
Piperine promotes ultraviolet (UV)-B-induced cell death in B16F10 mouse melanoma cells through modulation of major regulators of cell survival
Rather A. Rafiqa,
Bashir A. Ganaic and
Sheikh A. Tasduq*ab
aPK-PD and Toxicology Division, CSIR-Indian Institute of Integrative Medicine, Council of Scientific and Industrial Research (CSIR), Canal Road, Jammu Tawi, Jammu and Kashmir, India. E-mail: stabdullah@iiim.ac.in; tasduq11@gmail.com; Tel: +91-1912569000-10 ext. 332 Tel: +91-9419148712
bAcademy of Scientific and Innovative Research (AcSIR), New Delhi, India
cCentre of Research for Development (CORD), University of Kashmir, Srinagar, Jammu and Kashmir, India
First published on 8th January 2015
Abstract
An increase in the incidence of melanoma and its resistance to currently applied treatment regimes enhance the need for novel therapeutic agents and treatment modalities. In this study, we report that piperine, the most widely consumed dietary alkaloid, promotes cell death in ultraviolet (UV)-B-irradiated B16F10 mouse melanoma cells through the elevation of intracellular reactive oxygen species (ROS) formation, calcium homeostasis imbalance, and loss of mitochondrial membrane potential. Promotion of UVB-induced cell death by piperine was further revealed by caspase activations, poly(ADP) ribose polymerase cleavage, DNA fragmentation, and an increase in sub-G1 cells. Piperine promoted UVB-induced translocation of Bax from cytosol to mitochondria, accelerated an increase in the ratio of Bax to that of Bcl-2, and up-regulated the expression of apoptosis-inducing factor (AIF). These effects of piperine on UVB-irradiated cells were associated with apparent alterations in the expression of mitogen activated protein (MAP) kinase family proteins and PI3K–Akt survival signals. Piperine disrupted NF-κB signaling, inhibited UVB-induced nuclear translocation of NF-κB, and potentially reversed multi drug resistance (MDR) by reducing UVB-induced p-glycoprotein activity. Taken together, these results suggest the possibility of using piperine in combination with UVB as a possible therapeutic option for melanoma.
1. Introduction
Ultraviolet (UV)-B radiation-induced skin damage has been linked to a plethora of skin cancers, melanoma being highly lethal and aggressive among them.1 Median survival rates for patients with metastatic melanoma are extremely poor.2 Therefore, novel therapeutic agents and treatment modalities are needed to reduce this disease burden.Ultraviolet (UV)-B has high energy which causes DNA damage and cell death in several cell culture models as well as in human skin.3 The ability of skin cells to respond to and to mitigate UVB-induced DNA damage is crucial for skin cell homeostasis. Failure to repair the damaged DNA is the principal cause of skin cancer.4,5 UVB-induced cell death has been recognized as a complex process in which a variety of signaling pathways are involved.6 UV irradiation induces the formation of reactive oxygen species (ROS) which are highly detrimental for the mitochondrial physiology, contributing to the dissipation of mitochondrial membrane potential and release of pro-apoptotic triggers such as cytochrome c, Smac/DIABLO and apoptosis inducing factor (AIF).3 Further to these molecular events, Bcl-2 family proteins play a vital role in modulating apoptosis by regulating mitochondrial membrane permeabilization in response to many types of stress or death stimuli.7 Two additional signaling pathways known to be crucial in the response of cells to UVB irradiation include the PI3K–Akt pathway and mitogen activated protein (MAP) kinase pathway.8,9 NF-κB pathway is another pathway that melanoma tumors use to achieve survival, proliferation and apoptotic resistance.10 Cell cycle regulation is crucial for cell proliferation and in the maintenance of cellular homeostasis in response to exogenous genotoxic stressors.11
Piperine (1-piperoylpiperidine) is one the most important dietary alkaloids due to its occurrence in widely consumed black (Piper nigrum L.) and long (Piper longum L.) peppers as well as its biological activities (anti-inflammatory, anti-metastatic and anti-cancer activities).12–15 In addition, piperine has been reported as the potential functional food to improve mood and cognitive disorders.16 Recently, there has been a major focus on the use of dietary molecules in combination with UV light as possible therapeutic option for melanoma and other types of skin cancers. Indole-3-carbinol, a nutrient molecule found in cruciferous vegetables, has been reported to enhance the UVB-induced apoptosis by sensitizing SK-MEL-2 human melanoma cells.17 More recently, silibinin has been shown to act as a potent sensitizer of UVA radiation-induced apoptosis in human HaCaT keratinocytes via enhancing ROS generation and ER stress.18 Such agents that can enhance UVB-induced DNA damage and/or other biological events occurring following UVB exposure could of potential importance in killing tumor cells.
Here we have used B16F10 cells as a model to investigate the mechanistic basis for the pro-apoptotic effect of piperine in UVB-irradiated melanoma cells. We found that piperine, otherwise a safe alkaloid, acts as a potent UVB photosensitizer to cause programmed cell death in B16F10 murine melanoma cells.
2. Materials and methods
2.1 Reagents
Dulbecco's modified Eagle's media (DMEM), fetal bovine serum (FBS), penicillin–streptomycin, trypsin–EDTA, Dulbecco's phosphate buffer saline (Dulbecco's PBS), 3-(4,5-dimetylthiazol-yl)-diphenyl tetrazolium bromide (MTT), Hank's balanced salts modified, piperine, propidium iodide, rhodamine 123, 2,7-dichlorodihydrofluoresceindiacetate (H2DCF-DA), proteinase K, ribonuclease A (RNase A), β-nicotinamide adenine dinucleotide reduced disodium salt (β-NADH), sodium pyruvate, Pluronic F-127, anti-β-actin antibody, anti-VDAC antibody and Fluo-3 AM were obtained from Sigma-Aldrich Chemicals (St. Louis, MO). Antibodies against NF-κB, Bax, Bcl-2, cytochrome c, caspase-3, caspase-8, AIF, PARP, B-Raf, ERK, MEK, Akt, PI3K, and GAPDH were obtained from Santa Cruz Biotechnology (Santa Cruz Biotechnology, Inc). Histone H3 antibody was obtained from Cell Signaling Technology.Stock solution of piperine was prepared in dimethyl sulfoxide (DMSO) and diluted to final concentration in the culture medium. Final concentration of DMSO employed as vehicle never exceeded 0.1% (v/v) and had no discernible effects on B16F10 cells.
2.2 Cell culture
Mouse melanoma cell line, B16F10, human keratinocytes cell line, HaCaT, human fibroblast cell line, Hs68 and human epidermoid carcinoma cell line, A431 were obtained from American Type Culture Collection (ATCC, Rockville, MD, USA) and maintained in a monolayer culture in 95% air/5% CO2 at 37 °C in DMEM supplemented with 10% (v/v) fetal bovine serum (FBS), 0.022% sodium pyruvate, 0.26% sodium bicarbonate, 0.012% penicillin G and 0.027% streptomycin. Cells were treated with piperine at predetermined concentrations 24 h prior UVB irradiation and 24 h post UVB irradiation, except otherwise specified. For experiments the percentage of fetal bovine serum was reduced to 5%, except otherwise mentioned.2.3 UVB irradiation
UVB irradiation was performed as described previously19 using Daavlin UVA/UVB Research Irradiation Unit (Bryan, OH, USA) with UVB lamps (peak emission at 314 nm). UVB irradiation was performed in culture dishes containing a thin layer of prewarmed Dulbecco's PBS (pH 7.4). Control cells were identically processed but not irradiated. On the other hand, UVB control cells were irradiated with UVB but not exposed to piperine.2.4 MTT dye uptake method
The general viability of cells was determined by MTT assay as described previously.192.5 Lactate dehydrogenase leakage assay
LDH leakage was measured as described previously.20 LDH in the culture supernatant (130 μL) was measured with 0.2 mM β-NADH and 0.4 mM pyruvic acid upto 200 μL Dulbecco's PBS (pH 7.4). LDH concentration in the culture supernatant was proportional to the rate of NADH oxidation measured by the absorbance at 334 nm (OD/min) using a Multiskan Spectrum (Thermo Electron Corporation). LDH concentration in the culture medium was calculated as percentage fold change.2.6 DNA fragmentation assay
DNA fragmentation analysis was carried out as described previously.21 The cells were collected (including floating cells) and washed in Dulbecco's PBS containing 10 mM EDTA. The pellet was suspended in lysis buffer [10 mM Tris–HCl (pH 8.0), 100 mM NaCl, 5 mM EDTA, 5% triton X-100, 0.25% SDS, 400 μg mL−1 RNase] and incubated at 37 °C for 90 minutes, followed by incubation with proteinase K (200 μg mL−1) at 50 °C for further 1 hour. DNA was extracted with phenol–chloroform–isoamyl alcohol (25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :24:1); recovered from aqueous phase with chilled alcohol containing 0.3 M sodium acetate, washed in 80% alcohol, dried and dissolved in 50 μL Tris–EDTA buffer. The DNA pellet was 1.8% gel electrophoresed and visualized by ethidium bromide staining.
:24:1); recovered from aqueous phase with chilled alcohol containing 0.3 M sodium acetate, washed in 80% alcohol, dried and dissolved in 50 μL Tris–EDTA buffer. The DNA pellet was 1.8% gel electrophoresed and visualized by ethidium bromide staining.
2.7 Determination of intracellular ROS production
Reactive oxygen species (ROS) formation was determined as described previously.22 Equal number of B1610 cells were dispended per well of 96 well black plate. After being pretreated with piperine for 24 hours, cells were covered with a thin layer of Dulbecco's PBS and irradiated with 10 mJ cm−2 of UVB. Immediately after UV irradiation 5 μM H2DCF-DA reagent was added and formation of ROS was measured by the change in fluorescence due to the production of 2′,7′-dichlorofluorescein (DCF) at excitation and emission wavelengths of 488 and 525 nm respectively with Fluorescence Spectrometer (Perkin Elmer, LS, 55, USA).2.8 Measurement of intracellular Ca2+ levels
The measurement of intracellular Ca2+ was carried as described previously,23 with minor modifications. After being pre-treated with piperine for 24 hours, B16F10 cells were covered with a thin layer of Dulbecco's PBS and then irradiated with UVB. Immediately after UVB irradiation, cells were washed with Dulbecco's PBS and loaded with Fluo-3 AM (3 μM) in Hank's balanced salt solution (HBSS) containing 0.02% Pluronic F-127 and incubated at 37 °C for 20 minutes. After incubation, 1.5 mL of HBSS containing 1% FBS was added, and incubated at 37 °C for further 40 minutes. Cells were then washed with HEPES buffer (10 mM, pH 7.2) containing 0.1% BSA, trypsinized, collected and washed with Dulbecco's PBS. Cells were resuspended in Dulbecco's PBS and equal number of cells were transferred to 96 well black plate for the measurement of fluorescence (ex: 488 nm and em: 526 nm) in a spectrofluorimeter (Perkin Elmer LS, 55). Intracellular Ca2+ was expressed as fold change in fluorescent intensity (1 × 106 cells) relative to control.2.9 Measurement of mitochondria membrane potential
The changes in mitochondrial membrane potential (ΔΨM) were measured after staining of cells with rhodamine 123 (5 μM) for 15 minutes. Cells were harvested (including floating cells), washed in Dulbecco's PBS and resuspended in desired volume of Dulbecco's PBS. The intensity of fluorescence was measured by BD FACS Calibur Aria.2.10 Cell cycle analysis
Cell cycle analysis was performed as described previously.24 After a 90 minute incubation of cells in staining solution (50 μg mL−1 of propidium iodide in Dulbecco's PBS containing 10 μg mL−1 DNase free RNase A), propidium iodide (PI) fluorescence was measured using a BD FACS Calibur Aria.2.11 Western blotting
Whole cell lysates were prepared as described previously25 using RIPA buffer (Sigma; R-0278). Methods used for subcellular fractionation were similar to the methods described in literature.26,27 Western blotting was performed using equal amounts of protein. After blocking the PVDF membranes (Immobilon P 0.45 μm, Millipore) with 5% skim milk or 3% BSA in Tris-buffered saline (pH 8.0) for 2 h at room temperature, membranes were reacted overnight with specific antibodies in the same blocking solution at 4 °C. After extensive washing for 30 minutes with Tris-buffered saline containing 0.05% Tween 20, membranes were reacted with different HRP conjugated secondary antibodies for detection of desired proteins. Finally, blots were assayed by Super Signal- (West Femto Maximum Sensitivity Substrate, Thermo Scientific) mediated chemiluminescence by ChemiDoc™ XRS+ (Bio Rad). Analysis of differential protein expression was achieved by densitometry of specific bands with the Image Lab™ Software version 3 (Bio Rad). The correct protein loading was determined after stripping the membrane with Restore Western Blot Stripping Buffer (Thermo Scientific, #21059) for 15 minutes at 37 °C and then reprobing the membrane for appropriate loading control. Normalization of different bands against their respective loading controls was done with Image Lab™ software.2.12 Rhodamine 123 efflux assay
Rhodamine 123 (RH 123) efflux assay was performed as described previously.28 Briefly, the culture medium was changed to Hank's balanced salt solution and the cells were incubated at 37 °C for 30 minutes. After incubating with RH 123 (20 μM) for 90 minutes, the cells were washed thrice with chilled Dulbecco's PBS (pH 7.4) and lysed in lysis buffer. The fluorescence of the RH 123 cell lysates was measured at excitation and emission wavelengths of 480 and 540 nm respectively. Fluorescence values were normalized to the total protein content of each lysate and are presented as the ratio to control values.2.13 Statistical analysis
Numerical data were presented as mean ± standard error (SE). Statistical differences between control (untreated) versus all other groups (*) and UVB control (UVB-alone treatment) versus piperine plus UVB-treatments (#) were analyzed by using Student's t-test (SlideWrite V7.01). A p value <0.05 was considered statistically significant.3. Results
3.1 Piperine increases UVB-induced cell death in B16F10 melanoma cells
We first studied the cytotoxic effects of UVB radiations on B16F10 cells. B16F10 cells were irradiated with different doses of UVB (1, 5, 10, 20, 30, and 40 mJ cm−2) for 24 hours and cell viability was analyzed by MTT assay. UVB (1 mJ cm−2) had an insignificant effect on cell viability, whereas UVB (40 mJ cm−2) produced a highly cytotoxic effect and induced cell death in 82% of cell population (Fig. 1A). After proper titration of various irradiation doses, we selected a sub-lethal UVB dose of 10 mJ cm−2 for further experiments. UVB (10 mJ cm−2) induced cell death in around 40% of irradiated cell population. While lower concentrations of piperine (5 and 20 μM) had little effect on the cell viability of B16F10 cells, a moderate growth inhibition was observed at higher concentrations (30 and 40 μM) (Fig. 1B). To show the selectivity or specificity in the pro-apoptotic effects of piperine, HaCaT keratinocytes, Hs68 fibroblasts and A431 epidermoid carcinoma cells were treated with piperine, but no significant growth inhibition was observed in any case (Fig. 1C–E). Further, piperine caused no significant enhancement of cell death in UVB-irradiated HaCaT, Hs68 or A431 cells (Fig. 1F–H). Interestingly, treatment of UVB-irradiated B16F10 cells with piperine resulted in enhanced and dose dependent decrease in cell viability. 5, 20, 30 and 40 μM piperine added at 24 hour prior and subsequent to UVB irradiation caused 47%, 46%, 61% and 73% cell killing respectively (Fig. 1I). These results indicate that piperine selectively promotes cell death in UVB-irradiated B16F10 melanoma cells, without causing or promoting UVB-induced cell death in non-tumorigenic skin cells and non-melanoma cells. Next, to determine the effect of piperine on UVB-induced necrotic death, LDH leakage analysis was done. Piperine (5 μM) had little effect on LDH activity, whereas 20, 30, and 40 μM piperine documented moderate but insignificant increase in LDH activity (Fig. 1J). It is interesting to note that the treatment of UVB-irradiated B16F10 cells with 5, 20, 30, and 40 μM piperine showed 1.4-, 1.8-, 2- and 2.1-fold increase in LDH activity (Fig. 1J). A very low increase in LDH activity precludes necrosis as a potential mode of cell death and suggests the engagement of other kinds of cell destruction. | ||
| Fig. 1 Piperine promotes UVB-induced cell death. (A) Analysis of cell viability using the MTT assay at 24 h post-UVB in B16F10 cells treated with different doses of UVB (mJ cm−2). *P < 0.05 for control versus UVB treatments. (B) Analysis of cell viability using the MTT assay in B16F10 cells treated with indicated concentrations of piperine. *P < 0.05 for control versus piperine treated. (C) Analysis of cell viability using the MTT assay in HaCaT cells treated with different concentrations of piperine. (D) Analysis of cell viability in Hs68 cells treated with different concentrations of piperine. (E) Analysis of cell viability in A431 cells treated with different concentrations of piperine. (F) Analysis of cell viability in HaCaT cells using the MTT assay treated with piperine at 24 h pre- and post-UVB irradiation. (G) Analysis of cell viability in Hs68 cells treated with piperine at 24 h pre- and post-UVB irradiation. (H) Analysis of cell viability in A431 cells treated with piperine at 24 h pre- and post-UVB irradiation. (I) Analysis of cell viability using the MTT assay in B16F10 cells treated with piperine at 24 h pre- and post-UVB irradiation. *P < 0.05; **P < 0.01 for control versus treated; #P < 0.05; ##P < 0.01 for UVB-alone treatment versus UVB + piperine treatments. Un-irradiated control taken as 100% viable is not shown. (J) Effect of piperine on UVB-induced membrane disintegration by LDH leakage assay in cells treated with piperine at 24 h pre- and post-UVB irradiation. To detect LDH leakage into the culture supernatant, serum free medium was used. *P < 0.05 for control versus all other treatments; #P < 0.05 for UVB-alone treatment versus UVB + piperine treatment. (K) Effect of piperine on UVB-induced DNA fragmentation treated with piperine at 24 h pre- and post-UVB irradiation. (L) Immunoblot analysis of caspase-3/caspase-8 activation and PARP cleavage in cells treated with piperine at 24 h pre- and post-UVB irradiation. GAPDH was used as loading control. The experiment was repeated thrice independently and representative blot was displayed. Relative band intensities of cleaved forms of caspase-3 (M), caspase-8 (N) and PARP-1 protein (O) were quantified using Image Lab™ Software Version 3.0 (BioRad). *P < 0.05; **P < 0.01 for control versus all treatments; #P < 0.05; ##P < 0.01 for UVB-alone treatment versus UVB + piperine treatments. | ||
Next we performed DNA fragmentation analysis to determine whether the decrease in cell viability is caused by DNA fragmentation, an important hallmark of apoptosis. Piperine induced no cleavage of chromosomal DNA. However, treatment of UVB-irradiated cells with piperine caused remarkable DNA fragmentation with banding pattern almost similar to UVB-alone treated cells (Fig. 1K). Next, we examined the effect of piperine on UVB-induced expression of pro-apoptotic markers such as cleaved caspases and cleaved PARP. Western blot analysis showed that piperine at different doses increased UVB induced caspase-3/caspase-8 activation and PARP cleavage (Fig. 1L–O). These data suggest the enhanced pro-apoptotic effects of piperine on UVB irradiated B16F10 cells.
3.2 Piperine promotes UVB-induced mitochondrial membrane potential (ΔΨM) dissipation and favors release of cytochrome c and apoptosis inducing factor (AIF)
Mitochondrial physiology is impaired in cells undergoing either apoptosis or necrosis, often leading to the release of cytochrome c, a key regulator of caspases.29 However, the effects of piperine on mitochondrial physiology of UVB-irradiated B16F10 cells are largely unknown. UVB (10 mJ cm−2) caused 4.85-fold increase in the percentage of ΔΨM low cells relative to control. It is interesting to note the treatment of UVB-irradiated B16F10 cells with 5, 20 and 30 μM piperine induced a 3.53-, 3.38- and 2.79-fold increase in the percentage of ΔΨM low cells relative to 5, 20, and 30 μM piperine-alone treated cells respectively (Fig. 2A and B). Further, we found a marked accumulation of cytosolic cytochrome c and apoptosis-inducing factor (AIF) in cells treated with piperine and UVB than the cells receiving either UVB or piperine only (Fig. 2C–E). AIF is known to induce DNA fragmentation and chromatin condensation independent of caspase activation. | ||
| Fig. 2 Piperine promotes UVB-induced mitochondrial membrane potential dissipation. (A) Flow cytometric analysis of mitochondrial membrane potential (ΔΨM) dissipation using rhodamine 123 in cells treated with piperine at 24 h pre- and post-UVB (10 mJ cm−2) irradiation. (B) Represents the fold change in the percentage of ΔΨM low cells relative to control and piperine-alone treated cells as specified in the bar graph. (C) Cell lysates were prepared from various treatment groups and subjected to immunoblot analysis for cytosolic cytochrome c and AIF and quantified by densitometric analysis (D & E). *P < 0.05; **P < 0.01 for control versus all other treatments; #P < 0.05; ##P < 0.01 for UVB-alone treatment versus UVB + piperine treatments. | ||
3.3 Piperine increases sub-G1 population of UVB-irradiated cells
Here, we have studied the effects of piperine on cell cycle progression in UVB-irradiated B16F10 cells. UVB irradiation of B16F10 cells caused a 13-fold increase in the percentage of cells entering apoptosis (sub-G1 cells) relative to control. The sub-G1 cells arise as a result of DNA degradation in the late stages of apoptotic cell death. However, the treatment of UVB-irradiated B16F10 cells with 5, 20, 30 and 40 μM piperine caused around 7.16-, 6.37-, 6.12- and 5.61-fold increase in the percentage of sub-G1 cells relative to 5, 20, 30, and 40 μM piperine-alone treated cells respectively (Fig. 3A and B). These results indicate that piperine is fairly cytotoxic to B16F10 cells, and addition of piperine at 24 h pre- and post-UVB irradiation induced many fold increase in the percentage of sub-G1 cells. | ||
| Fig. 3 Piperine increases sub-G1 population affected by UVB irradiation. (A) Flow cytometric analysis of cell cycle progression in cells treated with piperine at 24 h pre- and post-UVB irradiation. Cells were harvested, stained with propidium iodide and analyzed for DNA content by BD FACS Calibur Aria. (B) Represents the fold change in the percentage of sub-G1 cells relative to control and piperine-alone treated cells as indicated in the analyzed bar graph. | ||
3.4 Piperine promotes reactive oxygen species (ROS) generation and elevation of intracellular free calcium in UVB-irradiated cells
To determine whether the difference in apoptotic cell death was associated with reactive oxygen species (ROS) generation, we analyzed reactive oxygen species production by fluorescence spectrometry. UVB (10 mJ cm−2) caused 2.6-fold increase in ROS generation relative to un-irradiated control cells. It is interesting to note that the treatment of B16F10 cells with 5, 20, 30, and 40 μM piperine prior to UVB irradiation caused 4.36-, 4.05-, 4.65-, and 5.99-fold increase in UVB-induced ROS generation respectively (Fig. 4A). Since intracellular Ca2+ has been reported as the first trigger that stimulates ROS formation in UVB-irradiated cells,30 we next analyzed the effect of piperine on intracellular free calcium. UVB-irradiation induced a 3.98-fold increase in intracellular free calcium relative to control cells. However, the treatment of UVB-irradiated B16F10 cells with 5, 20, 30, and 40 μM piperine caused 5.33-, 7.22-, 8.13-, and 9.37-fold increase in intracellular free calcium respectively (Fig. 4B). These results suggested the involvement of oxidative stress in the pro-apoptotic effects of piperine in UVB-irradiated cells. Further to these results, we found that cells treated with piperine prior and subsequent to UVB irradiation depicted a moderate decrease in the expression of NF-κB while the cells that received piperine treatment only exhibited almost a similar level of expression (Fig. 4C). We, however, further found that treatment of cells with piperine markedly inhibited the nuclear translocation of NF-κB induced by UVB irradiation (Fig. 4F and G). This is a condition likely to render cells susceptible to apoptosis.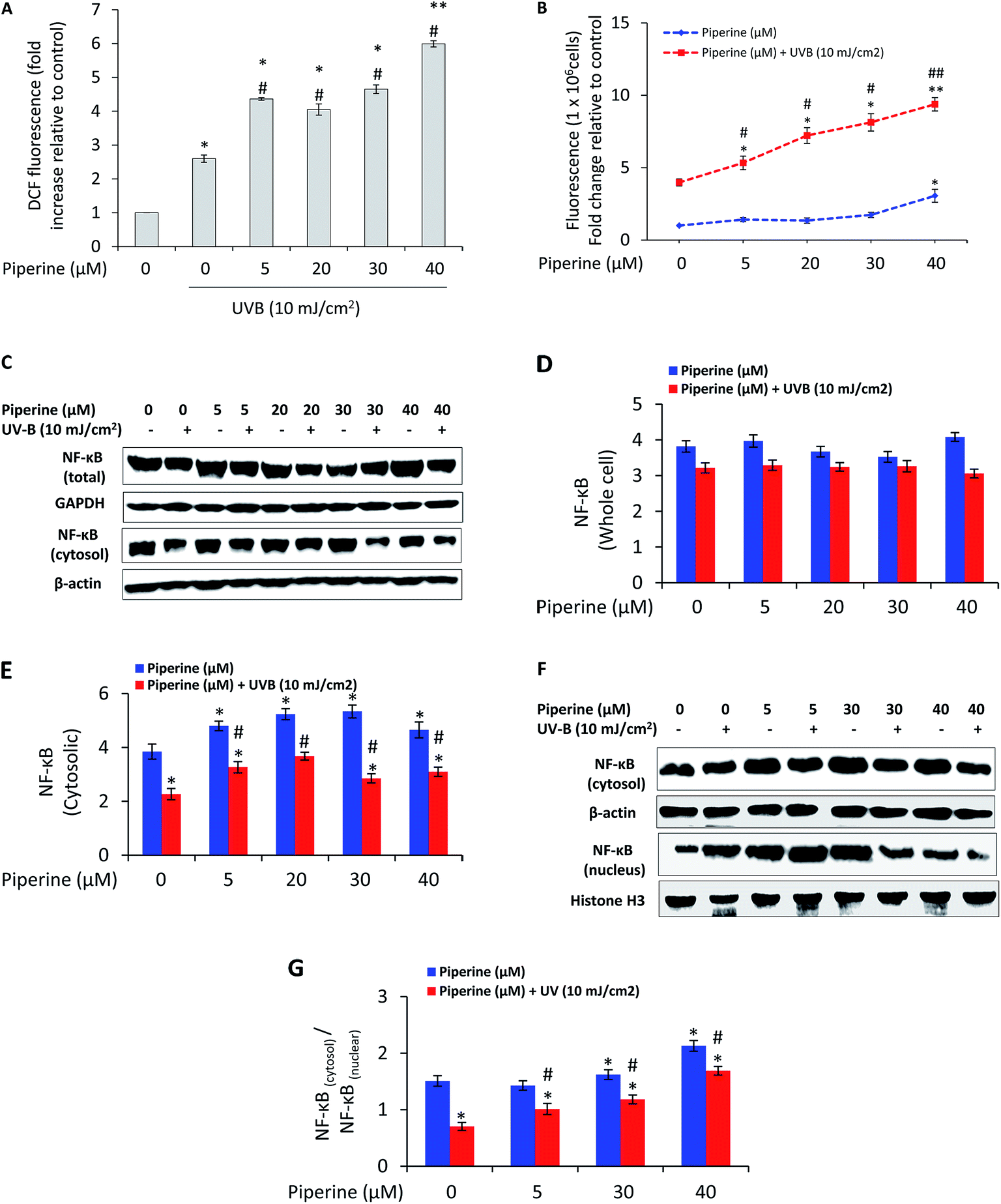 | ||
| Fig. 4 Piperine synergizes the effect of UVB in elevating intracellular Ca2+ levels and the production of ROS. (A) Measurement of intracellular ROS production in cells immediately after exposure to UVB pre-treated with or without piperine. Immediately after UVB exposure, 5 μM H2DCFDA reagent was added and incubated for 20 minutes at 37 °C. The generation of ROS was measured by the change in fluorescence due to the production of 2′,7′-dichlorofluorescein (DCF). Fluorescence intensity was read at excitation and emission wavelengths of 488 and 525 nm respectively. *P < 0.05; **P < 0.01 for control versus all other treatments; #P < 0.05 for UVB-alone treatment versus UVB + piperine treatments. (B) Measurement of intracellular calcium levels in cells immediately after exposure to UVB pre-treated with or without piperine. Soon after UVB irradiation, cells were loaded with Fluo-3 AM (3 μM) in HBSS containing 0.02% Pluronic F127 and incubated at 37 °C for 20 minutes. After incubation, HBSS containing 1% FBS was added to cells, and incubated at 37 °C for further 40 minutes. Cells were then washed with HEPES buffer containing 0.1% BSA, trypsinized, collected and washed with Dulbecco's PBS. Cells were resuspended in Dulbecco's PBS and equal numbers of cells were transferred to 96 well black plate for the measurement of fluorescence (ex: 488 nm and em: 526 nm) in a spectrofluorimeter (Perkin Elmer LS55). Intracellular Ca2+ was expressed as fold change of fluorescence relative to control. *P < 0.05 for control versus all other treatments; #P < 0.05 for UVB-alone treatment versus UVB + piperine treatment. (C) Immunoblot analysis and quantification of total NF-κB (D), and cytosolic NF-κB (E). (F) Immunoblot analysis of cytosolic and nuclear NF-κB. Signals were quantified using Image Lab™ Software Version 3.0 (BioRad) and expressed as ratio of NF-κB(cytosol) to NF-κB(nucleus) (G) for each treatment. *P < 0.05; **P < 0.01 for control versus all other treatments; #P < 0.05 for UVB-alone treatment versus UVB + piperine treatments. | ||
3.5 Piperine modulates the expression profile and translocation pattern of Bcl-2 family proteins
UVB irradiation has been reported to induce translocation of Bax from cytosol to mitochondria.31 However, the effects of piperine on translocation of Bax from cytosol to mitochondria in UVB-irradiated B16F10 cells are largely unknown. We measured this event by western blotting and our results suggested that piperine enhanced the UVB-induced Bax translocation from cytosol to mitochondria (Fig. 5C and D). Further, a moderate upregulation of Bax was detected in cells treated with piperine prior and subsequent to UVB irradiation than the cells receiving all other treatments (Fig. 5A). We further studied the effects of piperine on Bcl-2, an anti-apoptotic multidomain member of Bcl-2 family. Our results indicate that treatment of UVB-irradiated cells markedly reduced the expression of Bcl-2 and its cytosolic accumulation compared to all other treatments (Fig. 5A, E and F). Overall, these results suggest an increment in the ratio of Bax to that of Bcl-2 (Fig. 5A and B) accounting for the pro-apoptotic effects of piperine in UVB irradiated B16F10 cells. | ||
| Fig. 5 Piperine promotes the UVB-induced translocation of Bcl-2 family proteins and impairs the major protein regulators of cell survival. (A) Immunoblot analysis of Bax (total) and Bcl-2 (total) in cells at 24 h post-UVB (10 mJ cm−2) previously treated with or without piperine. Signals were quantified and expressed as Bax (total) to Bcl-2 (total) ratio (B). (C) Immunoblot analysis of cytosolic and mitochondrial Bax in cells at 24 h post-UVB pretreated with or without piperine at indicated concentrations. Signals were quantified using Image Lab™ Software Version 3.0 (BioRad) and expressed as ratio of Bax(mitochondria) to Bax(cytosol) (D) for each treatment. (E) Immunoblot analysis and quantification (F) of cytosolic Bcl-2 in cells treated with piperine at 24 h pre- and post-UVB irradiation. (G) Immunoblot analysis of PI3K, phospho-Akt and Akt in cells treated with piperine at 24 h pre- and post-UVB (10 mJ cm−2) irradiation. Signals were quantified for PI3K (H) and phospho-Akt (I) and normalized against the expression of GAPDH and total Akt respectively. (J) Immunoblot analysis of B-Raf, phospho-MEK, MEK, phospho-ERK and ERK in cells treated with piperine at 24 h pre- and post-UVB irradiation. Signals for B-Raf (K), p-MEK (L) and p-ERK (M) were quantified using Image Lab™ Software Version 3.0 (BioRad) and normalized against GAPDH, total MEK and total ERK respectively. | ||
3.6 Piperine influences PI3K–Akt and MAPK survival signals in UVB-irradiated B16F10 melanoma cells
PI3K–Akt pathway impacts on cell growth and survival and has recently been a focus of intense research.32 We studied the effects of piperine on the expression of PI3K and Akt in UVB-irradiated cells. Our results indicate that in UVB-irradiated cells, piperine dramatically reduced the expression of PI3K and its downstream effector phospho-Akt than the cells receiving all other treatments (Fig. 5G–I). In cells stimulated to die in response to UVB-irradiation, Ras–Raf–MEK–ERK signaling pathway plays a determinant role. In our experiment, we found that the treatment of UVB-irradiated B16F10 cells with piperine reduced the expression of B-Raf, without altering the total MEK and ERK levels (Fig. 5J–K). However, piperine reduced the expression of phospho-MEK and phospho-ERK in UVB-irradiated B16F10 cells than the cells receiving either UVB or piperine-alone treatment (Fig. 5J, L and M).3.7 Piperine inhibits the natural action of p-glycoprotein in UVB-irradiated cells
Highly lethal phenotype associated with malignant melanoma is, in part, due to p-glycoprotein, a 170 kDa membrane anchored protein implicated as the primary cause of multidrug resistance (MDR) in tumors where it acts as an efflux pump for chemotherapeutic agents.33 Here, we determined the effect of piperine on p-glycoprotein activity using rhodamine 123 efflux assay. UVB irradiation stimulated p-glycoprotein activity (Fig. 6A). It is interesting to note that treatment of cells with piperine reduced the UVB-induced p-glycoprotein activation by about 0.85-, 0.93-, 0.96- and 1.008-fold respectively. Our results indicate that piperine may act as a useful agent to reverse the UVB-induced p-glycoprotein activity. | ||
| Fig. 6 Piperine reverses the UVB-induced p-glycoprotein activity. (A) Analysis of p-glycoprotein activity using the RH 123 efflux assay in B16F10 cells treated with piperine at 24 h pre- and post-UVB irradiation. Intracellular RH 123 retention was expressed as fold change of fluorescence intensity normalized by mg protein against control cells. *P < 0.05 for control versus treatments; #P < 0.05 for UVB-alone treatment versus UVB + piperine treatments. | ||
4. Discussion
In the present study, piperine was explored for its potential to promote cell death in UVB-irradiated B16F10 melanoma cells. In melanoma therapy terms, it is important to specifically kill tumor cells while sparing normal cells to achieve maximum therapeutic benefit. Therefore, we considered normal human fibroblast cell line, Hs68, which represent an ubiquitous cell type, and normal human keratinocytes cell line, HaCaT, which represent a major cell type of the skin, as a more appropriate model for non-tumorigenic skin cells. We also studied the key effects of piperine in UVB-irradiated A431 epidermoid carcinoma cells, a form of non-melanoma skin cancer. We found that piperine specifically promoted cell death in UVB-irradiated B16F10 melanoma cells, without inducing cell death in non-tumorigenic skin cells and non-melanoma cells. In a therapeutic perspective, these results are important considering that targeting cancerous cells should not significantly damage normal cells. In order to investigate the mechanism(s) supporting the apoptotic process, we analyzed reactive oxygen species (ROS) production and intracellular Ca2+ homeostasis. Intracellular ROS and Ca2+ represent the most important signaling molecules that cross-talk to participate in the regulation and integration of diverse cellular functions, including cell survival and death.34,35 Our results indicated that piperine potentiated UVB-induced oxidative stress and potentially elevated intracellular free Ca2+ in UVB-irradiated B16F10 cells (Fig. 4A and B). This pro-oxidant effect of piperine may be one of the factors responsible for the anti-proliferative activity of piperine in UVB-irradiated B16F10 cells. These results are in consistence with the literature citing ROS as a key factor in triggering cell death36 and the pro-oxidant behavior of piperine.37 There are also indications that calcium mediated ROS production causes ΔΨM dissipation and subsequent activation of downstream caspase cascade.38 Interestingly, we observed that piperine substantially decreased the ΔΨM in UVB-irradiated cells (Fig. 2A). Further, when UVB-irradiated B16F10 cells were treated with piperine, a significant increase in the percentage of sub-G1 cells was observed (Fig. 3A), which also corroborated with the enhancing effect of piperine on UVB-induced cell death.The proteins of the Bcl-2 family are critical regulators of mitochondrial outer membrane permeabilization,7 and are primarily composed of two types of proteins: (i) the prosurvival Bcl-2 proteins, such as Bcl-2 itself, Bcl-2-like 1 (Bcl-XL) and myeloid cell leukemia-1 (Mcl-1), which block cell death through the direct interactions with the proapoptotic members; and (ii) the executioners Bax and Bak, which are thought to participate in mitochondrial outer membrane permeabilization during apoptosis.39 The balance between positive and negative apoptotic regulators is critical to turning on and off the cellular apoptotic machinery. Based on the results and observations in the present study, it may be suggested that piperine modulated the protein expression as well as altered the translocation of Bcl-2 family proteins in response to UVB-irradiation in deciding its pro-apoptotic effect in B16F10 cells (Fig. 5). Furthermore, the observation that piperine promoted ΔΨM dissipation in UVB-irradiated cells is in agreement with the enhancing effect of piperine on UVB-induced activation of caspases (Fig. 1L) and accumulation of cytosolic cytochrome c (Fig. 2C and D). Consistent with other results, we also observed that piperine caused a marked increase in the expression of apoptosis inducing factor (AIF) in UVB-irradiated B16F10 cells (Fig. 2C and E). Together, these results suggest the probable involvement of caspase-dependent and caspase-independent mechanisms in the pro-apoptotic effects of piperine in UVB-irradiated B16F10 cells.
We further determined the effect of piperine on nuclear factor-kappa B (NF-κB). NF-κB is a nuclear transcription factor that primarily resides in the cytoplasm of a cell and translocates to the nucleus when activated. Its activation is induced by a wide variety of agents including UV radiation-induced oxidative stress.10,40 NF-κB is responsible for the induction of anti-apoptotic proteins such as Bcl-2 and Bcl-XL depending upon the cellular context.41 In our experiment, the expression of NF-κB decreased following treatment of UVB-irradiated B16F10 cells with piperine, which together with the inhibition of NF-κB nuclear translocation may interfere with expression of Bcl-2, thereby contributing to inducing cell death in B16F10 cells (Fig. 5C–G). These results are quite interesting considering that inhibition of NF-κB is a promising option for anti-cancer therapeutics.10
Since PI3K–Akt pathway impacts on cell growth and survival, therefore inhibiting this pathway is likely to enhance tumor cell killings.32 It was found that piperine potentially attenuated PI3K–Akt signaling in UVB-irradiated cells (Fig. 5G–I). These results are even more interesting considering that PI3K–Akt pathway regulates apoptosis resistance in melanoma.32 We next studied the effects of piperine on MAPK family proteins in UVB-irradiated cells. Classically, the role of B-Raf–MEK–ERK signaling has been suggested as protective.9 Therefore, inhibition of MEK–ERK signaling axis is anticipated to enhance the therapeutic efficacy of UVB irradiation. When B16F10 cells were treated with piperine prior and subsequent to UVB-irradiation, a notable decrease in the expression of phospho-MRK and phospho-ERK was observed (Fig. 5J–M), which also confirmed the pro-apoptotic effects of piperine in UVB-irradiated B16F10 cells.
Taken together, our findings add new molecular insights into the pro-apoptotic effects of piperine in UVB-irradiated B16F10 cells and encourage further mechanistic studies to investigate the therapeutic efficacy of piperine in UVB-irradiated in vivo melanoma models.
Conflict of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This study was supported by Council of Scientific and Industrial Research (CSIR), New Delhi, India, under its 12th Five Year Plan Project titled, “Towards Understanding Skin Cell Homeostasis” (TOUCH), Project no. BSC 0302. We acknowledge the help in flow cytometry based experiments by Dr Shashak K. Singh, Senior Scientist, Cancer Pharmacology Division, CSIR-Indian Institute of Integrative Medicine, Jammu. Financial assistance to RAR by Council of Scientific and Industrial Research (CSIR), Human Resource Development Group (HRDG), Pusa, New Delhi, India is highly acknowledged.References
- Z. A. Abdel-Malek, A. L. Kadekaro and V. B. Swope, Pigm. Cell Melanoma Res., 2010, 23, 171–186 CrossRef CAS PubMed.
- V. Gray-Schopfer, C. Wellbrock and R. Marais, Nature, 2007, 445, 851–857 CrossRef CAS PubMed.
- S. Salucci, S. Burattini, M. Battistelli, V. Baldassarri, M. C. Maltarello and E. Falcieri, Int. J. Mol. Sci., 2012, 14, 532–546 CrossRef PubMed.
- L. M. Davids and B. Kleemann, Cancer Treat. Rev., 2011, 37, 465–475 CAS.
- D. Kulms and T. Schwarz, Photodermatol., Photoimmunol. Photomed., 2000, 16, 195–201 CAS.
- D. Kulms, E. Zeise, B. Poppelmann and T. Schwarz, Oncogene, 2002, 21, 5844–5851 CrossRef CAS PubMed.
- J. K. Brunelle and A. Letai, J. Cell Sci., 2009, 122, 437–441 CrossRef CAS PubMed.
- M. A. Lawlor and D. R. Alessi, J. Cell Sci., 2001, 114, 2903–2910 CAS.
- T. Wada and J. M. Penninger, Oncogene, 2004, 23, 2838–2849 CrossRef CAS PubMed.
- G. Madonna, C. D. Ullman, G. Gentilcore, G. Palmieri and P. A. Ascierto, J. Transl. Med., 2012, 10, 53 CrossRef CAS PubMed.
- K. Vermeulen, Z. N. Berneman and D. R. Van Bockstaele, Cell Proliferation, 2003, 36, 165–175 CrossRef CAS PubMed.
- J. J. Lu, J. L. Bao, X. P. Chen, M. Huang and Y. T. Wang, J. Evidence-Based Complementary Altern. Med., 2012, 2012, 485042 Search PubMed.
- S. Duessel, R. M. Heuertz and U. R. Ezekiel, Clin. Lab. Sci., 2008, 21, 151–157 Search PubMed.
- L. H. Lai, Q. H. Fu, Y. Liu, K. Jiang, Q. M. Guo, Q. Y. Chen, B. Yan, Q. Q. Wang and J. G. Shen, Acta Pharmacol. Sin., 2012, 33, 523–530 CrossRef CAS PubMed.
- C. R. Pradeep and G. Kuttan, Clin. Exp. Metastasis, 2002, 19, 703–708 CrossRef CAS PubMed.
- J. Wattanathorn, P. Chonpathompikunlert, S. Muchimapura, A. Priprem and O. Tankamnerdthai, Food Chem. Toxicol., 2008, 46, 3106–3110 CrossRef CAS PubMed.
- D. S. Kim, Y. M. Jeong, S. I. Moon, S. Y. Kim, S. B. Kwon, E. S. Park, S. W. Youn and K. C. Park, Cell. Mol. Life Sci., 2006, 63, 2661–2668 CrossRef CAS PubMed.
- S. Narayanapillai, C. Agarwal, C. Tilley and R. Agarwal, Photochem. Photobiol., 2012, 88, 1135–1140 CrossRef CAS PubMed.
- M. D. Adil, P. Kaiser, N. K. Satti, A. M. Zargar, R. A. Vishwakarma and S. A. Tasduq, J. Ethnopharmacol., 2010, 132, 109–114 CrossRef PubMed.
- N. Uchide, K. Ohyama, T. Bessho and H. Toyoda, Intervirology, 2009, 52, 164–173 CrossRef CAS PubMed.
- D. Fishman, B. Irena, S. Kellman-Pressman, M. Karas and S. Segal, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1740–1744 CrossRef CAS.
- U. Wolfle, P. R. Esser, B. Simon-Haarhaus, S. F. Martin, J. Lademann and C. M. Schempp, Free Radical Biol. Med., 2011, 50, 1081–1093 CrossRef PubMed.
- M. Ferreira, L. S. Assuncao, F. B. Filippin-Monteiro, T. B. Creczynski-Pasa and M. M. Sa, Eur. J. Med. Chem., 2013, 70, 411–418 CrossRef CAS PubMed.
- Y. Fernandez, M. Verhaegen, T. P. Miller, J. L. Rush, P. Steiner, A. W. Opipari Jr, S. W. Lowe and M. S. Soengas, Cancer Res., 2005, 65, 6294–6304 CrossRef CAS PubMed.
- M. Rieber and M. Strasberg Rieber, Int. J. Cancer, 2000, 86, 462–467 CrossRef CAS.
- X. D. Zhang, X. Y. Zhang, C. P. Gray, T. Nguyen and P. Hersey, Cancer Res., 2001, 61, 7339–7348 CAS.
- W. Yu, B. G. Sanders and K. Kline, Cancer Res., 2003, 63, 2483–2491 CAS.
- D. Anglicheau, N. Pallet, M. Rabant, P. Marquet, B. Cassinat, P. Meria, P. Beaune, C. Legendre and E. Thervet, Kidney Int., 2006, 70, 1019–1025 CrossRef CAS PubMed.
- L. A. Gillies and T. Kuwana, J. Cell. Biochem., 2014, 115, 632–640 CrossRef CAS PubMed.
- H. Masaki, Y. Izutsu, S. Yahagi and Y. Okano, J. Invest. Dermatol. Symp. Proc., 2009, 14, 50–52 CrossRef CAS PubMed.
- C. A. Bivik, P. K. Larsson, K. M. Kagedal, I. K. Rosdahl and K. M. Ollinger, J. Invest. Dermatol., 2006, 126, 1119–1127 CrossRef CAS PubMed.
- K. D. Courtney, R. B. Corcoran and J. A. Engelman, J. Clin. Oncol., 2010, 28, 1075–1083 CrossRef CAS PubMed.
- M. Colone, A. Calcabrini, L. Toccacieli, G. Bozzuto, A. Stringaro, M. Gentile, M. Cianfriglia, A. Ciervo, M. Caraglia, A. Budillon, G. Meo, G. Arancia and A. Molinari, J. Invest. Dermatol., 2008, 128, 957–971 CrossRef CAS PubMed.
- Y. Yan, C. L. Wei, W. R. Zhang, H. P. Cheng and J. Liu, Acta Pharmacol. Sin., 2006, 27, 821–826 CrossRef CAS PubMed.
- P. S. Brookes, Y. Yoon, J. L. Robotham, M. W. Anders and S. S. Sheu, Am. J. Physiol.: Cell Physiol., 2004, 287, C817–C833 CrossRef CAS PubMed.
- J. P. Fruehauf and V. Trapp, Expert Rev. Anticancer Ther., 2008, 8, 1751–1757 CrossRef CAS PubMed.
- C. Martin-Cordero, A. J. Leon-Gonzalez, J. M. Calderon-Montano, E. Burgos-Moron and M. Lopez-Lazaro, Curr. Drug Targets, 2012, 13, 1006–1028 CrossRef CAS PubMed.
- G. Petrosillo, F. M. Ruggiero, M. Pistolese and G. Paradies, J. Biol. Chem., 2004, 279, 53103–53108 CrossRef CAS PubMed.
- J. Eberle and A. M. Hossini, Curr. Genomics, 2008, 9, 409–419 CrossRef CAS PubMed.
- R. van den Berg, G. R. Haenen, H. van den Berg and A. Bast, Br. J. Nutr., 2001, 86(suppl. 1), S121–S127 CrossRef CAS PubMed.
- M. Karin and A. Lin, Nat. Immunol., 2002, 3, 221–227 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |