
Stereocomplex crystallite network in poly(D,L-lactide): formation, structure and the effect on shape memory behaviors and enzymatic hydrolysis of poly(D,L-lactide)
Yi Liab,
Shuangyang Xina,
Yijie Biana,
Qinglin Donga,
Changyu Han*a,
Kun Xua and
Lisong Donga
aKey Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China. E-mail: cyhan@ciac.ac.cn; Tel: +86-431-85262244
bCollege of Material Science and Engineering, Jilin Jianzhu University, Changchun 130118, China
First published on 2nd March 2015
Abstract
Stereocomplex crystallization is a very interesting crystal modification formed between enantiomeric polymers, such as poly(L-lactic acid) (PLLA) and poly(D-lactic acid) (PDLA). Herein, biodegradable poly(D,L-lactide) (PDLLA) and stereocomplex-poly(L- and D-lactide) (sc-PLA) blends were prepared by solution blending at various sc-PLA loadings ranging from 2.5 to 10 wt%. Wide-angle X-ray diffraction and differential scanning calorimetry results verified that complete stereocomplex crystallites without any evidence of the formation of homocrystallites in the PDLLA could be achieved. By a rheological approach, a transition from the liquid-like to solid-like viscoelastic behaviour was observed for the stereocomplex crystallites reserved PDLLA melt, and a frequency-independent loss tangent at low frequencies appeared at a sc-PLA concentration of 5 wt%, revealing the formation of stereocomplex crystallite network structure. By a delicately designed dissolution experiment, the structure of the formed network structure was explored. The results indicated that the network structure were not formed by stereocomplex crystallites connected directly with each other or by bridging molecules, but by the interparticle PDLLA chains which were significantly restrained by the crosslinking effect of sc-PLA. Accordingly, the mechanical properties of PDLLA were greatly enhanced after blending with sc-PLA. Moreover, the most intriguing result was that the shape memory behaviors of PDLLA had been improved obviously in the blends than in neat PDLLA, especially when a percolation network structure had formed, which may be of great use and importance for the wider practical application of PDLLA. Finally, it was found that the enzymatic hydrolytic degradation rates had been retarded in the blends than in neat PDLLA. The erosion mechanism of neat PDLLA and the blends was further discussed.
Introduction
Poly(lactic acid) (PLA) bioplastic has attracted much attention because it is biodegradable, biocompatible, producible from renewable resources, and nontoxic to the human body and the environment.1 Recent innovation on the production process has lowered significantly the production cost, which further stimulates the investigation on its properties and potential applications.2 Due to the presence of a chiral carbon in the skeletal chain, two enantiomers of PLA, namely poly(L-lactide) (PLLA) and poly(D-lactide) (PDLA), have been synthesized. The polymerization of racemic lactide (or lactic acid) or meso lactide results in the formation of atactic poly(D,L-lactide) (PDLLA), which is an amorphous polymer.1 The resulting amorphous material has more desirable mechanical characteristics and is more easily degraded than PLLA or PDLA, allowing its use in resorbable plating, artificial cartilage or bone, low strength scaffolding material for tissue regeneration, chemotherapeutics implants, and shape memory polymer in minimally invasive surgery applications,3–5 but its physical and mechanical properties were still needed to be further improved to meet wide applications.6 Consequently, the incorporation of bioactive ceramics (e.g. hydroxyapatite) or other biodegradable polymers into PDLLA matrix was an available method to further improve its physical properties.7–10 For examples, the PDLLA/hydroxyapatite hybrid nanocomposites with various hydroxyapatite proportions were prepared via solution blending or in situ grafting polymerization.7,8 It was found that the nanocomposites exhibit excellent shape memory performances with incorporation of hydroxyapatite. Drzal et al. prepared cellulose-nanofiber-reinforced (CNF) PDLLA composites prepared by a water-based approach.9 Increases in the modulus and strength (up to 58% and 210%, respectively) demonstrated the load-bearing capability of the CNF network in the PDLLA matrix. Moreover, porous scaffolds for tissue engineering applications based on PDLLA/poly(ε-caprolactone) blends has been described.10 The efficiency of scaffolds obtained with PDLLA based blends containing 30% by weight of poly(ε-caprolactone) as dispersed phase toward hepatocytes was tested by several biological assays and the authors found that they were able to promote a perfect adhesion, proliferation and growth of cells.Usually, PLA homopolymer (PLLA or PDLA) is a semi-crystalline polymer, which can form three kinds of crystal modifications (α, β and γ) depending on different crystallization processes.11–13 Recently, the existence of another crystal modification, the α′ form or disorder α on the basis of X-ray powder diffraction and infrared spectroscopic study has also been proposed.14,15 The stereocomplex of PLLA and PDLA (sc-PLA) is another important crystal modification of PLA. Since the pioneer works of Ikada et al.,16 PLLA and its enantiomeric opposite, PDLA, have been known to form specific stereocomplexes upon mixing in solution or in bulk conditions.17 More recently, Yang and coworkers succeeded in preparing stereocomplex crystallites with high crystallinity and melting temperature by a novel and interesting low temperature approach using high-molecular-weight PLA that was difficult to achieve using conventional melt blending and solution casting methods.18,19 The results demonstrated that exclusive stereocomplex crystallites without homocrystallites of high-molecular-weight PLA could be formed at processing temperature as low as 160 °C, either at equimolar PLLA/PDLA blends or non-equimolar PLLA/PDLA (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40) blends. Stereocomplex formation can be easily characterized by differential scanning calorimetry (DSC) and wide-angle X-ray scattering (WAXS). In particular, the stereocrystal melting point is in the range of 220–250 °C, higher by approximate 30–60 °C than that of the original parent polymer components.16 Their fast crystallization from the melt state, their nucleating effect in neat PLLA or PDLA,20 their better mechanical performance, their superior heat resistance properties and their lower thermal and hydrolytic degradation rates are the most fascinating properties that clearly highlight the high potential of PLA stereocomplexes to develop novel semi-crystalline PLA materials with improved performances for long-lasting applications.21,22 Moreover, stereocomplexed hydrogels containing star block copolymers of eight-arm poly(ethylene glycol)-poly(L-lactide) and poly(ethylene glycol)-poly(D-lactide) were also synthesized.23 These stereocomplexed hydrogels with high storage moduli (up to 14 kPa) were promising for use in biomedical applications, including drug delivery and tissue engineering, because they were biodegradable and the in situ formation allowed for easy immobilization of drugs and cells.
:40) blends. Stereocomplex formation can be easily characterized by differential scanning calorimetry (DSC) and wide-angle X-ray scattering (WAXS). In particular, the stereocrystal melting point is in the range of 220–250 °C, higher by approximate 30–60 °C than that of the original parent polymer components.16 Their fast crystallization from the melt state, their nucleating effect in neat PLLA or PDLA,20 their better mechanical performance, their superior heat resistance properties and their lower thermal and hydrolytic degradation rates are the most fascinating properties that clearly highlight the high potential of PLA stereocomplexes to develop novel semi-crystalline PLA materials with improved performances for long-lasting applications.21,22 Moreover, stereocomplexed hydrogels containing star block copolymers of eight-arm poly(ethylene glycol)-poly(L-lactide) and poly(ethylene glycol)-poly(D-lactide) were also synthesized.23 These stereocomplexed hydrogels with high storage moduli (up to 14 kPa) were promising for use in biomedical applications, including drug delivery and tissue engineering, because they were biodegradable and the in situ formation allowed for easy immobilization of drugs and cells.
Herein, we presented a simple but novel method to enhance rheological, mechanical properties and shape memory properties of PDLLA through solution blending with sc-PLA. Our results demonstrated that the stereocomplex crystals could be formed in the blends of PDLLA with equimolar PLLA and PDLA, as investigated by WAXD and DSC. By a delicately designed dissolution experiment, the structure of the prepared blends was tried to explore. We emphasized the effect of the stereocomplex crystals on the rheological, mechanical properties, shape memory properties and enzymatic hydrolysis of PDLLA. To the best of our knowledge, this is the first report on improving the physical properties of PDLLA through blending with sc-PLA. The results would be interesting to the polymer materials community which is driving to develop new sustainable blend materials and technology completely derived from renewable resources and degraded to benign byproducts at the end of their useful lifetime.
Experimental
Materials and samples preparation
PDLLA was purchased from Aldrich. It exhibited a weight-average molecular weight (Mw) of 8.5 × 104 g mol−1, polydispersity of 1.6 (GPC analysis). PLLA (4032D) was a product of NatureWorks LLC. It exhibited a Mw of 2.1 × 105 g mol−1, polydispersity of 1.7. D-Isomer content of PLLA was approximately 2.0%. PDLA was supplied by Zhejiang Hisun Biomaterials Co. Ltd. (Taizhou, China). It exhibited a Mw of 1.8 × 105 g mol−1, polydispersity of 1.8.Ternary blends comprising PLLA, PDLA, and PDLLA were prepared by solution casting using chloroform as a common solvent. In ternary blends, ratios of PLLA/PDLA in blends were fixed at 1/1, with the PDLLA contents ranging from 90/10, 92.5/7.5, 95/5 to 97.5/2.5 in mass ratios (the first number referring to the mass percentage of PDLLA). The prepared solutions were cast onto Petri dishes placed horizontally, and then the solvent was allowed to evaporate at room temperature for 12 h. The samples were further dried at 80 °C under vacuum for 7 days to remove the solvent completely. For comparison, neat PDLLA was treated using the same procedure. Then all the samples were hot-pressed at 130 °C for 2 min followed by cold press at room temperature to form the sheets with various thicknesses for characterization. For convenience, the samples with 2.5 wt%, 5 wt%, 7.5 wt% and 10 wt% sc-PLA in the blends were denoted as PDLLA2.5, PDLLA5, PDLLA7.5 and PDLLA10, respectively.
Characterizations
Thermal analysis was performed using a TA Instruments differential scanning calorimeter (DSC) Q20 with a Universal Analysis 2000. Samples weights were in the range of 5–8 mg. The heat enthalpy and temperature of DSC were calibrated with standard indium. All specimens were heated from −60 °C to 250 °C at a heating rate of 20 °C min−1 under nitrogen purge. It was worth noting that all recorded melt enthalpies of sc-PLA were normalized according to the amount of sc-PLA within the blends.Wide-angle X-ray diffraction (WAXD) experiments were performed on a D8 advance X-ray diffraction meter (Bruker, Germany) in the range of 5–40° with a scanning rate of 3° min−1. The Cu Kα radiation (λ = 0.15418 nm) source was operated at 40 kV and 200 mA.
Rheological measurements were carried out on a rheometer (AR2000EX, TA Instruments-Waters LLC, USA) equipped with a parallel plate geometry using 25 mm diameter plates. The sheet samples in thickness of 1.0 mm were molten at 130 °C under N2 atmosphere in the fixture and experienced dynamic frequency sweep. The oscillatory frequency swept ranging from 100 to 0.01, with a fixed strain of 0.5%.
The hydro dynamic diameter (Dh) of the sc-PLA in the blends was determined using a Malvern Zetasizer Nano ZS90 with a He–Ne laser (633 nm) and 90° collecting optics. The solutions of the blends for light scattering were prepared with acetone. Before testing, the samples were placed at room temperature for 48 hours. All measurements were performed at a sample concentration of 0.25 g L−1.
Dynamic mechanical analysis (DMA) was performed on the samples of 20.0 × 4.0 × 1.0 mm3 in size using a dynamic mechanical analyzer from Rheometric Scientific under tension mode in a temperature range of 0 to 100 °C at a frequency of 1 Hz and 3 °C min−1.
The enzymatic hydrolysis of PDLLA and the blend films was carried out in phosphate buffer (pH 7.4) containing proteinase K at 37 °C with shaking at 140 rpm. As already found, proteinase K can catalyze the hydrolytic degradation of L-lactyl chains in amorphous regions.24,25 The tie chains and chains with along free end in amorphous regions can be enzymatically cleaved, whereas the folding chains and the chains with a short free end are highly resistant to enzymatic cleavage.26 Therefore, the enzymatic hydrolysis reaction was occurred mainly in the amorphous PDLLA in the blends. Sample films (10 × 10 × 0.25 mm3) were placed in small glass bottles filled with phosphate buffer containing approximate 2.0 μg mL−1 of proteinase K. After the reaction was allowed to continue for a period of time, the films were removed, washed with distilled water, and dried to constant weight in vacuum before weight analysis. For each sample, three films were used and the average value of their weight loss was reported. The weight of released sc-PLA was excluded from the weight loss of PDLLA in evaluating hydrolysis. Control tests which were carried out for all samples in buffer solution free from the proteinase K, showed no appreciable weight losses over the time scale of the experiments.
Results and discussion
Stereocomplex formation
Formation of the stereocomplex crystals in the blends was characterized by WAXD and DSC. The WAXD profiles of the blends containing various content of sc-PLA are showed in Fig. 1. All WAXD patterns did not exhibit diffraction peaks of PLLA or PDLA homocrystallites at 2θ values of 16°, 18.4°, and 21.8°,11 indicating no homocrystallites existed, and neat PDLLA was amorphous. The most intense peaks of the blended samples were observed at 2θ values of 12, 21, and 24° attributed to the spacing d110, d300 and/or d030 and d006 of stereocomplex crystals.17 These peaks were for the stereocomplex crystallized in a pseudo-trigonal unit cell of dimensions: a = 0.916 nm, b = 0.916 nm, c = 0.870 nm, α = 109.2°, β = 109.2°, and γ = 109.8°, in which L-lactide and D-lactide segments were packed parallel taking 31 helical conformations.17 These peaks became stronger when the contents of PLLA and PDLA increased. | ||
| Fig. 1 WAXD profiles of the blends containing various content of sc-PLA. | ||
The melting peaks of sc-PLA crystallites at around 218 °C could be observed in Fig. 2 for the blends; the area of the melting endotherm for the stereocomplex increased as the level of PLLA and PDLA in the blend increased. It was in agreement with the results of WAXD. Moreover, the values of degree of crystallinity (Xc) were estimated from the relation Xc = ΔHc/ΔHom × 100%, where ΔHom = 155 J g−1 was the melting enthalpy of 100% sc-crystals.27 Thus, calculated Xcs for sc-PLA around 38.1–45.1% were obtained for the blends. The Xc depended on the optical purity and the molecular weight of PLLA and PDLA. 38.1–45.1% is a relatively high stereocomplex formation efficiency compared with the reported results.17–19 These results clearly indicated that sc-PLA had a relatively high stereocomplex formation in PDLLA. Besides the melting of sc-PLA crystallites, the glass transition temperature (Tg) could also be observed for PDLLA and the blends during the heating scans. The Tg values of neat PDLLA was around 55 °C, which was higher to the values reported previously.3 The difference between these values could be attributed to the different molecular weights and heating rate during DSC characterization. Moreover, it was seen that there existed only one Tg for each blend within the composition range, and the Tg for the blend shifted to high temperature. This result suggested the miscibility of the components in the blends, which was in agreement with previous reports that the blends of sc-PLA or PLLA with PDLLA were miscible at all compositions.11,28,29
 | ||
| Fig. 2 DSC melting profiles of the samples. | ||
Rheological behaviors and stereocomplex crystallite structure in the blends
From the results of WAXD and DSC aforementioned, it was confirmed that the stereocomplex crystals could be formed in the blends. To explore the effect of formed stereocomplex crystallites on the melt rheological properties of the blends, oscillatory shear rheological measurements were carried out at 130 °C. Here, the melting point of stereocomplex crystallites was around 218 °C (as shown in Fig. 2); thus, at 130 °C only PDLLA were melted, and stereocomplex crystallites were reserved in the melt of the blends. Fig. 3 and 4 shows the frequency dependences of storage modulus (G′), loss modulus (G′′), loss tangent (tanδ), and complex viscosity (|η*|) for neat PDLLA and its blends with different concentrations of sc-PLA.
 | ||
| Fig. 3 Variation of (a) storage modulus (G′) and (b) loss modulus (G′′), as functions of frequency for the blends with different sc-PLA concentrations. | ||
 | ||
| Fig. 4 Variation of (a) loss tangent (tanδ) and (b) complex viscosity (|η*|), as functions of frequency for the blends with different sc-PLA concentrations. | ||
In Fig. 3(a) and (b), at low frequencies, G′ and G′′ increased with increasing sc-PLA content, and the change of G′ was more significant than that of G′′. Neat PDLLA chains relaxed fully and exhibited the typical terminal behavior with the scaling law of approximate G′ ∝ ω2 and G′′ ∝ ω1. With increasing sc-PLA content, the slopes of the modulus curves of the blends at low frequencies decreased. The more obvious nonterminal behavior for the blends than that of neat PDLLA indicated that a slower relaxation behavior in the melt of the blends, which could be attributed to the formed stereocomplex crystallites that restrained the long-range motions of polymer chains.30,31
The loss tangent (tanδ = G′′/G′) is an important parameter characterizing relaxation behavior of the viscoelastic materials and is regarded more sensitive to the relaxation changes than G′ and G′′.32 It can be seen in Fig. 4(a) that tanδ of neat PDLLA decreased with increasing frequency, which was a typical behavior for viscoelastic liquid. With increasing sc-PLA content, tanδ decreased gradually, reflecting that the elastic response of the melt became more significant when the formed stereocomplex crystallites increased. When sc-PLA concentration reached 5 wt%, a frequency-independent loss tangent appeared for sample PDLLA5 at low frequencies, while when PDLLA concentration was above 5 wt%, a tanδ peak, an indicator of a dominant elastic response of the melt, could be observed.33 These results obviously indicated that a network structure had been formed in the melt of the blends with increasing content of the reserved stereocomplex crystallites. According to the approach proposed by Winter et al.,34 it was clear that a critical physical gel was formed at a sc-PLA concentration of 5 wt%. The transition from the liquid-like to solid-like viscoelastic behaviors at low frequencies also demonstrated that the long-range polymer chains motion was restrained by the formed stereocomplex crystallite network structure significantly.20
The complex viscosity (|η*|) was also used to reveal the effect of the reserved stereocomplex crystallites on the melt rheological behaviors. It can be seen from Fig. 4(b) that the typical Newtonian plateau at low frequencies was observed for neat PDLLA and the blends with a sc-PLA concentration lower than 5 wt%. However, for the blends with higher concentration of sc-PLA, the Newtonian plateau disappeared and shear thinning could be observed at low frequencies. Furthermore, for sample PDLLA2.5, the viscosity increased slightly compared with neat PDLLA, but for sample PDLLA5 a sharp increase of viscosity could be observed. These results indicated that the formed network structure significantly reinforced the melt.
It is necessary to discuss how the stereocomplex crystallites affect the melt rheological behaviors of PDLLA. For PDLLA melt embedded with stereocomplex crystallites, stereocomplex crystallites have two distinct effects on the rheological responses of the melt. First, stereocomplex crystallites with high modulus can act as dispersed solid particles in the melt and reinforce the melt, referred to as filler effect here. Second, stereocomplex crystallites formed from the co-crystallization of PLLA and PDLA chains may act as physical crosslinking points of PDLLA chains.20 Due to the miscibility between PDLLA and sc-PLA, the PDLLA chains may be trapped in the phase of sc-PLA, resulting in an increase in the apparent molecular weight of PDLLA.35 This is referred to as the crosslinking effect here. As solid fillers, stereocomplex crystallites can reinforce the melt and increase the elastic response of the melt as aforementioned. As a result of the physical crosslinking effect of stereocomplex crystallites, a slower relaxation structure is introduced into the melt and can also enhance the melt rheology. The enhancement in rheology has been reflected obviously by the increase of G′ and |η*| and the decrease of tanδ. Besides these two effects, the contribution of the three-dimensional network structure, formed in the melt when the sc-PLA concentration reaches 5 wt%, should be addressed. The formation of such a network structure can enlarge sharply these reinforcing effects, resulting in the transition from the liquid-like to solid-like viscoelastic behaviors.20
Structure of the formed network structure
As discussed above, there exists a network structure in the PDLLA melt embedded with stereocomplex crystallites. However, its structure is still unknown. Similar to the structures of the gel formed during polymer crystallization process proposed by Horst and Winter,34,36 there may be three probabilities existed for the structure of the formed network structure here: (i) immediate contact between stereocomplex crystallite units, (ii) a network structure of bridging PDLLA molecules which have segments in neighboring phase of the sc-PLA, or (iii) impingement of amorphous PDLLA chains, immobilized by their segment attachment within the sc-PLA phase, with similarly immobilized chains from adjacent structures. To give a clearer illustration of the probable structures of the formed network structure, a schematic diagram was presented in Fig. 5. The structures of A–C, D, and E stand for the probabilities of (iii), (ii), and (i), respectively. As is shown, with the structure evolution from A to E the distance between neighboring sc-PLA should decrease. The possibilities of (i) and (ii) have been ruled out by Coppola and co-workers by the proof that a relatively low crystallinity, of the order of 1%, was obtained at the gel transition.36 | ||
| Fig. 5 Schematic diagram for the probable structures of the formed sc-PLA network structure. | ||
Here, a dissolution experiment was delicately designed to reveal the structure of the formed network structure in PDLLA. It is based on the fact that sc-PLA cannot be dissolved in acetone while acetone is a good solvent for PDLLA.1 For the structures of D and E, neighboring sc-PLAs are immediately connected by each other and by the bridging molecules, respectively. So, after the PDLLA chains are dissolved in acetone, sc-PLAs are not dissolved and still connected by each other by these bridging molecules which have segments in neighboring sc-PLAs for the cases of D and E. While for the cases of A–C, neighboring sc-PLAs are connected by the amorphous PDLLA chains, which can be dissolved in acetone. So after these chains dissolved in acetone, the neighboring sc-PLAs will be separated with each other in the solvent for the cases of A–C.
It can be seen from Fig. 6 that the solution was transparent for neat PDLLA while becoming opaque for samples PDLLA2.5, PDLLA5, PDLLA7.5 and PDLLA10 due to the undissolved sc-PLAs. In addition, the sc-PLAs were not connected together as a whole but were separated from each other and suspended homogeneously in the solvent for the samples PDLLA2.5, PDLLA5, PDLLA7.5 and PDLLA10, thus excluding the possibilities (i) and (ii). This result was in agreement with the conclusions of Coppola et al.36 It should be noted that the structures of (i) and (ii) may still exist locally, especially for the blends containing high level of sc-PLAs, but from the view of three-dimensional spaces, sc-PLAs were connected by the amorphous PDLLA chains through entanglements.20 As aforementioned, sc-PLAs can act as a physical crosslinking point of PDLLA chains, which make these interactional chains relax in a much slower way and take part in the formation of network structure. Based on the discussion above, it can be deduced that the existed network structure should be formed by the rigid sc-PLA particles and the PDLLA chains which are significantly restrained by the crosslinking effect of sc-PLAs.
 | ||
| Fig. 6 The photos of the solutions of the prepared samples for neat PDLLA, PDLLA2.5, PDLLA5, PDLLA7.5, and PDLLA10, after dissolving in acetone without stirring. | ||
Moreover, the size of the sc-PLAs dispersed in acetone was determined by dynamic light scattering (DLS), which is shown in Fig. 7. A hydrodynamic diameter (Dh) of 758 nm was obtained for PDLLA2.5, 944 nm for PDLLA5, 1136 nm for PDLLA7.5 and 1521 nm for PDLLA10, respectively. There exist more PLLA and PDLA chains for stereocomplex crystallites to grow to a larger size with increasing the content of PLLA and PDLA. It should be noted that the lamellar thickness (lc) of stereocomplex crystallites in the asymmetric PLLA/PDLA blends has been determined previously by small angle X-ray scattering, and a lc of around 20 nm was obtained.37 There is almost 2 order of magnitude different between Dh and reported lc, which indicates the stereocomplex crystallites in acetone are agglomerated together. Moreover, the contribution of the amorphous PDLLA segments trapped in the sc-PLAs (as shown in Fig. 5) to the increase of Dh should be noted. These amorphous PDLLA segments surround the sc-PLAs and can expand in the acetone, resulting in the increase of Dh.
 | ||
| Fig. 7 The size distribution of the sc-PLAs dispersed in acetone determined by DLS. | ||
To further investigate the dispersion of sc-PLA in the blends, the fracture surfaces of the blends were observed by SEM. The morphology of the samples was shown in Fig. 8. However, the fracture surfaces of the blends with various sc-PLA loadings were smooth and featureless. The structure of the formed network structure (i, ii, or iii) was not observed. It was believed that the result was due to the miscibility between these component, which was also contributed to the improved rheological and mechanical properties of the blends as discussed aforementioned.
 | ||
| Fig. 8 SEM microphotographs of the fractured surfaces of (a) PDLLA2.5, (b) PDLLA5, (c) PDLLA7.5, and (d) PDLLA10. | ||
Dynamic mechanical properties
Dynamic mechanical spectroscopy was also used to analyse the samples. Fig. 9 plots the storage modulus and tanδ changes of the blends with different sc-PLA proportions against temperature. For the storage modulus curves, as shown in the temperature range of T < Tg, the investigated samples were glassy with an almost unchanged tensile storage modulus, which was only dependent on sc-PLA contents. It is known that introducing fillers into the polymer matrix is one of the common methods for increasing the elastic modulus of materials.38 Our results showed a consistent tendency that storage moduli (E′) increased with an increasing proportion of sc-PLA in the blend. One of the reasons could be attributed to the reinforced interaction forces between the sc-PLA and PDLLA polymer chains in the blends. Besides, stereocomplex crystallites took over some free volume and thus hindered the mobility of the polymer chains.8 The above explanations accounted for the increment of the elastic moduli of the blends. When the temperature was higher than Tg, the increasing molecular mobility allowed for the stored elastic energy to be released as a mechanical restoring force and for the material to recover its permanent original shape.7 It was reported that a higher elastic modulus in the glassy state was better for the shape fixation of thermal induced shape memory polymers (SMPs) at low temperature.8 Comparisons of our results suggested that the storage modulus loss of PDLLA10 (3083/6.6 MPa) was more obvious than that of pure PDLLA (2150/1.4 MPa), which was in accordance with the report that a larger storage modulus loss value was beneficial to the shape memory performance.8
 | ||
| Fig. 9 Plots of tensile storage modulus (E′) and tanδ against temperature for neat PDLLA and the blend samples. | ||
In correspondence to the modulus step, the dependence of tanδ on temperature is also shown in Fig. 9(b). Owing to the well-known frequency effect,39 the glassy transition temperature observed through the tanδ–T curves did not correspond to the Tg data obtained by DSC analysis. However, the tanδ–T curves corresponded with the E′ and showed a single Tg for each blend and a general tendency that the Tg values raised with increasing the proportion of sc-PLA, indicating the miscibility of the components in the blends. These results are consistent with the results of DSC.
Shape memory properties
SMPs can respond through a mechanical reaction triggered by an external stimulus. By controlling the environmental temperature, the SMPs possess the ability to memorize a permanent shape that can substantially differ from their initial temporary shape. Being a sort of thermally induced SMP, PDLLA has inspiring prospects in minimally invasive surgery applications.8 Therefore, the shape memory behaviors of amorphous PDLLA in the presence of sc-PLA and/or the network structure are attractive and worth to be investigated. Fig. 10 shows a visual comparison of the shape recovery capacity of the PDLLA blends with different sc-PLA proportions (0, 2.5, 5, 7.5 and 10%). A typical procedure is described here. The investigated samples with thicknesses of 1.0 mm were bent into a temporary helix shape at 70 °C and the deformed shape was fixed at −20 °C for 30 min. When it was put in hot water of 70 °C, it recovered the original shape within 20 seconds. It can be learnt from the macroscopic results that the pure PDLLA without a sc-PLA component and the sample PDLLA2.5 failed to recovery to its permanent shape, whereas the other samples with sc-PLA component all performed well. The intuitive results demonstrated that the presence of sc-PLA played a key role in maintaining the permanent shape when shape deformation occurred, and thus improved the shape memory performance. In order to further investigate the effect of temperature on the shape memory performances of the blends, all the samples with dimensions of 40 × 4 × 1 mm were evaluated at a temperature range of 50–80 °C. Fig. 11 shows the plots of the shape recovery ratio as a function of temperature. All the investigated samples were incubated at 70 °C for 10 min and then elongated to Ld. Then the deformed samples were fixed by placing at −20 °C for 30 min. The shape memory performances at different temperatures with 20 seconds were observed. The shape memory ability of each sample was quantified by the shape recovery ratio parameter (Rr), defined by the following equation:| Rr = (Ld − Lf)/(Ld − L0) × 100% | (1) |
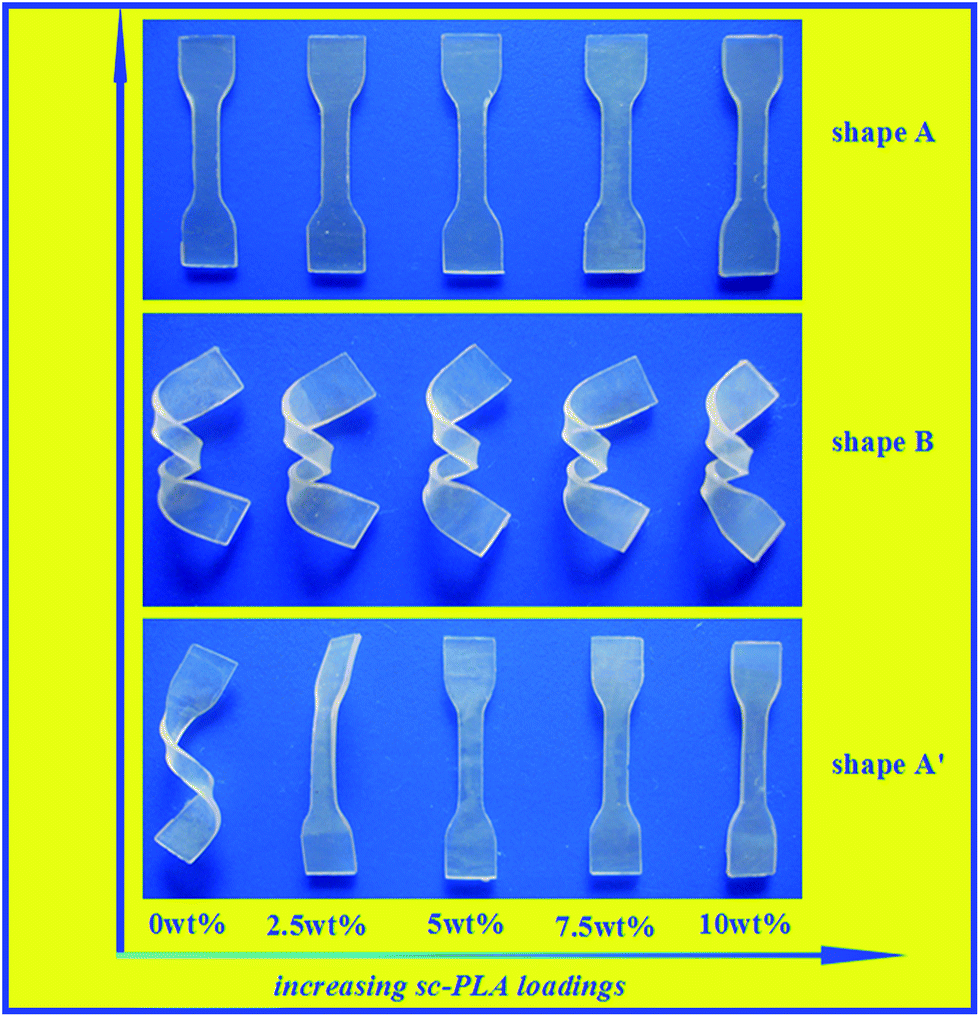 | ||
| Fig. 10 Photographs of the shape recovery of neat PDLLA and the blend samples. | ||
 | ||
| Fig. 11 The effect of temperature on the shape memory ratio of neat PDLLA and the blend samples. | ||
The general mechanism of thermal-plastic SMPs has been discussed previously.40,41 A SMP exists in the form of polymer networks, in which the net points being connected by chain segments determine the permanent shape.42 Typically, two components play key roles in the shape memory mechanism: one is “hard segments” acting as cross-linkers determining the permanent shape; and the other is “soft segments” acting as a continuous phase to fix the temporary shape at temperatures below the transition temperature.43 According to the above investigations, the PDLLA/sc-PLA blends demonstrated an improved shape recovery performance compared to the pure PDLLA polymer. The explanation to account for this phenomenon lied in the physical crosslinking between the sc-PLA and PDLLA. Sc-PLA served as “hard segments” effectively determined the permanent shape. In comparison, the shape fixing of amorphous PDLLA polymer depends on a random winding of molecular chains.44 Consequently, the amorphous PDLLA showed poor shape recovery behaviour as reported previously.7,8
Enzymatic hydrolysis
In the above sections, the effects of sc-PLA on the rheological behaviour, dynamic mechanical properties and shape memory properties of PDLLA were studied in the PLLA/sc-PLA blends. As reported previously, morphology (crystalline and amorphous) played an important role in affecting the enzymatic hydrolysis of PLA.45 Thus, the enzymatic hydrolysis behaviors of amorphous PDLLA in the presence of sc-PLA and/or the network structure are attractive and worth to be investigated. Fig. 12 shows the variation of weight loss of neat PDLLA and its blends with exposed time during enzymatic hydrolysis test. The values of weight loss increased with prolonging exposed time for both neat PDLLA and its blends. As shown in Fig. 12, mass loss started from the beginning of each degradation experiment and showed almost linear variation of weight loss with exposed time until 4 days. Since the weight loss showed almost linear increase with enzymatic hydrolytic time, the enzymatic hydrolytic rates of neat PDLLA and the PLLA/sc-PLA blend were obtained from the slopes of the plots of variation of weight loss with enzymatic hydrolytic time. For neat PDLLA, the enzymatic hydrolytic rate was around 1.79 mg per cm2 per day; however, the enzymatic hydrolytic rates were decreased to be around 1.71, 1.25, 1.01 and 0.81 mg per cm2 per day for the PDLLA/sc-PLA blends with increasing the sc-PLA loading from 2.5 to 10 wt%. It was obvious that the blends degraded slower than neat PDLLA, indicating the loading of sc-PLA decelerated the enzymatic hydrolytic of PDLLA in the blends; moreover, as shown in Fig. 12, the hydrolysis rate was rapidly decreased with increasing concentration of sc-PLA for the blends with a concentration of sc-PLA up to 5 wt%, indicating the hydrolysis rate of PDLLA could be greatly retarded when the sc-PLA formed a network structure. The results that the rate of enzymatic degradation of PLLA decreased with the increase in crystallinity have also been reported in the literatures.45 A threshold was observed when the degree of crystallinity was less than 22%. The sc-PLA dispersed in the PDLLA matrix may decrease the diffusion of water and molecular chains movement. Moreover, the presence of sc-PLA may also disturb the adsorption or cleavage process of proteinase K.17 These factors may contribute to the reduced hydrolysis rate of PDLLA in the presence of sc-PLA. When the sc-PLA network structure was formed in the PDLLA matrix, its effect on hydrolytic became more profound.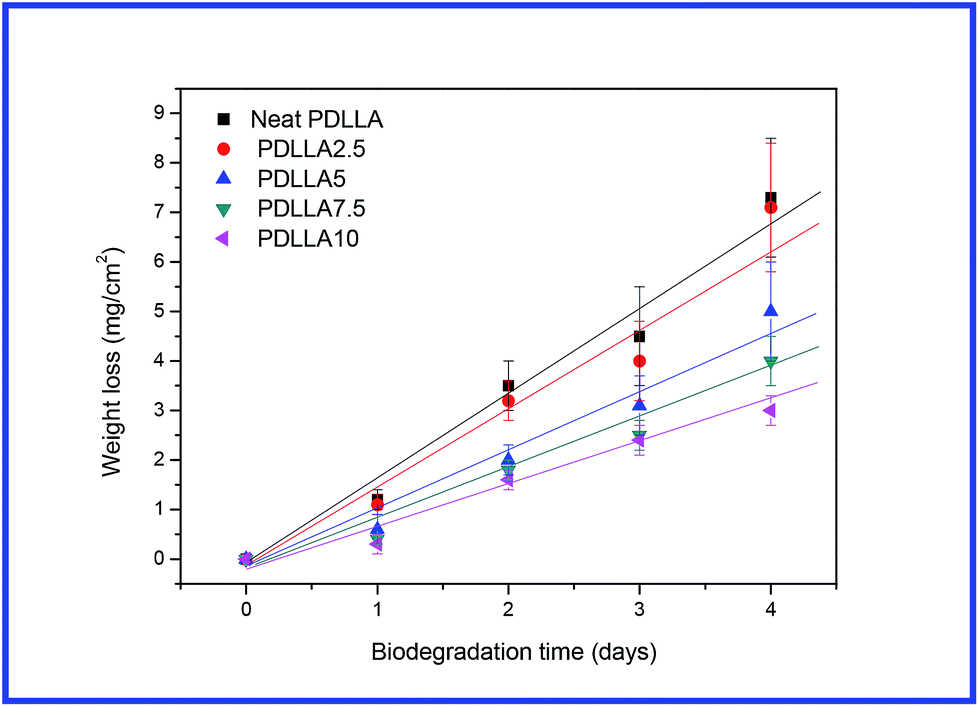 | ||
| Fig. 12 Variation of weight loss with enzymatic hydrolytic time for neat PDLLA and the blend samples. | ||
In order to further investigate the erosion mechanism, the surface and cross-section images of neat PDLLA and the PLLA/sc-PLA blends were studied with SEM. Parts (a)–(d) of Fig. 13 illustrate the SEM images of neat PDLLA before and after enzymatic hydrolysis. Fig. 13(a) and (b) show the surface and cross-section images of neat PDLLA prior to enzymatic hydrolysis, while Fig. 13(c) and (d) show the surface and cross-section images of neat PDLLA after an enzymatic hydrolysis of 2 days. It was obvious from parts (a) and (c) of Fig. 13 that the surface of neat PDLLA was very smooth before degradation while that of degraded neat PDLLA became blemished and appeared cell-like spots structure. On the contrary, no apparent morphology change took place in the inside of the films from the cross-section images study as shown in parts (b) and (d) of Fig. 13; however, the film thickness of neat PDLLA decreased after a enzymatic hydrolysis, indicating that the enzymatic hydrolysis of neat PDLLA may proceed via surface erosion mechanism. Similarly, the morphological changes of the surface and cross-section images for the PDLLA/sc-PLA blends were also studied for comparison. Parts (e) and (f) of Fig. 13 show the corresponding surface and cross-section SEM images of PDLLA5 sample after an enzymatic hydrolysis of 2 days as an example. The surface of the PDLLA5 sample is shown in Fig. 13(e). No apparent cell-like structure as seen in neat PDLLA was observed. The sc-PLA particles with an average size of 0.9 μm can be clearly seen on the surface of the enzymatic hydrolysis sample. The size of the sc-PLA was in agreement with the size determined by DLS. Moreover, the films tended to become thicker in the PDLLA5 sample than in neat PDLLA if we compared the cross-section images shown in Fig. 13(d) and (f) after a hydrolytic degradation. The variation of the film thickness suggested that surface erosion may still occur despite the presence of sc-PLA in the PDLLA/sc-PLA blend. Moreover, SEM images showing the morphology of surface and cross-section for the other blends samples are shown in Fig. 14 for comparison.
 | ||
| Fig. 13 SEM images showing the morphology of surface and cross-section for neat PDLLA and PDLLA5 samples: (a) surface of neat PDLLA before hydrolysis; (b) cross section of neat PDLLA before hydrolysis; (c) surface of neat PDLLA after a hydrolysis degradation of 2 days; (d) cross section of neat PDLLA after a hydrolysis degradation of 2 days; (e) surface of PDLLA5 after a hydrolysis degradation of 2 days; (f) cross section of PDLLA5 after a hydrolysis degradation of 2 days. | ||
 | ||
| Fig. 14 SEM images showing the morphology of surface and cross-section for the blends samples: (a) surface of PDLLA2.5 after a hydrolysis degradation of 2 days; (b) cross section of PDLLA2.5 after a hydrolysis degradation of 2 days; (c) surface of PDLLA7.5 after a hydrolysis degradation of 2 days; (d) cross section of PDLLA7.5 after a hydrolysis degradation of 2 days; (e) surface of PDLLA10 after a hydrolysis degradation of 2 days; (f) cross section of PDLLA10 after a hydrolysis degradation of 2 days. | ||
From the above studies, it can be deduced that the hydrolytic degradation of both neat PDLLA and the PDLLA/sc-PLA blends occurred at the surface of the films. Such results are consistent with the previous research conclusion that the erosion of PDLLA process proceeds via surface erosion.46 The hydrolytic degradation rates are slower in the PDLLA/sc-PLA blends than in neat PDLLA and decrease with the sc-PLA loading; however, the exact reason is still unknown and needs further investigation.
Conclusions
In this work, PDLLA and sc-PLA blends were prepared by solution blending. Wide-angle X-ray diffraction and differential scanning calorimetry results verified that complete stereocomplex crystallites without any evidence of the formation of homocrystallites in the PDLLA could be achieved. Investigation on the rheological properties of the melts embedded by stereocomplex crystallites showed that the presence of the formed stereocomplex crystallites can reinforce the PDLLA melt as a result of the filler effect and crosslinking effect of stereocomplex crystallites. Moreover, a transition from the liquid-like to solid-like viscoelastic behaviors was observed, which exactly indicated the stereocomplex crystallite network structure was formed in the PDLLA melt. With the formation of the network structure, the reinforcing effect of stereocomplex crystallite was obviously increased. From a dissolution experiment, it can be deduced that the formed network structure should be formed by the rigid sc-PLA particles and the inter particle polymer chains which were significantly restrained by the cross-linking effect of stereocomplex crystallites. Accordingly, the E′ increased with an increasing level of sc-PLA in the blend due to the reinforced interaction forces between the sc-PLA and PDLLA polymer chains. Moreover, the shape memory behaviors of PDLLA had been improved obviously in the blends than in neat PDLLA, especially when a percolation network structure had formed, which may be of great use and importance for the wider practical application of PDLLA. Finally, it was found that the enzymatic hydrolytic degradation rates had been retarded in the blends than in neat PDLLA. The erosion mechanism of PDLLA and its blends was further discussed, and the enzymatic hydrolysis of neat PDLLA and its blends may proceed via surface erosion.Acknowledgements
This work was supported by the National Science Foundation of China (50703042).Notes and references
- A. Södergård and M. Stolt, Prog. Polym. Sci., 2002, 27, 1123–1163 CrossRef.
- Y. Y. Zhao, Z. B. Qiu and W. T. Yang, J. Phys. Chem. B, 2008, 112, 16461–16468 CrossRef CAS PubMed.
- R. Langer and J. P. Vacanti, Science, 1993, 260, 920–926 CAS.
- H. M. Powell, O. Ayodeji, T. L. Summerfield, D. M. Powell, D. A. Kniss, D. L. Tomasko and J. L. Lannutti, Biomaterials, 2007, 28, 5562–5569 CrossRef CAS PubMed.
- R. Steendam, M. J. van Steenberger, W. E. Hennink, H. W. Frijlink and C. F. Lerk, J. Controlled Release, 2001, 70, 71–82 CrossRef CAS.
- S. Aslan, L. Calandrelli, P. Laurienzo, M. Malinconico and C. Migliaresi, J. Mater. Sci., 2000, 35, 1615–1622 CrossRef CAS.
- S. B. Zhou, X. T. Zheng, X. J. Yu, J. X. Wang, J. Weng, X. H. Li, B. Feng and M. Yin, Chem. Mater., 2007, 19, 247–253 CrossRef CAS.
- K. Du and Z. H. Gan, J. Mater. Chem. B, 2014, 2, 3340–3348 RSC.
- T. Wang and L. T. Drzal, ACS Appl. Mater. Interfaces, 2012, 4, 5079–5085 CAS.
- L. G. Calandrelli, A. Calarco, P. Laurienzo, M. Malinconico, O. Petillo and G. Peluso, Biomacromolecules, 2008, 9, 1527–1534 CrossRef CAS PubMed.
- P. J. Pan, Z. C. Liang, B. Zhu, T. Dong and Y. lnoue, Macromolecules, 2009, 42, 3374–3380 CrossRef CAS.
- B. Eling, S. Gogolewski and A. J. Pennings, Polymer, 1982, 23, 1587–1593 CrossRef CAS.
- L. Cartier, T. Okihara, Y. Ikada, H. Tsuji, J. Puiggali and B. Lotz, Polymer, 2000, 41, 8909–8919 CrossRef CAS.
- J. Zhang, Y. Duan, H. Sato, H. Tsuji, I. Noda, S. Yan and Y. Ozaki, Macromolecules, 2005, 38, 8012–8021 CrossRef CAS.
- T. Kawai, N. Rahman, G. Matsuba, K. Nishida, T. Kanaya, M. Nakano, H. Okamoto, J. Kawada, A. Usuki, N. Honma, K. Nakajima and M. Matsuda, Macromolecules, 2007, 40, 9463–9469 CrossRef CAS.
- Y. Ikada, K. Jamshidi, H. Tsuji and S. H. Hyon, Macromolecules, 1987, 20, 904–906 CrossRef CAS.
- H. Tsuji, Macromol. Biosci., 2005, 5, 569–597 CrossRef CAS PubMed.
- R. Y. Bao, W. Yang, W. R. Jiang, Z. Y. Liu, B. H. Xie, M. B. Yang and Q. Fu, Polymer, 2012, 53, 5449–5454 CrossRef CAS PubMed.
- R. Y. Bao, W. Yang, W. R. Jiang, Z. Y. Liu, B. H. Xie and M. B. Yang, J. Phys. Chem. B, 2013, 117, 3667–3674 CrossRef CAS PubMed.
- X. F. Wei, R. Y. Bao, Z. Q. Cao, W. Yang, B. H. Xie and M. B. Yang, Macromolecules, 2014, 47, 1439–1448 CrossRef CAS.
- H. Tsujiand and Y. Ikada, Polymer, 1999, 40, 6699–6708 CrossRef.
- S. Ndersson, M. Hakkarainen, S. Inkinen, A. Sodergård and A. Albertsson, Biomacromolecules, 2010, 11, 1067–1073 CrossRef PubMed.
- D. G. Abebe and T. Fujiwara, Biomacromolecules, 2012, 13, 1828–1836 CrossRef CAS PubMed.
- H. Tsuji and S. Miyauchi, Polym. Degrad. Stab., 2001, 71, 415–424 CrossRef CAS.
- H. Tsuji and S. Miyauchi, Polymer, 2001, 42, 4463–4467 CrossRef CAS.
- H. Tsuji and K. Ikarashi, Polym. Degrad. Stab., 2004, 85, 647–656 CrossRef CAS PubMed.
- D. Sawai, Y. Tsugane, M. Tamada, T. Kanamoto, M. Sungil and S. H. Hyon, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 2632–2639 CrossRef CAS.
- Y. Li, C. Y. Han, X. Zhang, Q. L. Dong and L. S. Dong, Thermochim. Acta, 2013, 573, 193–199 CrossRef CAS PubMed.
- Y. Li, C. Y. Han, Y. J. Bian, Q. L. Dong, H. W. Zhao, X. Zhang, M. Z. Xu and L. S. Dong, Thermochim. Acta, 2014, 580, 53–62 CrossRef CAS PubMed.
- F. Du, R. C. Scogna, W. Zhou, S. Brand, J. E. Fischer and K. I. Winey, Macromolecules, 2004, 37, 9048–9055 CrossRef CAS.
- Z. Xu, Y. Niu, Z. Wang, H. Li, L. Yang, J. Qiu and H. Wang, ACS Appl. Mater. Interfaces, 2011, 3, 3744–3753 CAS.
- Z. Xu, Y. Niu, L. Yang, W. Xie, H. Li, Z. Gan and Z. Wang, Polymer, 2010, 51, 730–737 CrossRef CAS PubMed.
- C. Liu, J. Zhang, J. He and G. Hu, Polymer, 2003, 44, 7529–7532 CrossRef CAS PubMed.
- R. H. Horst and H. H. Winter, Macromolecules, 2000, 33, 7538–7543 CrossRef CAS.
- H. Yamane, K. Sasai and M. J. Takano, J. Rheol., 2004, 48, 599–609 CrossRef CAS.
- S. Coppola, S. Acierno, N. Grizzuti and D. Vlassopoulos, Macromolecules, 2006, 39, 1507–1514 CrossRef CAS.
- J. Narita, M. Katagiri and H. Tsuji, Macromol. Mater. Eng., 2013, 298, 270–282 CrossRef CAS.
- I. A. Rousseau, Polym. Eng. Sci., 2008, 48, 2075–2089 CAS.
- E. Zini, M. Scandola, P. Dobrzynski, J. Kasperczyk and M. Bero, Biomacromolecules, 2007, 8, 3661–3667 CrossRef CAS PubMed.
- M. Behl and A. Lendlein, Mater. Today, 2007, 10, 20–28 CrossRef CAS.
- H. Zhang, H. Wang, W. Zhong and Q. Du, Polymer, 2009, 50, 1596–1601 CrossRef CAS PubMed.
- M. Behl, M. Y. Razzaq and A. Lendlein, Adv. Mater., 2010, 22, 3388–3410 CrossRef CAS PubMed.
- A. Alteheld, Y. K. Feng, S. Kelch and A. Lendlein, Angew. Chem., Int. Ed., 2005, 44, 1188–1192 CrossRef CAS PubMed.
- T. Ohki, Q. Q. Ni, N. Ohsako and M. Iwamoto, Composites, Part A, 2004, 35, 1065–1073 CrossRef PubMed.
- H. Cai, V. Dave, R. A. Cross and S. P. Mccarthy, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 2701–2708 CrossRef CAS.
- Y. Li, C. Y. Han, J. J. Bian, L. J. Han, L. S. Dong and G. Gao, Polym. Compos., 2012, 33, 1719–1727 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |