DOI:
10.1039/C5RA07405C
(Paper)
RSC Adv., 2015,
5, 48916-48927
Catalytic oxidation of 1,2-dichloroethane over Al2O3–CeO2 catalysts: combined effects of acid and redox properties†
Received
23rd April 2015
, Accepted 27th May 2015
First published on 27th May 2015
Abstract
The lower temperature catalytic combustion of 1,2-dichloroethane (DCE) over Al2O3, CeO2 and Al2O3–CeO2 catalysts were studied. An apparent deactivation for pure Al2O3 and CeO2 catalyst was observed at 300 °C, which was ascribed to the formation of coke for pure Al2O3 while the strong adsorption of Cl species at active sites for pure CeO2. However, Al2O3–CeO2 mixed oxides exhibited a high stable activity compared with the pure Al2O3 and CeO2, which indicated that suitable acidity and excellent redox performance was equally important for total oxidation of DCE. TPSR experiments indicated that ClCH2CHO was the intermediate product and vinyl chloride (VC) was the main by-product for DCE catalytic oxidation over Al2O3–CeO2 catalyst, ClCH2CHO and VC were the competitive products of the C–H-scission ethoxides, which generated by α- and β-CH-scission respectively. Based on in situ DRIFTS study, a simple reaction pathway was proposed: Firstly, DCE adsorbed on surface hydroxyl groups or Lewis acid sites via the C–Cl bond and was attacked by the basic site (O2−), and then the intermediates such as aldehyde species and by-product VC formed. Sequentially, the aldehyde species were converted into H2O, COx and HCl via further Cl abstraction and oxidation process, by contrast, VC was not easy to be further decomposed because of its stable structure, the weak adsorption and difficult dissociation on acid sites.
1. Introduction
Chlorinated volatile organic compounds (CVOCs), such as 1,2-dichloroethane (DCE), trichloroethylene (TCE), vinyl chloride (VC), and chlorobenzene (CB), are widely used as cleaning solvents in garment industries, electronic industries and automotive/aerospace industries, extraction agents, herbicides, additives paints and adhesives, and in the synthesis of pesticides and rubber ingredients.1–4 Because of their inertness and wide-spread application in industry combined with their threat to the air and groundwater when discharged into the environment, CVOCs have been always considered as one of the most hazardous organics. So the elimination of CVOCs becomes more and more important and catalytic oxidation is considered as the most effective method and widely applied due to its low operation temperature and high selectivity.
DCE is one of the main pollutants generated in VC monomer plants and also found in groundwater air-stripping emissions admixture with other chlorinated solvents such as TCE, DCM and 1,2-dichloroethylene.5–13 Generally, two kinds of catalysts were studied in the total oxidation of DCE, metal oxides and solid acid catalyst. Seiichiro et al.6 reported the decomposition of DCE over several metal oxides supported on titania–silica and found that Cr catalysts showed the best catalytic activity, however, this kind of catalyst was easy to form volatile metal oxychloride (CrOClx) and leaded to the reduction in the activity of the catalyst. Recently, Gutierrez-Ortiz et al.7–13 suggested that suitable acidity and excellent redox performance were very important for total oxidation of DCE over ceria–zirconia mixed oxides (Ce/Zr solid solutions). Among them, the Ce0.5Zr0.5O2 catalyst with relative mild acidity and the largest H2 consumption exhibited a higher activity than the Ce0.15Zr0.85O2 catalyst possessing the most total acidity and strong acidity sites, which indicated that the redox performance was more important than the acidity for the Ce/Zr solid solutions catalysts. Moreover, the major oxidation products were CO2, CO, Cl2 and HCl, whereas the formation of chlorine, generating from the deacon process (4HCl + O2 → 2H2O + 2Cl2), may lead to the production of polychlorinated by-products. Feijen-Jeurissen et al.1 studied the combustion behavior of DCE over γ-Al2O3 and found that a large amount of VC was formed at temperatures between 250 and 450 °C. They proposed that the first step of DCE elimination was the abstraction of HCl and formation of VC. Aranzabal et al.14 investigated the decomposition of DCE over the protonic acid catalysts (H-ZSM-5, H-MOR and H-BEA), which displayed high activity and selectivity to target products such as HCl, COx and H2O. Seiichiro et al.15 reported the catalytic combustion of DCE over several catalysts, and they found that the COx selectivity was only 33.5% over the zeolite Y at 400 °C while the selectivity to COx can reach up to as high as 97.8% over TiO2/SiO2. Gutierrez-Ortiz12 explored the causation of the deactivation over acidic catalysts (H-zeolites with a notable presence of strong Brønsted sites) for total oxidation of DCE, and the formation of coke originated from polymerisation of reaction intermediates such as VC and HCl was the primary cause.16–21 Therefore, the elimination or inhibition of coke was crucial for catalytic combustion of CVOCs over acidic catalysts, and the support of metal oxides were studied widely to improve the redox performance. Li et al. reported that (Cu–Mn)/MCM-41 can minimize coke formation.17 Kucherov et al. also reported that Rh/HZSM-5 and (Rh + Au)/HZSM-5 demonstrated high efficiency and reduced coke formation at 320 °C.22 Beatriz de Rivas and Zhou23,24 found that the addition of CeO2 can significantly improve the activity and stability of H-ZSM-5.
In this paper, DCE oxidation was investigated over Al2O3–CeO2 catalysts to study combined effects of acid and redox properties. The effects of preparation methods, Ce content, oxygen concentration and water vapor in the flow gas were studied detailed. Moreover, TPSR and in situ DRIFTS techniques were utilized to determine reaction intermediates, products and possible reaction pathways.
2. Experimental
2.1. Catalysts preparation
2.1.1 Co-precipitation method. CeO2 and Al2O3–CeO2 catalysts were prepared by precipitation and co-precipitation method using (NH4)2CO3 as the precipitating agent. In preparation of CeO2, 10 g of Ce(NO3)3·6H2O and 6.35 g of (NH4)2CO3 were dissolved in 100 mL of deionized water, respectively. Then, (NH4)2CO3 solution was quickly added to Ce(NO3)3·6H2O solution and stirred for 0.5 h at room temperature (RT). Subsequently, the mixture was placed statically for 18 h at RT; the precipitates were washed with distilled water and ethanol, then dried at 110 °C for 24 h and calcined in air at 550 °C for 3 h with a ramp of 2 °C min−1. For Al2O3–CeO2 catalysts, the same recipe except for the addition of aluminum precursor (Al(NO3)3·9H2O) was took. Hereafter the samples were marked as AlCeX/Y-NC, where X and Y referred to the molar percentage of Ce and Al, respectively.
2.1.2 Sol–gel method. Pluronic P123 (2.0 g) was dissolved in ethanol (40 mL) at RT. Citric acid (1.7 g), Al(NO3)3·9H2O (4.3 g), and Ce(NO3)3·6H2O (5 g) were then added to the P123 solution with vigorously stirring. The mixture was covered with polyethylene (PE) film, stirred at RT for at least 5 h, and then placed into a 60 °C drying oven to undergo the solvent evaporation process. After two days aging, the solution turned into a light yellow solid. Calcination was carried out by slowly increasing the temperature from RT to 550 °C (1 °C min−1) and heating at 550 °C for 3 h in air. The resulting catalysts were designated as AlCe50/50-P123.Hexadecyl trimethyl ammonium bromide (CTAB) (7.3 g) was dissolved in ethanol (80 mL) at RT. Citric acid (0.9 g), aluminum isopropoxide (Al(i-OPr)3) (3.7 g), and Ce(NH4)2(NO3)6 (10 g) were then added with vigorously stirring. The next steps were same with AlCe50/50-P123, and the obtained catalysts were designated as AlCe50/50-CTAB.
2.2. Catalytic activity measurement
Catalytic combustion reactions were carried out in a continuous flow microreactor constituted of a U-shaped quartz tube of 3 mm of inner diameter at atmospheric pressure. 150 mg catalyst was placed at the bottom of the U-shaped micro-reactor. The feed flow through the reactor was set at 40 cm3 min−1 and the gas hourly space velocity (GHSV) was maintained at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. Feed stream to the reactor was prepared by delivering liquid DCE with a syringe pump into dry air, which was metered by a mass flow controller. The injection point was electrically heated to ensure complete evaporation of the liquid reaction feeds. The concentration of DCE in the reaction feeds was set at 1000 ppm or 250 ppm. The temperature of the reactor was measured with a thermocouple located just at the bottom of the microreactor. The effluent gases were analyzed by an on-line gas chromatograph equipped with a FID detector. Catalytic activity was measured over the range 100–400 °C and conversion data were calculated by the difference between inlet and outlet concentrations. Additionally, mass spectrum (Hiden HPR 20) was used for the determination of the main intermediates and by-products.
000 h−1. Feed stream to the reactor was prepared by delivering liquid DCE with a syringe pump into dry air, which was metered by a mass flow controller. The injection point was electrically heated to ensure complete evaporation of the liquid reaction feeds. The concentration of DCE in the reaction feeds was set at 1000 ppm or 250 ppm. The temperature of the reactor was measured with a thermocouple located just at the bottom of the microreactor. The effluent gases were analyzed by an on-line gas chromatograph equipped with a FID detector. Catalytic activity was measured over the range 100–400 °C and conversion data were calculated by the difference between inlet and outlet concentrations. Additionally, mass spectrum (Hiden HPR 20) was used for the determination of the main intermediates and by-products.
3. Results and discussion
3.1. Catalytic activity and stability of Al2O3 and CeO2
Fig. 1a showed the initial catalytic performance of pure Al2O3 and CeO2 catalysts for catalytic oxidation of DCE. For pure CeO2-NC catalyst, DCE conversion displayed a valley within 300–400 °C, which was the same as the result for CB catalytic combustion over CeO2 catalyst.17,25 Moreover, the stability tests at 300 °C, as shown in Fig. 1b, clearly presented the conversion of DCE dropped sharply from 85% to 40% within 1 h. Our previous studies26 indicated that the deactivation was ascribed to the strong adsorption of Cl species produced during CVOCs decomposition. In contrast, pure Al2O3-NC catalyst exhibited a worse catalytic activity than pure CeO2, but, a slightly better activity was observed in the range of higher temperature and the DCE conversion reached 95% at 400 °C due to the relatively slow deactivation. However, the continuous deactivation of pure Al2O3 still was inevitable, and the conversion of DCE decreased from 70% to 10% within 7 h at 300 °C. Generally, the deactivation of Al2O3 was attributed to the blocking of the active sites originated from the accumulation of heavy products or coke formation on the surface.17–21,25 In order to clearly understand the decay of activity over pure Al2O3 and CeO2 catalysts, O2-TPO experiments were carried after the stability tests and the result was shown in Fig. 1c. An obvious peak corresponding to CO and CO2 was observed at about 500 °C over Al2O3-NC catalyst, which was a clear indication of the oxidative decomposition of the coke. By comparison, no perceptible COx was detected over the CeO2-NC catalyst. Totally, Al2O3 and CeO2 catalysts both demonstrated relative good catalytic activity for the oxidation of DCE, but the deactivation of both occurred inevitably owing to different reasons.
 |
| | Fig. 1 (a) Light-off curves of 1,2-dichloroethane over Al2O3 and CeO2 catalysts. DCE concentration: 1000 ppm; GHSV: 15000 h−1. (b) The stability of Al2O3 and CeO2 catalysts at 300 °C. (c) The O2-TPO of spent Al2O3 and CeO2 catalysts after the stability test. | |
3.2. Effect of preparation methods and Al/Ce ratio
The light-off curves of 1,2-dichloroethane over Al2O3–CeO2 catalysts prepared by different methods were displayed in Fig. 2a. The 50% conversion was achieved at the ranges between 175 and 230 °C (T50) over all catalysts and the temperature for complete removal (90% conversion, T90) was varied from 250 to 300 °C. The AlCe50/50-NC catalyst exhibited the best catalytic activity, with a T50 value of 175 °C, while the worst activity was observed over the AlCe50/50-CTAB catalyst and T50 value was about 230 °C. Sequentially, the effect of Al/Ce ratio on Al2O3–CeO2 catalysts prepared by co-precipitation method for the catalytic oxidation of DCE was studied, and the results were shown in Fig. 2b. The catalytic activity followed this trend: AlCe50/50-NC > AlCe25/75-NC > AlCe25/75-NC, especially AlCe50/50-NC catalyst presented a better activity between 150 and 250 °C.
 |
| | Fig. 2 Light-off curves of 1,2-dichloroethane over Al2O3–CeO2 catalysts prepared by different method (a) and with different Al/Ce ratios (b). DCE concentration: 250 ppm; GHSV: 15000 h−1; catalyst amount: 150 mg. | |
3.3. Catalyst characterization
The phase structures of Al2O3–CeO2 catalysts prepared by different methods and with different Al content were investigated by XRD and the patterns were shown in Fig. 3. A series of broad and weak peaks at 39°, 45° and 67° was observed for pure Al2O3 catalyst, which corresponded to γ-alumina phase. The Al2O3–CeO2 samples (b–g) exhibited only the characteristic peaks of the cubic fluorite structure of CeO2 at 28.7°, 33.1°, 47.4°, 56.3°, 69.7° and 76.9°, and no crystalline phase ascribed to Al2O3 can be found, indicating that Al2O3 was present as a non-crystalline phase or highly dispersed on surface. The crystallite sizes of CeO2 (111) were listed in Table 1 and fell within the ranges of 2–5.0 nm for all the Al2O3–CeO2 samples expect for the AlCe50/50-CTAB sample (8.0 nm), which was consistent with the results of activity tests (Fig. 2) that the AlCe50/50-CTAB sample presented the worst activity and other samples exhibited a similar catalytic performance. Moreover, the crystallite sizes of Al2O3–CeO2 prepared by using (NH4)2CO3 as the precipitating agent slightly increased with the increasing of Ce content, from 2.1 nm to 4.4 nm.
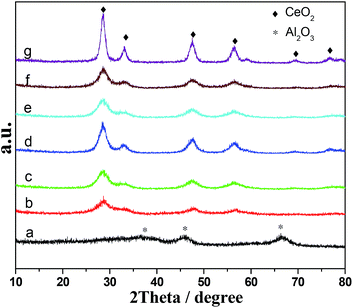 |
| | Fig. 3 XRD patterns of Al2O3–CeO2 catalysts. (a) Al2O3-NC; (b) AlCe75/25-NC; (c) AlCe50/50-NC; (d) AlCe25/75-NC; (e) CeO2-NC; (f) AlCe50/50-P123; (g) AlCe50/50-CTAB. | |
Table 1 Surface and structural properties of Al2O3–CeO2 catalysts
| Catalyst |
Ce/(Al + Ce)a |
Ce/(Al + Ce)b |
Crystallite sizec (nm) |
H2 consumptiond (μmol g−1 cat−1) |
TPR reduction temperatured (°C) |
| Ce and Al content (at.%) measured by EDS. Ce and Al content (at.%) measured by XPS. The crystallite sizes of the CeO2 (111) were calculated from X-ray diffraction line broadening using the Scherrer equation Calculated from TPR results. |
| AlCe75/25-NC |
|
|
2.1 |
270 |
515 |
| AlCe50/50-NC |
69 |
46 |
3 |
619 |
528 |
| AlCe25/75-NC |
77 |
— |
4.4 |
841 |
563 |
| CeO2-NC |
— |
— |
3.2 |
323 |
500 |
| AlCe50/50-P123 |
53 |
47 |
3.2 |
1013 |
550 |
| AlCe50/50-CTAB |
52 |
74 |
8 |
470 |
525 |
TPR profiles of prepared Al2O3–CeO2 catalysts were shown in Fig. 4 and the amounts of hydrogen consumption calculated based on peak area were summarized in Table 1. A broad reduction peak was achieved varied between 300 and 675 °C for the all Al2O3–CeO2 samples (Fig. 4b–h), and the redox process for these samples was found to be promoted with respect to that for the pure ceria (Fig. 4e). The reduction of CeO2 was well known to occur via a step wise mechanism, starting at lower temperature (400–550 °C) with the reduction of the outermost layer of Ce4+ (surface reduction), followed (above 700 °C) by reduction of inner/deeper Ce4+ layers (bulk reduction) at higher temperature. According to the reduction peak area and the reduce of copper oxide power as a reference, the H2 consumption was calculated and the results were listed in the Table 1, all catalysts except for the AlCe75/25-NC sample consumed more hydrogen consumption than that of the pure CeO2 sample, and the hydrogen consumption of AlCe50/50-P123 sample reached a maximum value and represented around three times than that of the pure CeO2. This result inferred that the interaction between CeO2 and Al2O3 may promote the mobility of the lattice oxygen when compared to the pure CeO2, which may be one of reasons that these catalysts exhibited better activity. Additionally, it can be found that the reduction temperature of Al2O3–CeO2-NC samples (b–d) increased with the increasing of the amount of Ce.
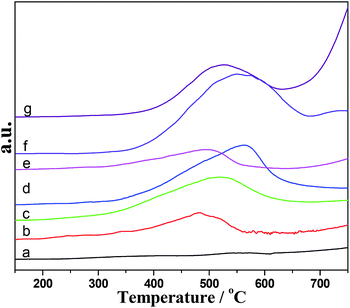 |
| | Fig. 4 H2-TPR profiles of catalysts. (a) Al2O3-NC; (b) AlCe75/25-NC; (c) AlCe50/50-NC; (d) AlCe25/75-NC; (e) CeO2-NC; (f) AlCe50/50-P123; (g) AlCe50/50-CTAB. | |
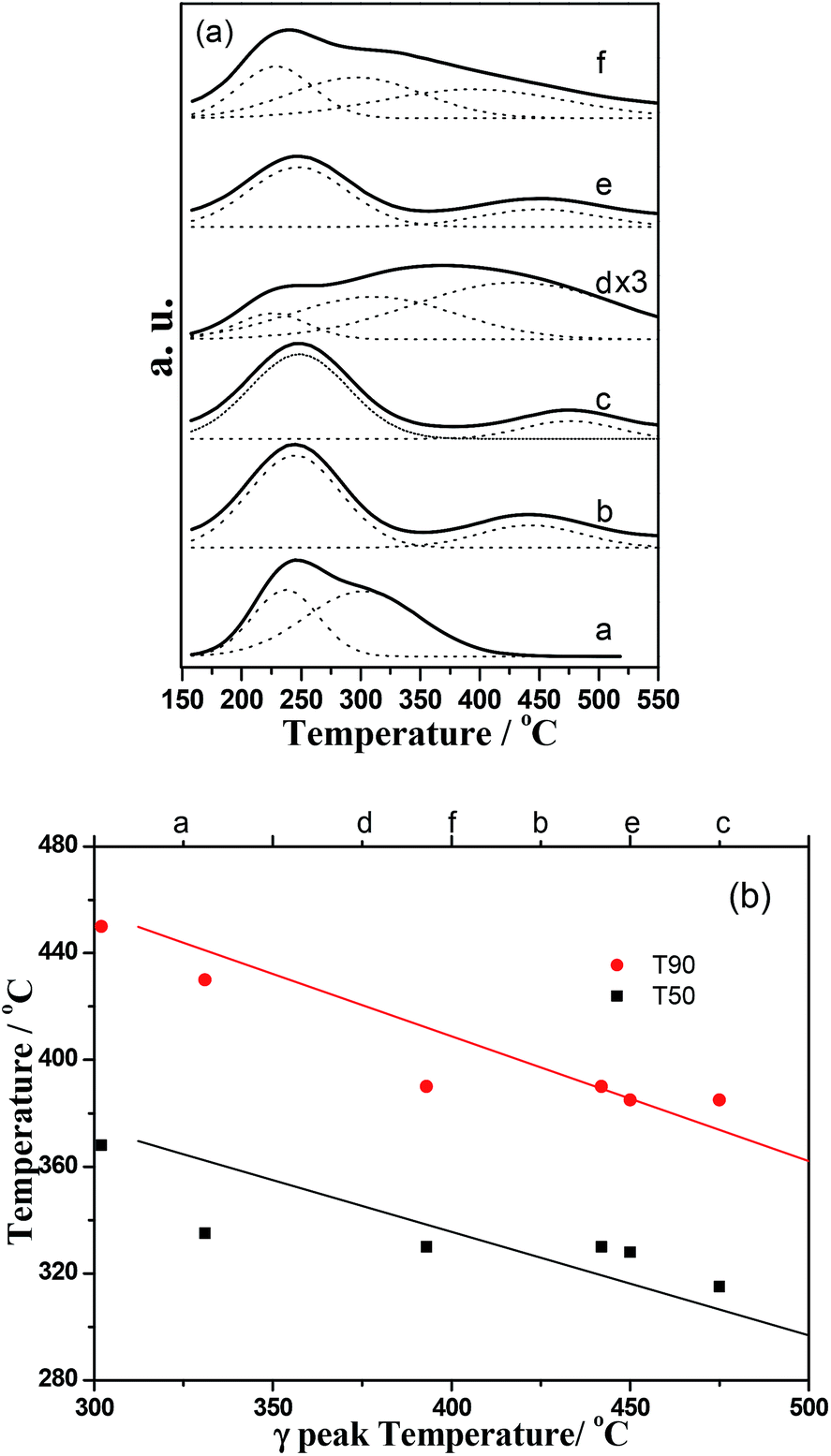
The NH3-TPD profiles, shown in Fig. 5, evidenced that acid properties of the mixed oxides notably differed from those of the pure oxides. Generally, the acid desorbed at temperatures lower than 250 °C was considered as weak acid sites.27–30 Thus, in Fig. 5a the profiles were simulated according to two function associated with desorption at low temperature (LT, around 230 °C) and high temperatures (HT, 430–475 °C), respectively. The desorption peak at high temperature shifted to higher temperature with increasing of cerium oxide. Hence, the AlCe50/50-NC sample also exhibited the strongest acid sites in these catalysts. As showed in Fig. 5b, a linear relationship between T50, T90 and the HT peak temperature had been observed. The T50 and T90 decreased as the temperature of the HT peak increases and thus the acid strength was crucial for DCE combustion. Total acidity of the synthesized catalysts was present in Table 2. The pure CeO2 possessed the lowest value and with the addition of Al, the total acidity of the catalysts markedly increased, and thus the AlCe25/75-NC held the highest acidity. Furthermore, it was noted the strong acidity made a greater contribution to total acidity for cerium-rich mixed oxides, which may hint that the strong acid sites was related with the ceria. Gutierrez-Ortiz et al.9 suggested that the suitable acidity and excellent redox performance was important for the total oxidation of DCE.
 |
| | Fig. 5 (a) NH3-TPD profiles of catalysts, a. Al2O3-NC; b. AlCe25/75-NC; c. AlCe50/50-NC; d. CeO2-NC; e. AlCe50/50-P123; f. AlCe50/50-CTAB; (b) T50 and T90 of Al2O3–CeO2 catalysts as a function of the γ peak temperature of the NH3-TPD. | |
Table 2 Amount of NH3 desorbed from Al2O3–CeO2 catalysts
| Sample |
Peak temp. (°C) |
Total acidity (mmol NH3 per g cat) |
Strong sites (mmol NH3 per g cat) |
Activitya (°C) |
Activitya (°C) |
| LT |
HT |
T50 |
T90 |
| The result of Fig. S2. |
| Al2O3-NC |
237 |
302 |
0.178 |
0.066(mid) |
368 |
450 |
| AlCe50/50-NC |
248 |
475 |
0.173 |
0.029 |
315 |
385 |
| AlCe25/75-NC |
240 |
433 |
0.183 |
0.038 |
328 |
385 |
| CeO2-NC |
226 |
331 |
0.08 |
0.048 |
335 |
430 |
| AlCe50/50-P123 |
247 |
450 |
0.116 |
0.023 |
330 |
390 |
| AlCe50/50-CTAB |
229 |
393 |
0.18 |
0.069 |
330 |
390 |
3.4. Effect of oxygen concentration
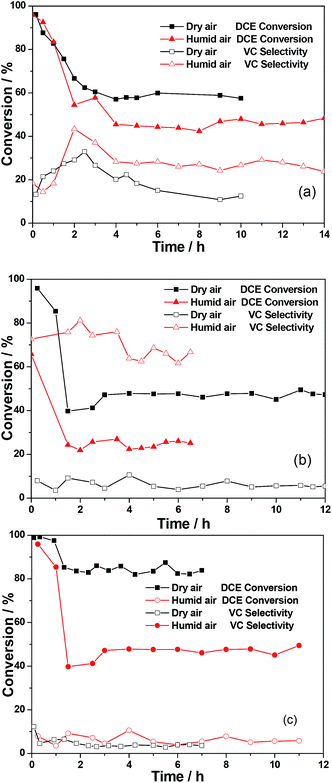
Fig. 6 described the stability activity and VC selectivity over CeO2-NC, Al2O3-NC and AlCe50/50-NC catalysts under different inlet oxygen concentration at 300 °C. The results showed that the deactivation was observed over all the catalysts during the initial stages (within 2–4 h), and then the conversion of DCE can be maintained at a constant value (within 10 h) dependent on catalysts components and oxygen concentration. In the presence of 20% oxygen, the AlCe50/50-NC catalyst exhibited the best stability activity, with about 85% DCE conversion, while the worst activity was detected over the Al2O3-NC catalyst and the DCE conversion was only remained at about 45%. Additionally, it can be found that AlCe50/50-NC catalyst presented the lowest VC selectivity (lower than 5%). By contrast, the pure Al2O3-NC and CeO2-NC catalysts exhibited a higher VC selectivity, ranging from 10 to 20%. Without the presence of oxygen, all catalysts demonstrated an obvious rapid deactivation within initial 2 h and was the same with that under 20% oxygen condition. Expectedly, the stable conversion of DCE was lower than that under condition with 20% oxygen, even over the AlCe50/50-NC and CeO2-NC catalysts possessing the highest active, their stable conversion can only reach up to 35%. Simultaneously, 100% and 80% of DCE was converted to VC over CeO2-NC and AlCe50/50-NC catalysts respectively. However, the selectivity to VC over the Al2O3-NC catalyst was only 35%. Therefore, the transformation of DCE under no-oxygen condition should really be considered as HCl abstraction, especially for CeO2 based catalysts.
 |
| | Fig. 6 The stability of the DCE and selectivity of VC over CeO2-NC (a), Al2O3-NC (b) and AlCe50/50-NC (c) catalysts under different inlet oxygen concentration conditions. DCE concentration: 250 ppm; GHSV: 15000 h−1; catalyst amount: 150 mg; balance N2; reaction temperature: 300 °C. | |
3.5. Effect of water
Fig. 7 showed the effect of water on the activity and VC selectivity of DCE catalytic oxidation over pure CeO2, Al2O3 and Al2O3–CeO2 catalysts at 300 °C. It was found that the addition of 3% (v/v) water apparently inhibited the catalytic decomposition of DCE over all catalysts and this negative effect can be explained by the competitive adsorption of water and DCE on the active sites. Ramanathan et al.31 reported the inhibitive effect of water on the conversion of DCE over the chromia catalyst, was probably due to blocking of active oxygen sites. Additionally, the presence of water increased obviously the VC selectivity over the Al2O3 and CeO2 catalysts, especially for the Al2O3, from 10% to 65%. However, the effect of water on the VC selectivity over AlCe50/50-NC catalyst was almost ignored. Mochida32 reported that the elimination reactions of the chlorinated compounds over the dry alumina were postulated to proceed through an E2-concerted mechanism where the chlorine and proton were eliminated almost simultaneously by the acidic and basic sites of the alumina. Water can decrease the number of Lewis acidic sites and increase the number of Brönsted acidic sites, however, the Brönsted acidic sites may not play an important role in this kind of elimination reaction, at the same time, and the number of basic sites may be unchanged because new basic sites created on the oxygen atom of adsorbed water. Thus, the addition of water resulted in a shift of the mechanism from E2-concerted into E1 mechanism, and then increased the selectivity of VC over Al2O3. Widely accepted, CeO2 demonstrated both Lewis acid and Lewis base, and basicity was more predominate. Additionally, the addition of CeO2 into Al2O3 greatly enhanced the surface basicity of the catalysts.33,34 Therefore, for CeO2 based catalysts (including pure CeO2 and AlCe50/50-NC catalysts), the elimination reactions of HCl occurred via E1 mechanism whether under dry or humid conditions, which led to a negligible change of VC selectivity.
 |
| | Fig. 7 The effect of water vapour (30000 ppm) in the feed on the catalytic oxidation of DCE over CeO2-NC (a), Al2O3-NC (b) and AlCe50/50-NC (c) catalysts. DCE concentration: 250 ppm; GHSV: 15000 h−1; catalyst amount: 150 mg; temperature: 300 °C. | |
3.6. XPS of fresh and spent catalysts
The fresh and spent (after 10 h reaction at 400 °C under Ar condition) catalysts were characterized by XPS, and the spectra of Ce 3d, O 1s and Cl 2p were presented in Fig. 8. As shown in Fig. 8a, Ce 3d spectra of all samples exhibited typical ten peaks,35–37 including the six peaks at about 882.4, 888.7, 897.6, 900.1, 907.7 and 916.6 eV arising from Ce4+ contributions and four peaks at about 880.2, 884.6, 898.7 and 902.6 eV assigned to Ce3+, which indicated the co-existence of Ce3+ and Ce4+ in all samples. The proportion of Ce3+ ions with regard to the total cerium was calculated from the ratio of the sum area of Ce3+ species to the sum area of total cerium species and the concentration of Ce3+ was estimated to be 22.5, 22.1, 22.3 and 22.1% for CeO2, spent CeO2, AlCe50/50 and spent AlCe50/50 (Table 3), respectively, and the changes of Ce 3d XPS spectra were negligible. Thus it can be seen that the introduction of Al and the enduring test did not cause the increase of Ce3+, which indicated that the CeO2 was stable and not chlorinated. The O 1s spectra clearly showed the presence of two kinds of oxygen species (Fig. 8b). For the CeO2-NC, the binding energy of 529.8 eV, denoted as Oα, was the index of lattice oxygen, and the peak at 531.7 eV was assigned to oxygen defects or surface oxygen species (Oβ) with low coordination.38–40 However, with the addition of Al, the binding energy of Oα shifted to the lower value (529.4 eV) and the banding energy of Oβ showed the same trend. The decrease of Oα binding energy indicated that part of Al was doped into the lattice of CeO2 which resulted in the substitution of Al3+ ions for Ce4+ ions. Additionally, the value Oβ/Oα increased from 0.13 to 0.99 and more oxygen defect or the surface oxygen species formed on the surface of Al2O3–CeO2, which was favor to obtain a high activity and stability. Lee41 reported the addition of Al2O3 in CeO2 would lead to the formation of oxygen vacancies because of charge compensation. Mao et al.42 reported that the enhanced stability of the Sn–MnCeLa catalysts can be attributed to the fact that the addition of Sn could increase the concentration of surface adsorbed oxygen species for removal of the adsorbed Cl species and inhibit the formation of MnOxCly, avoiding the deactivation of the MnCeLa catalyst. Moreover, the addition of Al2O3 may induce a large distortion of the surrounding lattice because Al3+ ion had much smaller size compared with that of Ce4+ ion. Generally, the high concentration of Oβ in favor to the total oxidation of VOCs, thus, the AlCe50/50 catalyst showed a better catalytic activity for the catalytic combustion of DCE than pure CeO2 catalysts. After the enduring test (the final conversion was maintained at about 20–40%), it could be found that the Oβ/Oα ratio obviously decreased from 0.13 to 0.02 for CeO2-NC and from 0.99 to 0.76 for AlCe50/50 and this decline was closely related to the adsorption of inorganic chlorine species on the oxygen vacancies (active sites for oxygen activation). In our opinion, the depletion and non-replenishment of surface oxygen species was responsible for the reduction of catalytic performance, especially for the complete oxidation performance. However, the amount of the inorganic chlorine species on CeO2-NC and AlCe50/50 catalysts surface (Table 3) was almost same (9.4% and 10%, respectively), thus, it can be speculated that the better stable activity of AlCe50/50 catalyst may benefit from other factors. Additionally, as shown in Fig. 8c, the binding energy of Cl 2p on AlCe50/50 (197.8 eV) was obvious lower than that on CeO2-NC (198.6 eV), and the possible reason was that the Cl species mainly adsorbed on Al3+ for the AlCe50/50 catalyst. Furthermore, the transfer of the Cl species to Al3+ also improved the exposure of oxygen vacancies and Ce3+/4+ active sites, and then enhanced the resistant ability to Cl poisoning and stability for DCE catalytic oxidation.
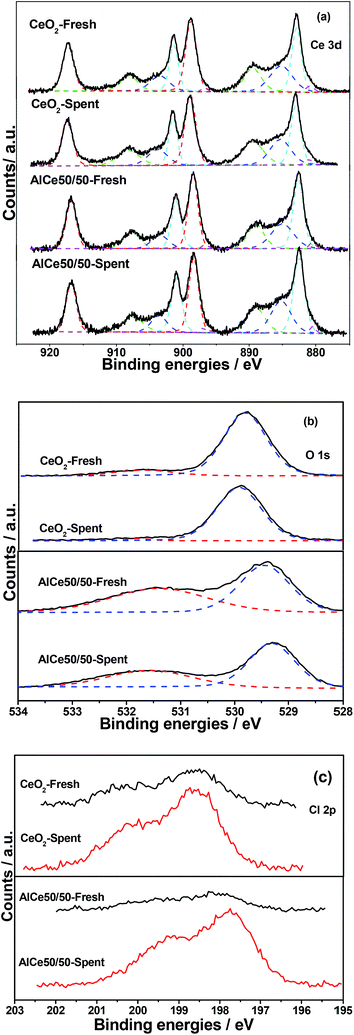 |
| | Fig. 8 XPS spectra of fresh and spent CeO2-NC and AlCe50/50-NC catalysts. | |
Table 3 XPS data of fresh and spent CeO2 and AlCe50/50 catalysts
| Sample |
Binding energy (eV) |
Oβ/Oα |
Ce3+ (at.%) |
Cl (at.%) |
| Olat. |
Osur. |
Cl |
| Cl content (at.%) measured by XPS. Cl content (at.%) measured by XRF. |
| Al2O3-NC |
531.2 |
— |
|
— |
— |
| Al2O3-NC-used |
531.1 |
198.9 |
— |
— |
1.45 |
| AlCe50/50-NC |
529.4 |
531.4 |
197.8 |
0.99 |
22.3 |
2a |
| AlCe50/50-NC-used |
529.3 |
531.6 |
199.2/197.7 |
0.76 |
22.1 |
10a/4.6b |
| CeO2-NC |
529.8 |
531.7 |
200.2/198.5 |
0.13 |
24.5 |
3.6a |
| CeO2-NC-used |
529.9 |
531.8 |
200.2/198.6 |
0.02 |
22.1 |
9.4a/4.2b |
3.7. In situ DRIFTS studies
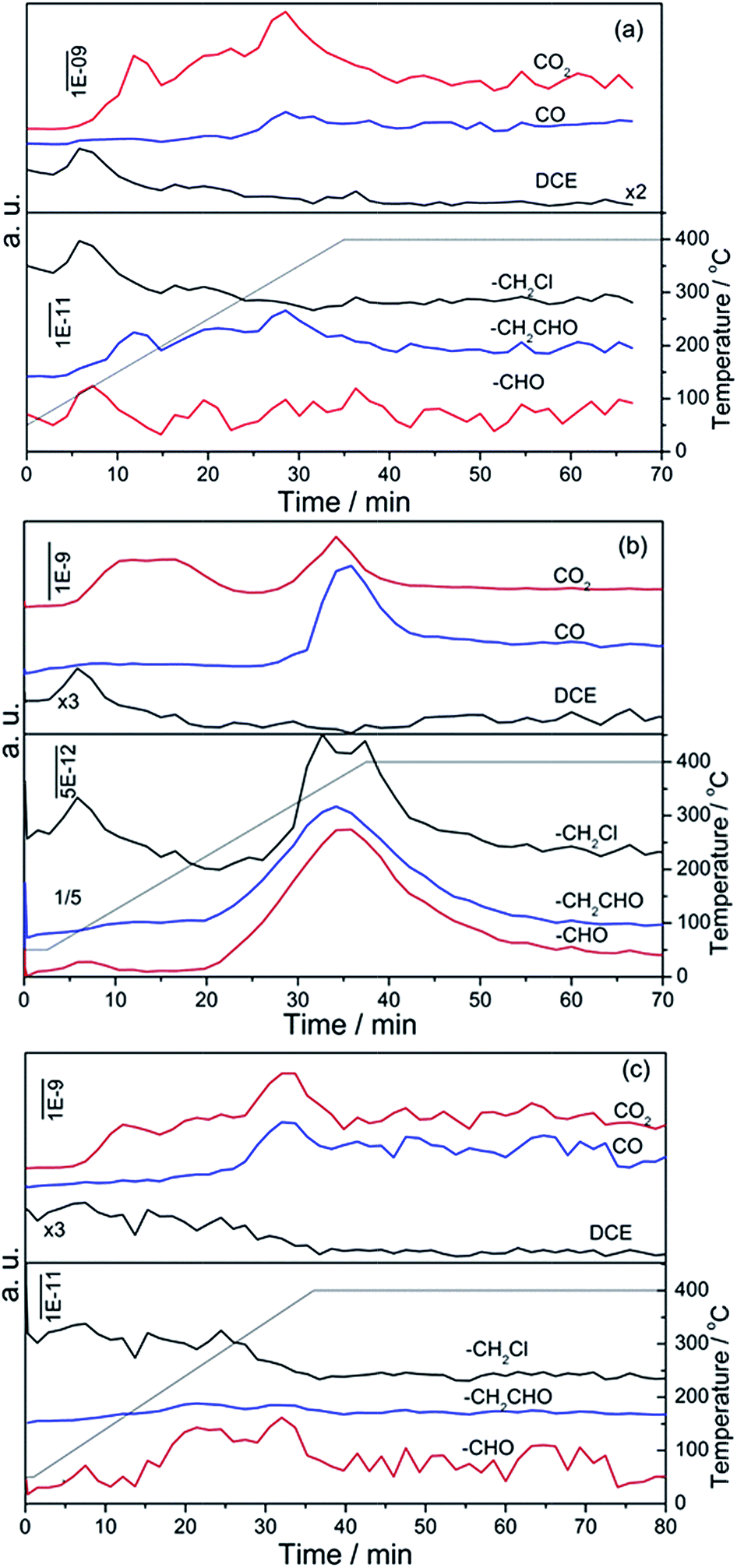
3.7.1 In situ DRIFTS of DCE adsorbed on CeO2 catalyst. Fig. 9a showed the DRIFTS of DCE adsorbed on pure CeO2 catalysts at different temperatures (from 50 to 400 °C) under O2/Ar atmosphere. In the range of OH group vibration, three negative bands in the subtraction spectra at around 3710, 3690 and 3660 cm−1, which should be assigned to the OH vibration mode of hydroxyl groups of CeO2, were observed even at 50 °C. The intensity of bands increased with the increasing of reaction temperature and presented a max value at 250 °C. The first band can be assigned to the isolated OH groups on CeO2 surface, and the latter corresponded to the bridged OH groups. Obviously, these sites were occupied by physisorption or chemisorption DCE. Moreover, some excepted bands at 1235, 1283, 1430 and 2960 cm−1 assigning to τCH2, ωCH2, δCH2, νsCH2 of DCE were observed at 50 °C, three new bands appear at 1115, 1189 and 2845 cm−1 (which was considered to be related with alcohol species), which suggested that chemisorption or dissociation of DCE on CeO2 surface had occurred. In order to further understand the adsorption behavior of DCE at 50 °C, Fig. 9b displayed the IR spectra of adsorbed DCE on CeO2 with the increase of the purging time at 50 °C. It can be found that the intensity of the peaks corresponding to DCE (such as 1235 and 2960 cm−1) decreased constantly, while the intensity of the peak corresponding to new species (such as 1115, 1189, 2845 and 2879 cm−1) enhanced with the prolonging of the purging time. Therefore, the dissociation of DCE on CeO2 could occur over at 50 °C, which was coherent with the high activity of CeO2 catalyst. When the temperature rose to 100 °C, the bands at 1115 and 1189 cm−1 assigning to alcohol species disappeared (Fig. 9a). To identify the relation of new bands and alcohol species, DRIFTS spectra of ethanol adsorbed on pure CeO2 catalysts were collected and presented in Fig. 3S.† Bands at 1118, 2840 and 2970 cm−1 were observed, which was ascribed to the νC–C–O, νsCH2 and νasCH3 of ethanol. Additionally, the adsorbed ethanol can be oxidized into aldehyde species even at 50 °C and the characteristic bands at 2935, 2853 and 2715 cm−1 were recorded. Therefore, the new bands observed for the adsorption of DCE on CeO2 at 50 °C could be certainly assigned to alcohol species, and these species were easy to be further dissociated or oxidized. Accompanied by the disappearance of alcohol species at 100 °C, the bands at 1577, 1545 and 1367 cm−1 assigning to carboxylate species were observed at 100 °C. George Socrates43 suggested that carboxylic acid salts had a very strong fingerprint band in the region 1695–1540 cm−1 due to the asymmetric stretching vibration of CO3−. The symmetric stretching vibration of this group gave rise to a band in the range 1440–1335 cm−1. Meanwhile, weak bands at 2715 and 2935 cm−1 assigning to aldehyde species were monitored, which was similar with the adsorption of ethanol on CeO2 catalyst (Fig. 4S†). With the reaction temperature was raised to 150 °C, the intensity of bands corresponded to aldehyde and carboxylate species significantly increased. Furthermore, in the range of 2000–2500 cm−1, four bands at 2096, 2132, 2265 and 2336 cm−1 appeared. The first two bands were assigned to the adsorption of CO,44,45 and the latter two can be ascribed to CO2 adsorbed on the surface of catalysts. The band appeared at about 1655 cm−1 could be ascribed to bicarbonate species, which came from the interaction between adsorbed CO2 and surface hydroxyl groups. Schubert46 observed the bicarbonate species (HCO3−, bands at 1658, 1435, 1304 and 1229 cm−1) during CO oxidation. However, the bicarbonate species were unstable, and the complete decomposition can occur at 200 °C. After the reaction temperature was higher than 200 °C, no new species generated and the carboxylic acid salts species disappeared at 300 °C. In summary, the formation of species on CeO2 surface and the transformation can be described in Fig. 10.
 |
| | Fig. 9 In situ DRIFTS of DCE adsorbed on pure CeO2 catalysts at different temperatures under O2/Ar atmosphere. | |
 |
| | Fig. 10 The formation and transformation of species on CeO2 surface during DCE catalytic oxidation. | |
3.7.2 In situ DRIFTS of DCE adsorbed on Al2O3 catalyst. Fig. 7S(a)† presented in situ DRIFTS spectra of DCE adsorbed on pure Al2O3 catalyst at different temperature. A negative peak at 3753 cm−1 was observed, which suggested that the surface OH groups were occupied by DCE physisorption or chemisorptions species. Additionally, the intensity of negative bands enhanced obviously with the increasing of reaction temperature in the range 50–300 °C and then decreased, which suggested that the adsorption of DCE on Al2O3 was enhanced with increasing temperature and then the occupied or consumed hydroxyl groups can be recovered partially after the oxidation of DCE was finished. However, except a very weak band at 2963 cm−1 (see the enlarged figure), attributed to νsCH2 of DCE, no bands associated with DCE or other new species were observed at 50 and 100 °C. Even if the reaction temperature was raised to 200 °C, any behavior of DCE adsorption had not been observed, but a new band at 1625 cm−1 appeared. Therefore, the adsorption of DCE on Al2O3 surface was difficult or weak, which verified the poor activity of Al2O3 for DCE catalytic combustion. In addition, the sharp bands located at 1625 cm−1 appeared, which should be related to VC and regarded as the adsorption of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond on Lewis acid sites of Al2O3 and the reasons were that: (1) activity tests showed that a large amount of VC generated on Al2O3 catalysts; (2) the adsorption experiment of VC (adsorption for 30 min at 50 °C, no sweeping) indicated that two bands at 1620 and 1610 cm−1 were observed (see Fig. 7S(b)†). After increasing the reaction temperature to 300 °C, new weak bands at 2285, 2340, 2394, 2720 and 2920 cm−1 were observed, the bands at 2285 and 2340 cm−1 were ascribed to CO2 adsorbed on Al2O3 surface and the bands at 2720 and 2920 cm−1 were assigned to aldehyde species. However, the assignment of the band at 2394 cm−1 was still ambiguous. According to Aines and Rossman's report, this band may be related to libration, sum and different motions of the CO2 molecule.47 Moreover, the intensity of band corresponding to VC increased obviously with the increasing of reaction temperature, which was consistent with the results of activity tests.
C double bond on Lewis acid sites of Al2O3 and the reasons were that: (1) activity tests showed that a large amount of VC generated on Al2O3 catalysts; (2) the adsorption experiment of VC (adsorption for 30 min at 50 °C, no sweeping) indicated that two bands at 1620 and 1610 cm−1 were observed (see Fig. 7S(b)†). After increasing the reaction temperature to 300 °C, new weak bands at 2285, 2340, 2394, 2720 and 2920 cm−1 were observed, the bands at 2285 and 2340 cm−1 were ascribed to CO2 adsorbed on Al2O3 surface and the bands at 2720 and 2920 cm−1 were assigned to aldehyde species. However, the assignment of the band at 2394 cm−1 was still ambiguous. According to Aines and Rossman's report, this band may be related to libration, sum and different motions of the CO2 molecule.47 Moreover, the intensity of band corresponding to VC increased obviously with the increasing of reaction temperature, which was consistent with the results of activity tests.
3.7.3 In situ DRIFTS of DCE adsorbed on CeO2–Al2O3 catalyst. In situ DRIFTS spectra of DCE adsorbed on AlCe50/50 catalyst were presented in Fig. 8S.† During the OH stretching frequency range, the spectra presented three obvious negative bands at 3760, 3690 and 3653 cm−1, which can be assigned to the consumption of OH group just like on Al2O3 and CeO2. Similar to pure CeO2, the formation of ethanol species was observed at 50 °C (2840 cm−1) accompanied with the adsorption of DCE (1236, 1286 and 2963 cm−1). After the reaction temperature was raised to 100 °C, bands ascribed to VC were observed, for examples, the bands at 1590–1630 cm−1 were assigned to the stretching of the CC double bond, the peak at 2910 cm−1 was attributed to the C–H stretching of –CH2, and the 1378, 1365 and 1345 cm−1 was assigned to methyl and methylenes. When the temperature further rose to 400 °C, the disappearance of these bands indicated that VC was almost completely desorbed or decomposed, and this phenomenon was highly consist with the results of activity tests (see Fig. 1S†). Aldehyde species, the characteristic bands of which appeared at 2933, 2855 and 2714 cm−1 were observed at 150 °C, and then followed by the formation of CO (2147 cm−1) and CO2 (2338 and 2392 cm−1) at 200 °C. Compared with pure CeO2 and Al2O3 catalysts, the adsorption behavior of DCE on AlCe50/50 catalyst was similar with pure CeO2, which suggested that the chemisorptions or dissociation of DCE mainly occurred on CeO2 active sites, however, the by-product VC was more inclined to adsorb on Lewis sites of Al2O3.To further investigate the role of acid sites in the reaction, inhibition of DCE decomposition on CeO2 and AlCe50/50 by using pre-adsorbed pyridine to block acid sites was shown in Fig. 6S.† The new bands at 1236 and 1284 cm−1 can be assigned to the DCE physical adsorption and these bands disappeared after sweeping 30 min at 50 °C. As the temperature increasing, the bands at 1220, 1440, 1594 cm−1 decreased and disappeared at 150 °C. At 300 °C, the bands at 1648 and 1622 cm−1 which can be ascribed to the HCO3− and VC species, which suggested that the acid site may be the active center for DCE elimination and dehydrochlorination reaction. Compared with the in situ DRIFTS result that the dissociation of DCE on CeO2 could occur over at 50 °C and 100 °C on CeO2 and AlCe50/50, respectively, only DCE physical adsorption was observed at 50 °C over CeO2 and AlCe50/50 pre-adsorbed pyridine. The conversion and in situ DRIFTS of VC were investigated over three catalysts, shown in Fig. S9,† and the result showed that the poor catalytic activity for vinyl chloride was due to the difficult adsorption/dissociation on catalysts surface.
3.8. TPSR
Fig. 11 described the decomposition of DCE over Al2O3-NC, CeO2-NC and AlCe50/50 catalysts under O2 (20 vol%)/Ar atmosphere using online MS to monitor possible intermediates and products. A desorption peak of DCE can be observed between 80 and 150 °C over Al2O3 and CeO2 catalysts, meanwhile, the decomposition of DCE was also observed above 180 °C (the formation of CO and CO2 was monitored by MS). Compared to desorption peak of DCE on Al2O3 catalyst, desorption on CeO2 catalyst was more obvious, however, appreciable desorption of DCE was not observed on AlCe50/50 catalyst. It can be found that the desorption behavior was consistent with catalytic activities, especially at lower temperature (Fig. 1S†). Moreover, the evolution of CO and CO2 was monitored over all catalysts, the peak temperature and the concentration of CO was as follows: CeO2 < AlCe50/50 < Al2O3, which was considered to be closely related to the oxidation performance of catalysts. The in situ DRIFTS experiments indicated that aldehyde species were observed over all catalysts, which could be the intermediates of DCE catalytic oxidation. Thus, the fragments with m/z = 29(–CHO, corresponding to ClCH2CHO or CH3CHO), 43(–CH2CHO, corresponding to ClCH2CHO) and 50(–CH2Cl, corresponding to DCE or ClCH2CHO) were expected to monitor during TPSR experiments with higher GHSV (30000 h−1, catalyst amount: 40 mg). These three fragments displayed apparent evolution peaks over Al2O3 catalyst in the range of 225 and 400 °C, which confirmed the formation of intermediate species ClCH2CHO. However, the evolution was negligible over CeO2 and AlCe50/50 catalysts. Therefore, in situ DRIFTS and TPSR experiments demonstrated that ClCH2CHO was the intermediate product of DCE catalytic oxidation, which can be further dissociated or oxidized into COx on CeO2. Sounak48 reported acetaldehyde was explained by cleavage of the α-CH bond on ethoxide. A concerted E2 pathway was identified as a low-energy pathway that can occur when ethanol was adsorbed on acidic oxides adjacent to a surface base oxygen and this pathway may be the source of the ethylene production from ethanol produced in this temperature range (E2 β-CH-scission).49 Combining the result of the oxygen concentration experiment, it can be assumed that acetaldehyde was produced by cleavage of the α-CH bond on ethoxide in the presence of enough oxygen, the acetate formation by α-CH scission of acetaldehyde and COx formation by decarboxylation of acetic acid, meanwhile, VC was generated by β-CH-scission with an adjacent surface base oxygen species (O2−) when oxygen was deficient.
 |
| | Fig. 11 TPSR profiles for DCE oxidation over CeO2-NC (a), Al2O3-NC (b) and AlCe50/50-NC (c) catalysts. DCE concentration: 1000 ppm; GHSV: 30000 h−1; catalyst amount: 40 mg (MS, DCE (m/z = 98), CO2 (44), CO (28), –CHO (29), –CH2CHO (43), –CH2Cl (50)). | |
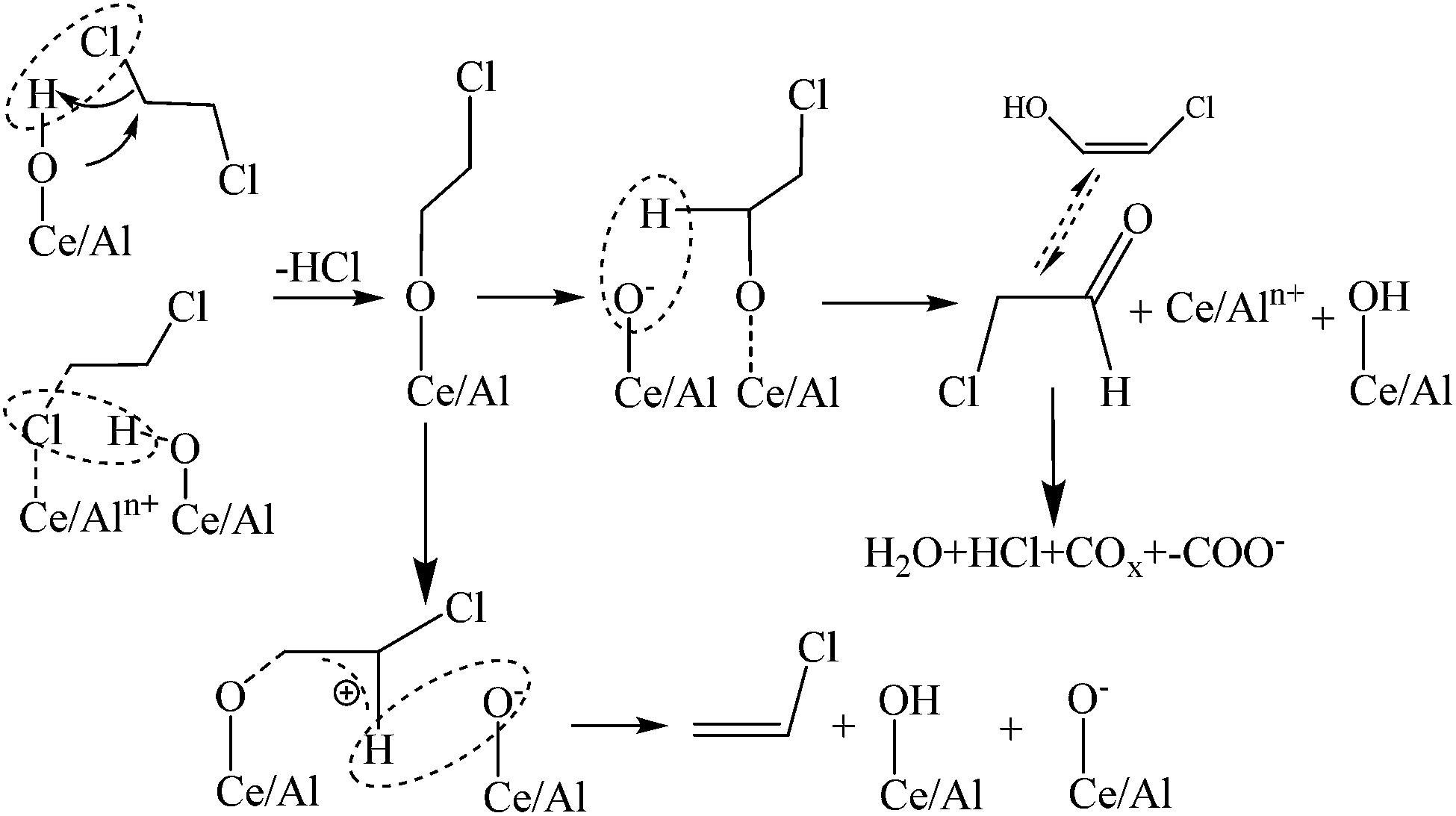
Based on the results of in situ DRIFTS, a simple reaction pathway of 1,2-dichloroethane oxidation over Al2O3–CeO2 catalysts was proposed and shown in Scheme 1. Firstly, a chlorine atom of 1,2-dichloroethane adsorbed on surface hydroxyl groups or Lewis acid sites (such as Al3+ or Ce3+/4+), and then a HCl molecular was abstracted directly (for the former) or via a reaction of the adsorbed Cl with another surface hydroxyl group (for the latter), meanwhile, the C–Cl bond was activated and dissociated. Afterwards, the dissociated DCE (Ce/Al–O–CH2–CH2Cl species) was attacked by the basic site (O2−), and this process included two pathways: (1) hydrogen of the activated –CH2– (Ce/Al–O–CH2–) was attacked and aldehyde species as intermediate species were formed; (2) hydrogen of the non-activated –CH2– (–CH2Cl) was nucleophilic attacked and then the by-product VC formed through an E2 elimination reaction. Finally, the aldehyde species were converted into H2O, COx and HCl via further Cl abstraction and oxidation, while most of VC as by-product removed from the surface of catalysts for its adsorption and dissociation were difficult.
 |
| | Scheme 1 The proposed reaction pathway of 1,2-dichloroethane oxidation over Al2O3–CeO2 catalysts. | |
4. Conclusions
The low temperature catalytic combustion of dichloroethane over Al2O3, CeO2 and Al2O3–CeO2 catalysts was investigated. The result of O2-TPO experiments after the stability tests showed that the deactivation of pure Al2O3 was attributed to the blocking of the active sites originated from the accumulation of heavy products or coke formation on the surface the deactivation, while the deactivation of pure CeO2 was ascribed to the strong adsorption of Cl species produced during DCE oxidation. Then the effects of preparation methods, Ce content, inlet oxygen concentration and water vapor were studied, and the AlCe50/50-NC catalyst presented a better catalytic performance due to the improvement of the redox and acid properties. The acid strength played more important role in the DCE catalytic elimination than the amount of acid and the transfer of the Cl species to Al3+ also improved the exposure of oxygen vacancies and Ce3+/4+ active sites, and enhanced the tolerant ability for Cl poisoning and stability for DCE catalyst oxidation. Therefore, the synergistic effects of acid and redox properties improved the DCE catalytic elimination process. According to the adsorption–desorption and reaction behavior of DCE, VC and ethanol on these catalysts were determined by in situ DRIFTS and TPSR experiments, a simple reaction pathway was proposed.
Acknowledgements
This research was supported by National Natural Science Foundation of China (nos 21307033, 21277047), Shanghai Natural Science Foundation (no. 13ZR1411000).
Notes and references
- M. M. R. Feijen-Jeurissen, J. J. Jorna, B. E. Nieuwenhuys, S. Gilles, P. Corinne and J. P. Hindermann, Catal. Today, 1999, 54, 65–79 CrossRef CAS.
- J. R. Gonzalez-Velasco, R. López-Fonseca, A. Aranzabal, J. I. Gutiérrez-Ortiz and P. Steltenpohl, Appl. Catal., B, 2000, 24, 233–242 CrossRef CAS.
- D. C. Kim and S. K. Ihm, Environ. Sci. Technol., 2001, 35, 222–226 CrossRef CAS.
- A. Aranzabal, J. A. González-Marcos, J. L. Ayastuy and J. R. Gonzalez-Velasco, Chem. Eng. Sci., 2006, 61, 3564–3576 CrossRef CAS PubMed.
- K. S. Go, Y. Kim, S. R. Son and S. D. Kim, Chem. Eng. Sci., 2010, 65, 499–503 CrossRef CAS PubMed.
- I. Seiichiro, T. Kenji, Y. Tomoyuki and U. Kazunori, J. Jpn. Pet. Inst., 2003, 46(2), 138–141 CrossRef.
- J. I. Gutierrez-Ortiz, B. de Rivas, R. Lopez-Fonseca and J. R. Gonzalez-Velasco, Appl. Catal., B, 2006, 65, 191–200 CrossRef CAS PubMed.
- B. de Rivas, J. I. Gutierrez-Ortiz, R. Lopez-Fonseca and J. R. Gonzalez-Velasco, Appl. Catal., A, 2006, 314, 54–63 CrossRef CAS PubMed.
- B. de Rivas, R. Lopez-Fonseca, J. R. Gonzalez-Velasco and J. I. Gutierrez-Ortiz, J. Mol. Catal. A: Chem., 2007, 278, 181–188 CrossRef CAS PubMed.
- B. de Rivas, R. Lopez-Fonseca, M. A. Gutierrez-Ortiz and J. I. Gutierrez-Ortiz, Appl. Catal., B, 2011, 104, 373–381 CrossRef CAS PubMed.
- B. de Rivas, R. Lopez-Fonseca, M. A. Gutierrez-Ortiz and J. I. Gutierrez-Ortiz, Waste Management and the Environment IV, 2008, 857–866 CAS.
- B. de Rivas, R. Lopez-Fonseca, M. A. Gutierrez-Ortiz and J. I. Gutierrez-Ortiz, Appl. Catal., B, 2011, 101, 317–325 CrossRef CAS PubMed.
- J. I. Gutierrez-Ortiz, B. de Rivas, R. Lopez-Fonseca and J. R. Gonzalez-Velasco, Appl. Catal., A, 2004, 269, 147–155 CrossRef CAS PubMed.
- A. Aranzabal, J. A. Gonzalez-Marcos, M. Romero-Saez, J. R. Gonzalez-Velasco, M. Guillemot and P. Magnoux, Appl. Catal., B, 2009, 88, 533–541 CrossRef CAS PubMed.
- I. Seiichiro, T. Hiroyuki and I. Shingo, Ind. Eng. Chem. Res., 1989, 28, 1449–1452 CrossRef.
- M. Paulis, L. M. Gandia, A. Gil, J. Sambeth, J. A. Odriozola and M. Montes, Appl. Catal., B, 2000, 26, 37–46 CrossRef CAS.
- W. B. Li, J. X. Wang and H. Gong, Catal. Today, 2009, 148, 81–87 CrossRef CAS PubMed.
- R. Lopez-Fonseca, B. de Rivas, J. I. Gutierrez-Ortiz, A. Aranzabal and J. R. Gonzalez-Velasco, Appl. Catal., B, 2003, 41, 31–42 CrossRef CAS.
- R. Lopez-Fonseca, J. I. Gutierrez-Ortiz, J. L. Ayastui, A. M. Gutierrez-Ortiz and J. R. Gonzalez- Velasco, Appl. Catal., B, 2003, 45, 13–21 CrossRef CAS.
- R. Lopez-Fonseca, J. I. Gutierrez-Ortiz and J. R. Gonzalez-Velasco, Catal. Commun., 2004, 5, 391–396 CrossRef CAS PubMed.
- R. Lopez-Fonseca, A. Aranzabal, P. Steltenpohl, J. I. Gutierrez-Ortiz and J. R. Gonzalez-Velasco, Catal. Today, 2000, 62, 367–377 CrossRef CAS.
- A. V. Kucherov, I. M. Sinev, S. Ojala, R. Keiski and L. M. Kustov, Stud. Surf. Sci. Catal., 2007, 170, 1129–1136 Search PubMed.
- B. de Rivas, C. Sampedro, R. Lopez-Fonseca, M. A. Gutierrez-Ortiz and J. I. Gutierrez-Ortiz, Appl. Catal., A, 2012, 417, 93–101 CrossRef PubMed.
- P. Yang, X. M. Xue, Zh. H. Meng and R. X. Zhou, Chem. Eng. J., 2013, 234, 203–210 CrossRef CAS PubMed.
- M. A. Peluso, E. Pronsato, J. Sambeth, E. Thomas, J. Horacio and G. Busca, Appl. Catal., B, 2008, 78, 73–79 CrossRef CAS PubMed.
- Q. G. Dai, H. Huang, Y. Zhu, W. Deng, S. X. Bai, X. Y. Wang and G. Z. Lu, Appl. Catal., B, 2012, 117, 360–368 CrossRef PubMed.
- N. Y. Topsøe, K. Pedersen and E. G. Derouane, J. Catal., 1981, 70, 41–52 CrossRef.
- V. H. Carmela, I. Hirofumi, H. Tadashi, N. Miki and M. Yuichi, J. Catal., 1984, 85, 362–369 CrossRef.
- B. de Rivas, R. Lopez-Fonseca, C. Sampedro and J. I. Gutierez-Ortiz, Appl. Catal., B, 2009, 90, 545–555 CrossRef CAS PubMed.
- B. Van den, R. W. Krzan, M. M. R. Feijen-Jeurissen, R. Louw and P. Mulder, Appl. Catal., B, 2000, 24, 255–264 CrossRef.
- K. Ramanathan and J. J. Spivey, Combust. Sci. Technol., 1989, 63, 247–255 CrossRef CAS PubMed.
- I. Mochida, A. Uchino, H. Fujitsu and K. Takeshita, J. Catal., 1978, 51, 72–79 CrossRef CAS.
- Q. G. Dai, S. X. Bai, J. W. Wang, M. Li, X. Y. Wang and G. Z. Lu, Appl. Catal., B, 2013, 142–143, 222–233 CrossRef CAS PubMed.
- X. Ge, S. H. Hu, Q. Sun and J. Y. Shen, J. Nat. Gas Chem., 2003, 12, 119–122 CAS.
- S. Klaus-Dieter, Surf. Sci., 1998, 399, 29–38 CrossRef.
- H. Huang, Q. G. Dai and X. Y. Wang, Appl. Catal., B, 2014, 158–159, 96–105 CrossRef CAS PubMed.
- E. Friedrich, F. Stefano, L. Zhou, M. Tiziano, A. Cristina, F. Paolo, C. Giovanni and R. Renzo, Science, 2005, 309, 752–755 CrossRef PubMed.
- X. H. Guo, C. C. Mao, J. Zhang, J. Huang, W. N. Wang, Y. H. Deng, Y. Y. Wang, Y. Cao, W. X. Huang and S. H. Yu, Small, 2012, 8, 1515–1520 CrossRef CAS PubMed.
- J. Qi, J. Chen, G. D. Li, S. X. Li, Y. Gao and Z. Y. Tang, Energy Environ. Sci., 2012, 5, 8937–8941 CAS.
- H. C. Yao and Y. F. Yao, J. Catal., 1984, 86, 254–265 CrossRef CAS.
- J. S. Lee, K. H. Choi, B. K. Ryu, B. C. Shin and I. S. Kim, Ceram. Int., 2004, 30, 807–812 CrossRef CAS PubMed.
- D. Mao, F. He, P. Zhao and S. Liu, RSC Adv., 2015, 5, 10040–10047 RSC.
- S. George, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, Wiley, 3rd edn, 2004 Search PubMed.
- T. Jin, Y. Zhou, G. J. Mains and J. M. White, J. Phys. Chem., 1987, 91, 5931–5937 CrossRef CAS.
- X. Y. Liu, A. Q. Wang, L. Li, T. Zhang, C. Y. Mou and J. F. Lee, J. Catal., 2011, 278, 288–296 CrossRef CAS PubMed.
- M. M. Schubert, H. A. Gasteiger and R. J. Behm, J. Catal., 1997, 172, 256–258 CrossRef CAS.
- R. D. Aines and G. R. Rossman, Am. Mineral., 1984, 69, 319–327 CAS.
- R. Sounak, M. Giannis, D. Y. Hong, D. G. Vlachos, A. Bhan and R. J. Gorte, ACS Catal., 2012, 2, 1846–1853 CrossRef.
- M. J. Li, Z. L. Wu and S. H. Overbury, J. Catal., 2013, 306, 164–176 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra07405c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.