In contrast, because of the larger moment of Cr3+(S = 3/2), the magnetic moment of Cr is increased (Table 4). In addition, the longer Cr–O bond length gives less hybridization with oxygen, which will also contribute to the enhancement of the Cr magnetic moment. However, the net magnetic moment of 4.00 μB per f.u. remains, which is a requirement for half metallic compounds.
Finally, strain often exists in films, which appears due to the mismatch between substrate and sample, and this will make the physical properties different from bulk. To simulate this effect, the electronic structure during compression as well as expansion of its equilibrium lattice constant is studied. In the calculation, parameter c is set to be compressed (elongated) by 2% and kept fixed, meanwhile atomic positions and parameters a and b are optimized. The magnetic coupling of Ni and Cr is not influenced by the strain effect, which remains ferromagnetic in both compressive and tensile cases. From the total DOS (Fig. 7), it is seen that half metallic character could be preserved in both compressive and tensile strain from the GGA + U method. However, in the GGA calculation, it is normal metal for the compressive case because the conduction band moves downward in the spin-down channel. This is understandable considering that the electronic wave function becomes more overlapped as the atomic distance gets closer. The situation is opposite for the case of tensile strain. Thus, a gap is formed in the spin-down channel with the conduction band moving upward. Therefore, a half metallic nature could be obtained in both GGA and GGA + U methods under tensile strain.
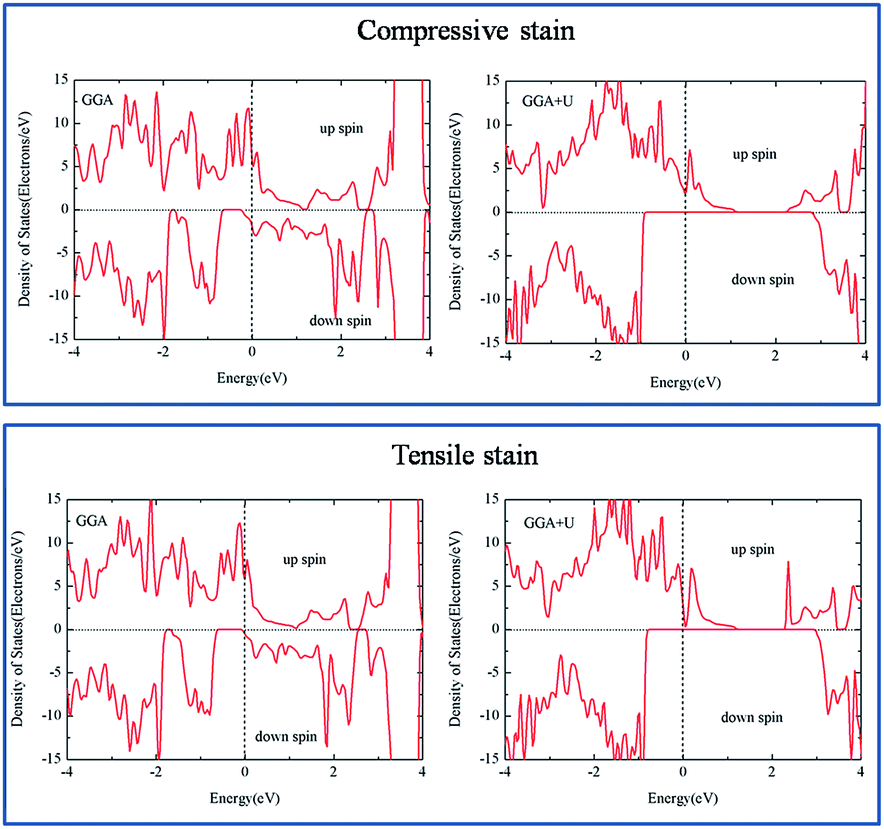 | ||
| Fig. 7 Total density of states of Ni/Cr along [001] arrangement under compressive and tensile strain from the GGA and GGA + U (UNi = 6.0 eV and UCr = 4.0 eV) methods. | ||
Conclusions
La2NiCrO6 with cation ordering in both [111] and [001] direction is investigated by density functional theory. The effects of electron correlation are important to decide both magnetic and electronic states. Within reasonable U values, a semiconductor to half-metal transition is expected with the Ni/Cr arrangement changing from [111] to [001]. Under both compressive and tensile strains, the half metallic character could be preserved. The lattice constant of SrTiO3 (3.905 Å) is close to the in-plane parameter of the [001] case (3.905√2 ≈ 5.515, close to a and b), which is usually chosen to be the substrate. It is interesting that the LaNiO3–LaCrO3 superlattice grown on SrTiO3 substrate could be of potential use in spintronics. Further experimental investigations are suggested from this theoretical study.Acknowledgements
The authors thank the National Natural Science Foundation of China (Grant nos 21403185, 21221061, 21201148, and 21303156), the Natural Science Foundation of Hebei (Grant no. B2015203268, B2011203121) and China Postdoctoral Science Foundation (Grant no. 2014M551048) for financial support.Notes and references
- N. S. Rogado, J. Li, A. W. Sleight and M. A. Subramanian, Adv. Mater., 2005, 17, 2225 CrossRef CAS PubMed.
- R. N. Mahato, K. Sethupathi and V. Sankaranarayanan, J. Appl. Phys., 2010, 107, 09D714 Search PubMed.
- M. Hashisaka, D. Kan, A. Masuno, M. Takano, Y. Shimakawa, T. Terashima and K. Mibu, Appl. Phys. Lett., 2006, 89, 032504 CrossRef PubMed.
- H. Das, U. V. Waghmare, T. Saha-Dasgupta and D. D. Sarma, Phys. Rev. Lett., 2008, 100, 186402 CrossRef.
- S. H. Lv, H. P. Li, Z. C. Wang, L. Han, Y. Liu, X. J. Liu and J. Meng, Appl. Phys. Lett., 2011, 99, 202110 CrossRef PubMed.
- S. Baidya and T. Saha-Dasgupta, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 035131 CrossRef.
- A. J. Barón-González, C. Frontera, J. L. García-Muñoz, J. Roqueta and J. Santiso, J. Phys.: Conf. Ser., 2010, 200, 092002 CrossRef.
- H. Z. Guo, A. Gupta, T. G. Calvarese and M. A. Subramanian, Appl. Phys. Lett., 2006, 89, 262503 CrossRef PubMed.
- R. I. Dass and J. B. Goodenough, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 014401 CrossRef.
- P. A. Joy, Y. B. Khollam and S. K. Date, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 8608 CrossRef CAS.
- R. I. Dass, J.-Q. Yan and J. B. Goodenough, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 064415 CrossRef.
- M. P. Singh, C. Grygiel, W. C. Sheets, P. Boullay, M. Hervieu, W. Prellier, B. Mercey, C. Simon and B. Raveau, Appl. Phys. Lett., 2007, 91, 012503 CrossRef PubMed.
- M. P. Singh, K. D. Truong, S. Jandl and P. Fournier, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 224421 CrossRef.
- M. Zhu, Y. Lin, E. W. C. Lo, Q. Wang, Z. J. Zhao and W. H. Xie, Appl. Phys. Lett., 2012, 100, 062406 CrossRef PubMed.
- W. E. Pickett, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 10613 CrossRef CAS.
- M. W. Lufaso and P. M. Woodward, Acta Crystallogr., Sect. B: Struct. Sci., 2001, 57, 725 CrossRef CAS PubMed.
- A. M. Glazer, Acta Crystallogr., Sect. B: Struct. Sci., 1972, 28, 3384 CrossRef CAS.
- A. M. Glazer, Acta Crystallogr., Sect. A: Found. Crystallogr., 1975, 31, 756 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- K. Schwarz and P. Blaha, Comput. Mater. Sci., 2003, 28, 259 CrossRef CAS.
- P. Blaha, K. Schwarz, G. K. H. Madsen, D. Kvasnicka and J. Luitz, WIEN2K, An Augmented Plane Wave and Local Orbitals Program for Calculating Crystal Properties, Technical University Wien, Vienna, 2001 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- A. I. Liechtenstein, V. I. Anisimov and J. Zaanen, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, R5467 CrossRef CAS.
- K. Miura and K. Terakura, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 63, 104402 CrossRef.
- H. H. Chen, A. J. Millis and C. A. Marianetti, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 111, 116403 Search PubMed.
- M. J. Han and M. van Veenendaal, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 195102 CrossRef.
- K.-W. Lee and W. E. Pickeet, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 180406(R) CrossRef.
| This journal is © The Royal Society of Chemistry 2015 |