
An “all-water” strategy for regiocontrolled synthesis of 2-aryl quinoxalines†
Abstract
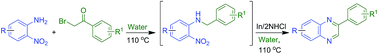
A new synthetic strategy of tandem N-aroylmethylation-nitro reduction–cyclocondensation has been developed for the first and generalized regioselective synthesis of 2-aryl quinoxalines adopting “all water chemistry.” Water plays the critical role through hydrogen bond driven ‘synergistic electrophile–nucleophile dual activation’ for chemoselective N-aroylmethylation of o-nitroanilines, that underlines the origin of the regioselectivity, as the use of organic solvents proved to be ineffective. Water also provides beneficial effects during the nitro reduction and the penultimate cyclocondensation steps.
 Please wait while we load your content...
Please wait while we load your content...

