
Palladium catalyzed ortho-halogenation of 2-arylbenzothiazole and 2,3-diarylquinoxaline†
Sourav Kumar Santra,
Arghya Banerjee,
Nilufa Khatun,
Asim Samanta and
Bhisma K. Patel*
Department of Chemistry, Indian Institute of Technology Guwahati, 781 039, Assam, India. E-mail: patel@iitg.ernet.in; Fax: +91-3612690762
First published on 14th January 2015
Abstract
A palladium catalysed ortho-halogenation strategy has been developed using benzothiazoles and quinoxalines as the directing substrates. This method provides mono-o-halogenated product at the other available ortho site of a mono-ortho substituted 2-arylbenzothiazole. However, ortho-unsubstituted 2-arylbenzothiazole afforded di-ortho halogenated product exclusively. The preformed (or installed) ortho-group is towards the sulphur side of the benzothiazole from energy minimised calculation. Thus the selective formation of di-ortho-halogenated products is due to favourable exposure of the second ortho site for subsequent halogenation. However the phenyl ring in 2,3-diarylquinoxalines can be selectively mono ortho halogenated. A plausible reaction mechanism has been proposed for this halogenation process.
Introduction
Transition metal-catalysed directed1 and non directed2 C–H functionalisation has emerged as an atom and step economic strategy for developing synthetically versatile intermediates via unprecedented disconnections. Besides other transition metals, the use of a palladium catalyst in chelation-directed C–H activation reactions are of interest due to its better efficacy and high turnover numbers.3 Of late a plethora of Pd-catalysed ortho C–H functionalisations have appeared in the literature using various rigid and flexible directing substrates.4 Although a number of transition metal-catalysed C–H functionalisation strategies are reported for the formation of carbon–carbon (C–C) bonds,5 formation of carbon–heteroatom (C–X) bonds, in particular carbon–halogen bonds, are relatively less explored. Aryl halides (Ar–X) are useful intermediates for synthetic organic chemistry and are valuable precursors for nucleophilic substitution reactions as well as for the synthesis of various organometallic reagents.6 Aryl halides are also used in transition metal-catalysed cross-coupling reactions such as Suzuki, Negishi and Heck type couplings to construct complex structures.7 However, preparation of aryl halides via classical halogenation suffer from some drawbacks such as poor regioselectivity and polyhalogenations, particularly for activated aromatics.8 As a solution to these problems, substantial endeavours have been made recently for the regioselective o-halogenation of arenes catalysed by transition metals. A few Pd-catalysed protocols have emerged recently for the selective installation of halo groups at the ortho site of various directing groups via arene sp2 C–H activation.9 2-Arylbenzothiazoles and 2,3-diarylquinoxalines bearing o-chelating moieties may provide cyclometallation at their proximal sites via the assistance of their nitrogen donor atoms and therefore could be employed for o-functionalisations. Recently our group and others have developed strategies for the ortho selective C–C and C–X bond formation using benzothiazole and quinoxaline as directing arenes.10 In continuation to these reports we envisaged that these two directing arenes viz. benzothiazole and quinoxaline could similarly be ortho halogenated. 2-Arylbenzothiazoles and 2,3-diarylquinoxalines are both privileged motifs present in many naturally occurring molecules and pharmaceuticals.11 Thus, further functionalisations of these important scaffolds may provide useful intermediates which may find potential applications. Herein, we describe a Pd-catalysed o-halogenation of 2-arylbenzothiazoles and 2,3-diarylquinoxalines using N-halosuccinamides NXS (X = Cl, Br and I) as halogen sources.Results and discussion
From the crystal-structure of 2-(benzo[d]thiazol-2-yl)phenyl benzoate (1) it was found that the ester group in 2-aryl ring is towards the sulfur side of benzothiazole and the two aromatic moieties are periplanar.12 This periplanar orientation with further assistance from N-atom of benzothiazole is favourable for cyclopalladation. Thus, 2-(benzo[d]thiazol-2-yl)phenyl benzoate (1) was chosen as the model substrate for ortho-bromination using N-bromosuccinimide as the source of bromine and Pd(OAc)2 as the catalyst. When (1) (1 equiv.) was reacted with N-bromosuccinimide (a) (1.2 equiv.) in the presence of Pd(OAc)2 (5 mol%) in toluene at 90 °C, mono-o-bromo product (1a) was obtained in 39% yield. Absence of any meta or para-bromo products suggest a substrate-directed regioselective o-bromination. Encouraged by this success, other reaction parameters such as solvents, catalysts, additives and their quantities were varied to maximise the product yield. Polar aprotic solvents such as DMF or DMSO (Table 1, entries 2 and 3) failed to give any trace of product whereas non polar solvents such as cyclohexane, o-xylene (Table 1, entries 4 and 5) provided very low yield of the product. Interestingly, by switching the solvent from toluene to 1,2-dichloroethane (DCE) (Table 1, entry 6) the product yield improved up to 58% under otherwise identical conditions. Other palladium salts such as Pd(TFA)2, PdCl2 and PdBr2 (Table 1, entries 7–9), were relatively less potent compared to Pd(OAc)2. In the absence of catalyst (Pd(OAc)2) no o-bromination occured (Table 1, entry 10). No significant improvement in the product yield was observed even when the catalyst loading was increased from 5 to 10 mol%, (Table 1, entry 11). In a pursuit to further improve the yield, acid additives such as pivallic acid (PivOH), para-toluenesulfonic acid (PTSA), CF3COOH (TFA) and CH3COOH (AcOH) were used during the reaction. After screening the reaction with these acid additives, it was found that the use of 50 mol% PTSA under otherwise identical conditions provided an improved yield (79%) of the desired o-brominated product (1a) (Table 1, entry 12). In the presence of other acid additives such as TFA, AcOH cleavage of ester group in (1) was observed along with the formation of o-bromo product (1a) (Table 1, entries 13 and 14) thereby effectively lowering the yield. The ester group survived when PivOH was used as the additive but was not so effective toward desired o-bromination (Table 1, entry 15). Thus, after a series of experimentations, 2-(benzo[d]thiazol-2-yl)phenyl benzoate (1) (1 equiv.), catalyst Pd(OAc)2 (5 mol%), additive PTSA (50 mol%) and N-bromosuccinimide (a) (1.2 equiv.) in 1,2 dichloroethane (2 mL) at 90 °C and a reaction time of 7 h was found to be the best conditions for this transformation (Table 1, entry 12).
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | Oxidant | Yield (%) |
| a Reaction conditions: 1 (0.25 mmol), NBS (0.3 mmol) at 90 °C for 7 h.b Isolated yield.c Additive (0.13 mmol). | ||||
| 1 | Pd(OAc)2 (5) | Toluene | NBS | 39 |
| 2 | Pd(OAc)2 (5) | DMF | NBS | 00 |
| 3 | Pd(OAc)2 (5) | DMSO | NBS | 00 |
| 4 | Pd(OAc)2 (5) | C6H12 | NBS | Trace |
| 5 | Pd(OAc)2 (5) | o-Xylene | NBS | Trace |
| 6 | Pd(OAc)2 (5) | DCE | NBS | 58 |
| 7 | Pd(TFA)2 (5) | DCE | NBS | 46 |
| 8 | PdCl2 (5) | DCE | NBS | 44 |
| 9 | PdBr2 (5) | DCE | NBS | 43 |
| 10 | — | DCE | NBS | 00 |
| 11 | Pd(OAc)2 (10) | DCE | NBS | 62 |
| 12 | Pd(OAc)2 (5) | DCE | NBS/PTSA | 79c |
| 13 | Pd(OAc)2 (5) | DCE | NBS/TFA | 68c |
| 14 | Pd(OAc)2 (5) | DCE | NBS/AcOH | 66c |
| 15 | Pd(OAc)2 (5) | DCE | NBS/PivOH | 63c |

Keeping the above optimised conditions in mind, this methodology was further applied to various substituted 2-(benzo[d]thiazol-2-yl)phenyl carboxylate with N-bromosuccinamide (a). 2-(Benzo[d]thiazol-2-yl)aryl benzoate containing various substituents at the 2-aryl ring such as p-Me (2), p-OMe (3), m-OMe (4) and p-OCOPh (5) gave their corresponding o-brominated products (2a), (3a), (4a) and (5a) in excellent yields (81–87%) (Scheme 1). It is clear from substrates (1–5) that no loss of regioselectivity and product yields were observed even with different substitution patterns on the aryl ring. Conversely, for substrates (6) and (7) the yields of their desired o-bromo products (6a) and (7a) were dropped marginally when substituents are present in the phenyl rings possessing the ester group (Scheme 1). Naphthylester bearing benzothiazole (8) also provided moderated yield of its corresponding o-bromo product (8a) (Scheme 1).
 | ||
| Scheme 1 Scope of o-halogenation of 2-arylbenzothiazoles. aReaction conditions: 2-arylbenzothiazole (1–10) (0.25 mmol), NXS (a–c) (0.30 mmol) and PTSA (0.13 mmol) in DCE (2.0 mL) at 90 °C, 7 h. bYields of isolated product. cTemperature: 110 °C, 10 h. dTemperature: 60 °C, 5 h. | ||
When analogous chlorination reaction of (1) with N-chlorosuccinimide (b) was carried out using the above optimised conditions, only 33% yield of o-chloro product (1b) was obtained. However by increasing the reaction temperature from 90 °C to 110 °C and prolonging the reaction time from 7 h to 10 h provided the o-chlorinated product (1b) in an improved yield of 72%. Benzothiazole (5) having ortho and para di-OCOPh group in its 2-aryl ring gave 77% of o-chlorinated product (5b). Then the strategy was further extended toward o-iodination of mono-o-protected benzothiazole (1) using N-iodosuccinamide. Substrates (9) and (10) bearing p-Cl and o-NO2 groups in their phenyl rings of the ester provided mono-o-iodo products (9c) and (10c) in 82% and 83% yields respectively. Interestingly, no substantial change in the product yield was observed even when the reaction was performed at lower temperature (60 °C). However, only trace amount of desired o-iodo product was formed when the reaction was carried out at 40 °C. From these observations it is evident that the rates of halogenation follow the order: iodination > bromination > chlorination.
To verify whether this selective mono-o-bromination strategy can be applied to 2-arylbenzothiazole in the absence of o-ester functionality, substrate (11) with two available ortho sites was reacted under the above optimised conditions. When 2-phenylbenzothiazole (11) (1 equiv.) was treated with NBS (1.2 equiv.) under otherwise identical conditions surprisingly rather than the expected mono-o-brominated product only ortho-di-bromo product (11a) was obtained in 41% yield. Typically substrate-directed o-halogenation provides mono-halogenated as the major product with trace of di-ortho-halogenated product in few cases.9a–c,f–h,13 In the present case exclusive formation of ortho-di-bromo product (11a) was rather surprising to us. After the initial o-mono bromination the benzothiazole and the 2-phenyl ring possibly adopt a similar periplanar orientation to that of substrate (1) exposing the other ortho site for subsequent bromination. Even the use of 2 equiv. of (NBS) provided no traces of mono-o-bromo product rather the yield of ortho-di-bromo product (11a) was enhanced to 53%.
2-Arylbenzothiazole having moderately electron-donating group p-Me (12) in its 2-aryl ring gave 59% yield of di-o-brominated product (12a) again giving no trace of mono-o-bromo product. If our assumption on periplanar orientation of benzothiazole and 2-aryl ring after first bromination is true then preformed mono ortho substituted benzothiazoles should react to provide better yields of bis-ortho substituted products. To verify our assumption o-Cl (13), o-OMe (14) and o-OCH2Ph (15) substituted 2-arylbenzothiazoles were employed for ortho bromination. All three substrates provided o-brominated products (13a), (14a) and (15a) in far better yields as shown in Scheme 2. Energy calculation performed using Gaussian 09 package14 at B3LYP-D3/6-31G (d,p) level of substrate 2-(2-chlorophenyl)benzo[d]thiazole (13) revealed the periplanar orientation of benzothiazole and 2-chloro phenyl ring to be the most stable conformer with the chloro group orienting toward the sulphur side (Fig. 1(a)). The extra stability of this conformer is due to sulphur (S)⋯chlorine (Cl) interaction. Similar chloro (halo) and sulphur atom interaction is reported in various organic and inorganic moieties.15 Such periplanar arrangement with the oxygen atom of the ester group orienting toward sulphur side is also observed in the energy minimised structure of 2-(benzo[d]thiazol-2-yl)phenyl benzoate (1) (Fig. 1(b)). Apparently, the energy minimised structure of (1) (Fig. 1(b)) matches exactly with its X-ray crystal structure.12 Thus this type of periplanar orientation is the most favourable for o-palladation leading to o-halogenation. These results demonstrate the conformational predominance of the substrates over their steric and electronic effects during directed bromination. In case of substrate 2-(naphthalen-1-yl)benzo[d]thiazole (16) possessing both ortho and peri C–H bonds, bromination occurred selectively at the ortho site giving product (16a) in 64% yield. When o-bromination strategy was applied to 2-phenethylbenzo[d]thiazole (17), interestingly bromination occurred at the β Csp3–H bond which happened to be a benzylic carbon as well. Now the query arises whether the bromination is due to classical radical benzylic bromination or a substrate directed metal-catalysed sp3 C–H functionalisation. When the reaction was performed in absence of a Pd-catalyst under otherwise identical conditions, no bromination was observed thereby suggesting the later possibility. The lower yield (29%) obtained for (17a) is possibly due to the free rotation of Csp3–Csp3 containing benzylic and its adjacent carbon thereby lowering the possibility of cyclopalladation. However other β Csp3–H bearing alkane substrates such as (18) and (19) failed to provide desired brominated products (Scheme 2). Thus, for substrate (17) due to the presence of an acidic benzylic β Csp3–H's the bromination was favourable compared to substrates (18) and (19). The success of this bromination strategy was then extended towards o-chlorination of substrates (11) and (12) using N-chlorosuccinimide (b) following the previous optimised conditions for chlorination, which provided o-di-chloro products (11b) and (12b) respectively in moderate yields (Scheme 2). Here again substrate (16) possessing both ortho and peri C–H bonds, the chlorination occurred regioselectively at the ortho site giving product (16b) in 55% yield. Analogous o-iodination of (12) and (13) using N-iodosuccinamide (c) gave o-di-iodo products (12c) and (13c) in 65% and 79% yields respectively under identical conditions for iodination. Here also the rates of o-di-halogenation (Scheme 2) follow the same order (iodination > bromination > chlorination) to that of mono-o-halogenation (Scheme 1) (Scheme 3).
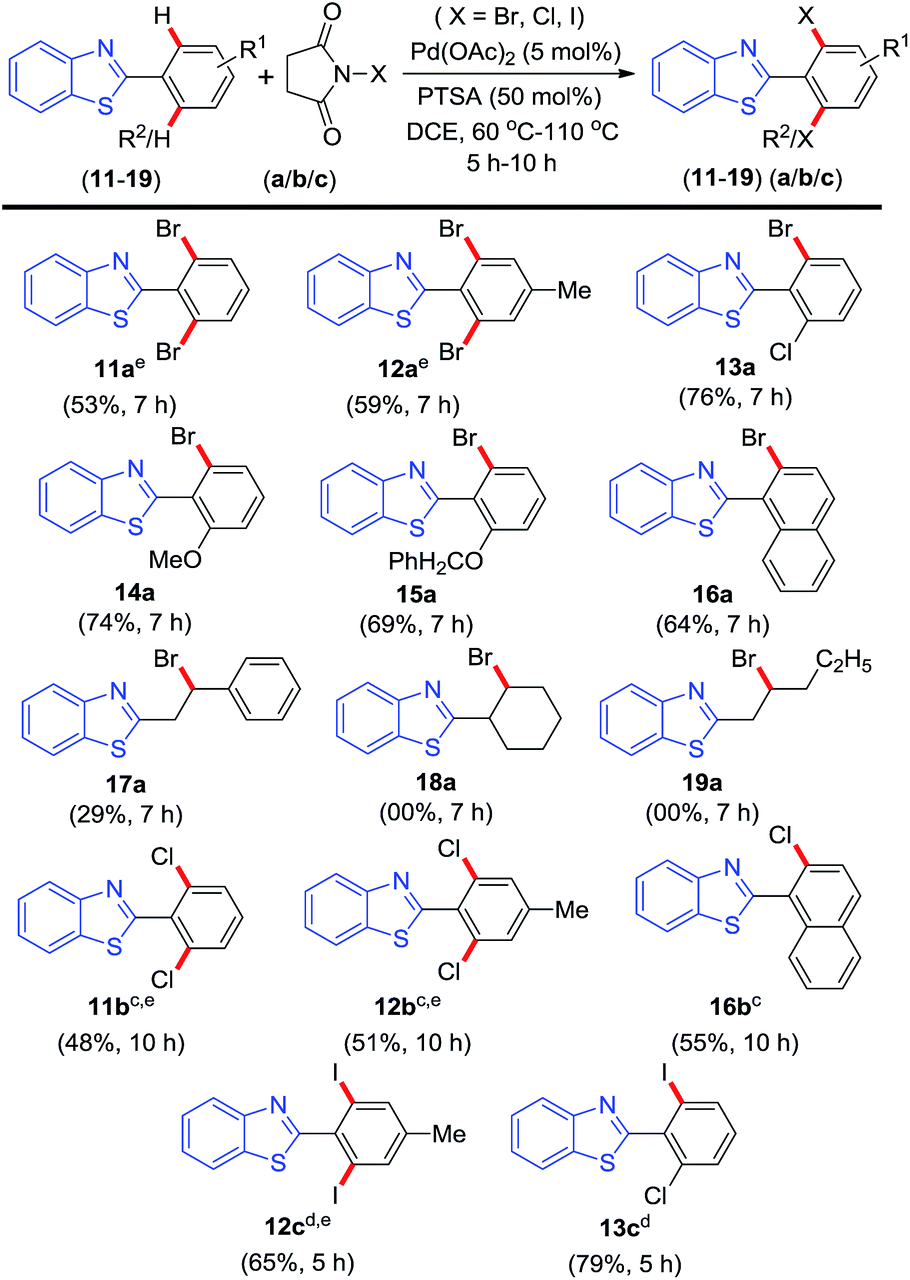 | ||
| Scheme 2 Scope of o-halogenation of 2-arylbenzothiazoles. aReaction conditions: 2-arylbenzothiazole (11–19), (0.25 mmol), NXS (a–c) (0.30 mmol) and PTSA (0.13 mmol) in DCE (2.0 mL) at 90 °C, 7 h. bYields of isolated product. cTemperature: 110 °C, 10 h. dTemperature: 60 °C, 5 h. eNXS (a–c) (0.50 mmol). | ||
 | ||
| Fig. 1 Energy minimised diagram of substrates 13 and 1. | ||
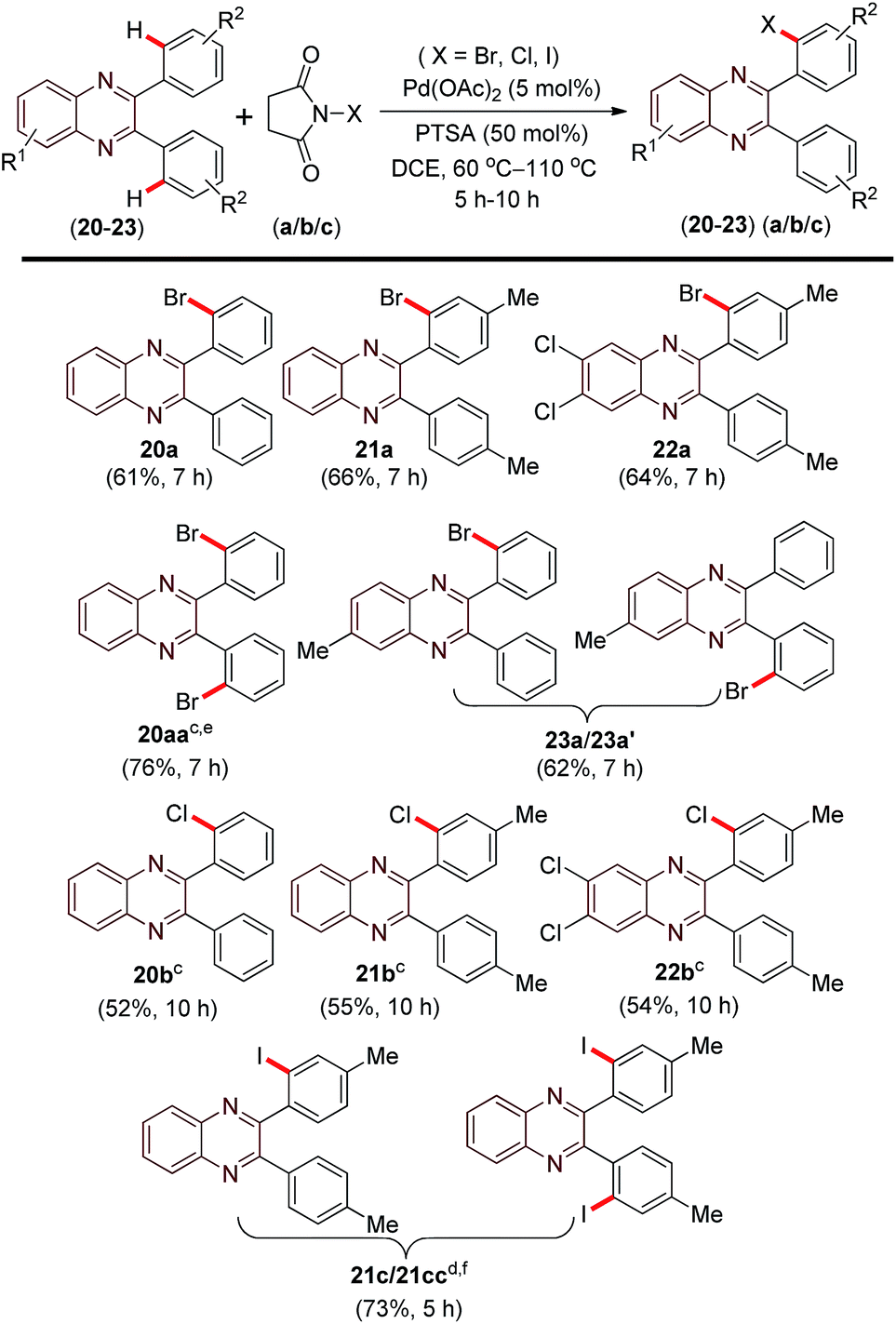 | ||
| Scheme 3 Scope of o-halogenation of 2,3-diarylquinoxalines. aReaction conditions: 2,3-diarylquinoxaline (20–23), (0.25 mmol), NXS (a–c) (0.30 mmol) and PTSA (0.13 mmol) in DCE (2.0 mL) at 90 °C, 7 h. bYields of isolated product. cTemperature: 110 °C, 10 h. dTemperature: 60 °C, 5 h. eNBS (a) (0.50 mmol). fYield calculated with respect to recovered of starting material. | ||
Unlike in 2-aryl benzothiazoles, 2,3-diarylquinoxaline moiety has four ortho sites for possible directed halogenations. Thus, it would be interesting to see during halogenation which of the following products would be formed selectively under a particular condition. A mono-halogenation in one of the phenyl ring, mono-halogenation in each of the phenyl ring; dihalogenation in one of the ring or a dihalogenation in both the rings are some of the possibilities. Thus the above optimised halogenation conditions were applied to 2,3-diarylquinoxalines for bromination, chlorination and iodination respectively. 2,3-Diphenyl quinoxaline (20), when reacted with NBS (a) (1.2 equiv.) at 90 °C, mono-o-bromo product (20a) was obtained in 61% yield along with a trace (<5%) of di-bromo (mono-bromination in each of the phenyl ring) product (20aa). However increasing the quantity of (NBS) to 2 equiv. the yield of the di-bromo product (20aa) was improved to 12%. Maintaining the (NBS) quantity to 2 equiv. and increasing the reaction temperature to 110 °C substantial improvement in the yield (76%) of the di-bromo product (20aa) was observed.
Presently, however we concentrate on achieving selective mono-bromination. 2,3-Diphenylquinoxaline (21) containing electron-donating group (–Me) in its aryl rings gave 66% of the mono-o-bromo product (21a). The substrate 6,7-dichloro-2,3-diphenylquinoxaline (22) on treatment with NBS (a) afforded moderate yield (64%) of the mono-o-bromo product (22a). Unsymmetrical 2,3-diphenylquinoxaline (23) having a –Me group at its 6th position when reacted with NBS provided an inseparable regioisomeric mono-o-bromo products 23a/23a′ in the ratio of 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :45 (as judged from its 1H and 13C NMR spectra) in 62% yield. So far chlorination is concerned under the optimised conditions various unsubstituted and substituted 2,3-diphenylquinoxaline such as (20), (21) and (22) all provided exclusive mono ortho-brominated products (20b), (21b) and (22b) respectively in good yields. Like previous cases iodination of substrate (21) provided an inseparable mixture of mono (21c) and di-iodo (21cc) products in a combined yield of 73%. In this system also the same reactivity trend (iodination > bromination > chlorination) to that of mono-o-halogenation was observed.
:45 (as judged from its 1H and 13C NMR spectra) in 62% yield. So far chlorination is concerned under the optimised conditions various unsubstituted and substituted 2,3-diphenylquinoxaline such as (20), (21) and (22) all provided exclusive mono ortho-brominated products (20b), (21b) and (22b) respectively in good yields. Like previous cases iodination of substrate (21) provided an inseparable mixture of mono (21c) and di-iodo (21cc) products in a combined yield of 73%. In this system also the same reactivity trend (iodination > bromination > chlorination) to that of mono-o-halogenation was observed.
A possible mechanism has been proposed for this palladium catalysed ortho-halogenation reaction as shown in Scheme 4. For o-bromination process initial cyclopalladation of substrate (1) leads to the formation of intermediate (I). This intermediate further undergoes oxidative addition with N-bromosuccinamide (NBS) forming either a dimeric Pd(III)16 or a monomeric Pd(IV)9b,17 intermediate (II). During the mass spectral analysis of the reaction mixture some of the monomeric Pd(IV) species have been detected (see ESI, Fig. S1†). However, the formation of a dimeric. Pd(III) during the reaction cannot be ruled out16 Subsequent reductive elimination of intermediate (II) leads to o-bromo product (1a) via C–Br bond formation and regenerating Pd(II) catalyst for the next cycle. The mono-ortho bromo product so formed orient favourably for subsequent bromination following similar mechanistic path leading to the formation of di-bromo product. Similar mechanism can be proposed for chlorination and iodination of 2-aryl benzothiazole and 2,3-diarylquinoaline.
 | ||
| Scheme 4 Plausible mechanism for o-bromination. | ||
Conclusion
In conclusion we have developed an ortho-halogenation strategy using palladium as the catalyst and N-halosuccinamide as the halogen source using benzothiazoles and quinoxalines as the directing substrates. This method provides mono-o-halogenated product at the other available ortho site of a mono-ortho substituted 2-arylbenzothiazole. Although ortho-unsubstituted 2-arylbenzothiazole afforded di-ortho halogenated product exclusively while ortho-unsubstituted 2,3-diarylquinoxaline afforded mono-o-halogenated products under identical reaction conditions.General procedure for the synthesis of 2-(benzo[d]thiazol-2-yl)-3-bromophenyl benzoate (1a) from 2-(benzo[d]thiazol-2-yl)phenyl benzoate (1) and N-bromosuccinamide (a)
To an oven-dried 25 mL round bottom flask were added 2-(benzo[d]thiazol-2-yl)phenyl benzoate (1) (0.083 g, 0.25 mmol), N-bromosuccinamide (0.053 g, 0.3 mmol), Pd(OAc)2 (0.003 g, 0.013 mmol), p-toluenesulfonic acid (0.024 g, 0.13 mmol) and 1,2-dichloroethane (2.0 mL). Then the reaction mixture was refluxed in an oil bath preheated to 90 °C. After completion of the reaction (7 h) excess solvent was evaporated under reduced pressure. The product was extracted with ethyl acetate (3 × 10 mL) and the combined organic layer was washed carefully with saturated sodium bicarbonate solution (2 × 5 mL), dried over anhydrous sodium sulphate, and evaporated under reduced pressure. The crude product so obtained was purified by silica gel column chromatography (hexane/ethyl acetate, 10:0.3) to give pure 2-(benzo[d]thiazol-2-yl)-3-bromophenyl benzoate (1a) (0.081 g, yield 79%). The identity and purity of the product was confirmed by spectroscopic analysis.
Acknowledgements
B. K. P acknowledges the support of this research by the Science and Engineering Research Board (SERB) (SB/S1/OC-53/2013), New Delhi, and the Council of Scientific and Industrial Research (CSIR) (02(0096)/12/EMR-II). S.K.S., A.B. and N.K. thank CSIR. We are thankful to Mr H.K. Sahu for Gaussian calculation of substrates.References
- (a) G. Dyker, Handbook of C–H Transformations: Application in Organic Synthesis, Weinheim, 2005 Search PubMed; (b) Activation and Functionalization of C–H Bond, ed. K. I. Goldberg and A. S. Goldman, ACS Symposium Series 885, American Chemical Society, Washington, DC, 2004 Search PubMed; (c) N. Chatani, Directed Metallation, Springer, Berlin, Germany, 2008 Search PubMed; (d) O. Daugulis, H. Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (e) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed.
- (a) J. Huang, L.-T. Li, H.-Y. Li, E. Husan, P. Wang and B. Wang, Chem. Commun., 2012, 48, 10204 RSC; (b) E. Shi, Y. Shao, S. Chen, H. Hu, Z. Liu, J. Zhang and X. Wan, Org. Lett., 2012, 14, 3384 CrossRef CAS PubMed; (c) J. Zhao, H. Fang, W. Zhou, J. Han and Y. Pan, J. Org. Chem., 2014, 79, 3847 CrossRef CAS PubMed; (d) A. Banerjee, S. K. Santra, A. Mishra, N. Khatun and B. K. Patel, Org. Biomol. Chem., 2015 10.1039/c4ob01962h; (e) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed; (f) J. A. Ashenhurst, Chem. Soc. Rev., 2010, 39, 540 RSC.
- (a) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (c) J. J. Li and G. W. Gribble, Palladium in Heterocyclic Chemistry, Pergamon, New York, 1999 Search PubMed; (d) G. Balme, E. Bossharth and N. Monteiro, Eur. J. Org. Chem., 2003, 4101 CrossRef CAS; (e) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644 CrossRef CAS PubMed.
- (a) Y.-J. Liu, H. Xu, W.-J. Kong, M. Shang, H.-X. Dai and J.-Q. Yu, Nature, 2014, 515, 389 CrossRef CAS PubMed; (b) M. D. K. Boele, G. P. F. V. Strijdonck, J. G. D. Vries and P. W. N. M. Leeuwen, J. Am. Chem. Soc., 2002, 124, 1586 CrossRef CAS PubMed; (c) H.-Y. Thu, W.-Y. Yu and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 9048 CrossRef CAS PubMed; (d) B. Xiao, Y. Fu, J. Xu, T. J. Gong, J. J. Dia, J. Yi and L. Liu, J. Am. Chem. Soc., 2010, 132, 468 CrossRef CAS PubMed; (e) Y. Lu, D.-H. Wang, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 5916 CrossRef CAS PubMed; (f) E. J. Yoo, S. Ma, T.-T. Mei, S. L. Chan and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 7652 CrossRef CAS PubMed; (g) A.-G. Rubia, R. G. Arrayas and J. C. Carretero, Angew. Chem., Int. Ed., 2011, 50, 10927 CrossRef PubMed; (h) P. Gandeepan and C. H. Cheng, J. Am. Chem. Soc., 2012, 134, 5738 CrossRef CAS PubMed; (i) H. Li, P. Li and L. Wang, Org. Lett., 2012, 15, 620 CrossRef PubMed.
- Y. Wu, J. Wang, F. Mao and F. Y. Kwong, Chem.–Asian J., 2014, 9, 26 CrossRef CAS PubMed and the references therein.
- (a) N. Sotomayor and E. Lete, Curr. Org. Chem., 2003, 7, 275 CrossRef CAS; (b) Handbook of Grignard Reagents, ed. G. S. Silverman and P. E. Rakita, Dekker, New York, 1996 Search PubMed; (c) M. R. Crampton, in Organic Reaction Mechanism, ed. A. J. Knipe, Wiley, New York, 2007 Search PubMed.
- (a) A. de Meijere and F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH Verlag GmbH & Co.KGaA, Weinheim, Germany, 2004 Search PubMed; (b) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed; (c) J. P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651 CrossRef CAS PubMed; (d) L. C. Campeau and K. Fagnou, Chem. Commun., 2006, 1253 RSC; (e) L. X. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133 CrossRef CAS PubMed; (f) G. P. McGlacken and I. J. S. Fairlamb, Eur. J. Org. Chem., 2009, 4011 CrossRef CAS.
- For electrophilic aromatic substitution, see: (a) R. Taylor, Electrophilic Aromatic Substitution, John Wiley, New York, 1990 Search PubMed; (b) J. Barluenga, G. Rasul and G. A. Olah, J. Am. Chem. Soc., 2004, 126, 15770 CrossRef PubMed; (c) E. B. Merkushev, Synthesis, 1988, 923 CrossRef CAS PubMed.
- (a) D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Org. Lett., 2006, 8, 2523 CrossRef CAS PubMed; (b) S. R. Whitfield and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 15142 CrossRef CAS PubMed; (c) B. Du, X. Jiang and P. Sun, J. Org. Chem., 2013, 78, 2786 CrossRef CAS PubMed; (d) J. J. Li, T. S. Mei and J. Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 6452 CrossRef CAS PubMed; (e) S. R. Whitfield and M. S. Sanford, Organometallics, 2008, 27, 1683 CrossRef CAS; (f) R. Giri, X. Chen and J. Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 2112 CrossRef CAS PubMed; (g) X. B. Wan, Z. X. Ma, B. J. Li, K. Y. Zhang, S. K. Cao, S. W. Zhang and Z. J. Shi, J. Am. Chem. Soc., 2006, 128, 7416 CrossRef CAS PubMed; (h) T. S. Mei, R. Giri, N. Maugel and J. Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 5215 CrossRef CAS PubMed; (i) X. Zhao, E. Dimitrijević and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 3466 CrossRef CAS PubMed; (j) T. S. Mei, D. H. Wang and J. Q. Yu, Org. Lett., 2010, 12, 3140 CrossRef CAS PubMed; (k) F. Kakiuchi, T. Kochi, H. Mutsutani, N. Kobayashi, S. Urano, M. Sato, S. Nishiyama and T. Tanabe, J. Am. Chem. Soc., 2009, 131, 11310 CrossRef CAS PubMed; (l) B. R. Song, X. J. Zheng, J. Mo and B. Xu, Adv. Synth. Catal., 2010, 352, 329 CrossRef CAS; (m) R. B. Bedford, M. F. Haddow, C. J. Mitchell and R. L. Webster, Angew. Chem., Int. Ed., 2011, 50, 5524 CrossRef CAS PubMed; (n) E. Dubost, C. Fossey, T. Cailly, S. Rault and F. Fabis, J. Org. Chem., 2011, 76, 6414 CrossRef CAS PubMed; (o) K. S. L. Chan, M. Wasa, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 9081 CrossRef CAS PubMed; (p) X.-C. Wang, Y. Hu, S. Bonacorsi, Y. Hong, R. Burrell and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 10326 CrossRef CAS PubMed; (q) L. Chu, X.-C. Wang, C. E. Moore, A. L. Rheingold and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 16344 CrossRef CAS PubMed; (r) L. Chu, K.-J. Xiao and J.-Q. Yu, Science, 2014, 346, 451 CrossRef CAS PubMed; (s) A. John and K. M. Nicholas, J. Org. Chem., 2012, 77, 5600 CrossRef CAS PubMed; (t) N. Kuhl, N. Schr€oder and F. Glorius, Org. Lett., 2013, 15, 3860 CrossRef CAS PubMed; (u) D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Tetrahedron, 2006, 62, 11483 CrossRef CAS PubMed.
- (a) A. Banerjee, S. K. Santra, S. Guin, S. K. Rout and B. K. Patel, Eur. J. Org. Chem., 2013, 1376 Search PubMed; (b) A. Banerjee, A. Bera, S. Guin, S. K. Rout and B. K. Patel, Tetrahedron, 2013, 69, 2175 CrossRef CAS PubMed; (c) S. K. Santra, A. Banerjee and B. K. Patel, Tetrahedron, 2014, 70, 2422 CrossRef CAS PubMed; (d) J. S. Yadav, K. Ramesh and B. V. S. Reddy, Synlett, 2011, 2, 169 Search PubMed; (e) S.-J. Lou, D.-Q. Xu, A.-B. Xia, Y.-F. Wang, Y.-K. Liu, X.-H. Du and Z.-Y. Xu, Chem. Commun., 2013, 49, 6218 RSC; (f) Y.-K. Liu, S.-J. Lou, D.-Q. Xu and Z.-Y. Xu, Chem.–Eur. J., 2010, 16, 13590 CrossRef CAS PubMed; (g) Y. Leng, F. Yang, W. Jhu, Y. Wu and X. Li, Org. Biomol. Chem., 2011, 9, 5288 RSC; (h) N. Khatun, A. Banerjee, S. K. Santra, A. Behera and B. K. Patel, RSC Adv., 2014, 4, 54532 RSC.
- (a) K. R. J. Thomas, M. Velusamy, J. T. Lin, C.-H. Chuen and Y.-T. Tao, Chem. Mater., 2005, 17, 1860 CrossRef CAS; (b) K. Serdons, C. Terwinghe, P. Vermaelen, K. Van Laere, H. Kung, L. Mortelmans, G. Bormans and A. Verbruggen, J. Med. Chem., 2009, 52, 1428 CrossRef CAS PubMed; (c) L. E. Seitz, W. J. Suling and R. Ceynolds, J. Med. Chem., 2002, 45, 5604 CrossRef CAS PubMed; (d) S. Louis, M. G. Marc, J. W. Jory and P. B. Joseph, J. Org. Chem., 2003, 68, 4179 CrossRef PubMed; (e) L. S. Jonathan, M. Hiromitsu, M. Toshihisa, M. L. Vincent and F. Hiroyuki, J. Am. Chem. Soc., 2002, 124, 13474 CrossRef PubMed; (f) S. M. Sondhi, N. Singh, A. Kumar, O. Lozach and L. Meijer, Bioorg. Med. Chem., 2006, 14, 3758 CrossRef CAS PubMed.
- S. K. Santra, A. Banerjee, N. Khatun and B. K. Patel, Eur. J. Org. Chem., 2015, 350 CrossRef CAS.
- P. Sadhu, S. K. Alla and T. Punniyamurthy, J. Org. Chem., 2013, 78, 6104 CrossRef CAS PubMed.
- http://www.gaussian.com/g_tech/g_ur/m_citation.htm.
- (a) H. D. Arman, R. L. Gieseking, T. W. Hanks and W. T. Pennington, Chem. Commun., 2010, 46, 1854 RSC and the references therein; (b) S. K. Sahoo, H. S. Jena, G. Majji and B. K. Patel, Synthesis, 2014, 46, 1886 CrossRef PubMed.
- (a) D. C. Powers, M. A. L. Geibel, J. E. M. N. Klein and T. Ritter, J. Am. Chem. Soc., 2009, 131, 17050 CrossRef CAS PubMed; (b) D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302 CrossRef CAS; (c) D. C. Powers and T. Ritter, Acc. Chem. Res., 2012, 45, 840 CrossRef CAS PubMed.
- (a) N. R. Deprez and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 11234 CrossRef CAS PubMed; (b) J. M. Racowski, A. R. Dick and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 10974 CrossRef CAS PubMed; (c) P. A. Sibbald, C. F. Rosewall, R. D. Swartz and F. E. Michael, J. Am. Chem. Soc., 2009, 131, 15945 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra. See DOI: 10.1039/c4ra15461d |
| This journal is © The Royal Society of Chemistry 2015 |