DOI:
10.1039/C4RA08195A
(Paper)
RSC Adv., 2015,
5, 988-1008
A facile electrochemical approach for the deposition of iron–manganese phosphate composite coatings on aluminium†
Received
6th August 2014
, Accepted 24th November 2014
First published on 24th November 2014
Abstract
A novel electrochemical approach to deposit an iron–manganese phosphate composite coating on aluminium, which would otherwise be difficult to achieve by a conventional chemical conversion coating method, is addressed. An electrolyte solution containing manganese and phosphate ions and, a steel anode, were used to deposit the iron–manganese phosphate composite coating on the aluminium cathode. During the electrochemical treatment, besides the aluminium cathode, coating deposition was also observed on the steel anode. The resultant coatings were characterized by Fourier-transform infrared spectroscopy, Raman spectroscopy, scanning electron microscopy, energy dispersive X-ray analysis, X-ray photoelectron spectroscopy and X-ray diffraction measurements to ascertain the type of functional groups, morphological features, chemical composition and phase constituents, respectively. The surface coverage of the coatings was estimated by immersion plating of copper. The corrosion resistance of the iron–manganese phosphate composite coated aluminium, in 3.5% NaCl, was evaluated by potentiodynamic polarization, current–time transient and electrochemical impedance spectroscopy studies. Optical micrographs were also acquired after corrosion testing to understand the corrosion mechanism. The novel electrochemical approach addressed in this study provides a new avenue to deposit an iron–manganese phosphate composite coating on aluminium that offers good corrosion protection for aluminium in 3.5% NaCl.
1. Introduction
The biggest challenge of the automotive industry is the ever increasing demand to reduce energy consumption and air pollution. Lightweight vehicles are considered ideal to improve fuel economy and to reduce greenhouse gas emissions.1,2 The high strength-to-weight ratio, better formability and recyclability make aluminium (Al) the perfect candidate material to replace steel towards the development of lightweight vehicles.3 Following this, the use of Al in automotive industry has steadily increased. Zinc phosphate, chromate and chromate–phosphate conversion coatings are widely used to improve the corrosion resistance of Al as well as to serve as a paint base.4–6 Since the use of Al and Al alloys are extended to the make engine blocks, pistons and cylinder heads, that require a combination of good corrosion and wear resistance, the type of surface treatments have been extended to plasma electrolytic oxidation, electroless Ni–P–SiC composite coatings, etc.7–10
Manganese phosphate coating is well known for its ability to provide good corrosion resistance and lubricant properties. The uniformity, compactness, high hardness and ability to retain lubricants enable manganese phosphate coatings to offer superior corrosion and wear resistance.11–15 It is commonly applied over ferrous metals for brake and clutch assemblies, engine components, leaf or coil springs, drill bits, screws, nuts and bolts, washers, anti-vibration washers, tools, magnet cores, casting interiors, etc. It facilitates a low coefficient of friction and eliminates scuffing and galling of engine, gears and power transmission systems. In addition, its ability to deform slightly under pressure helps to reduce stress concentration.11,14
The deposition of manganese phosphate coating by chemical conversion method, besides on ferrous metals, has also been carried out on non-ferrous metals such as AZ91D and AZ31 Mg alloys.16,17 However, the predominant species deposited on them is different from those deposited on ferrous metals, viz., iron and manganese hureaulites. During manganese phosphating of ferrous metals, the base metal is the only source of Fe2+ ions that subsequently react with Mn2+ and PO43− ions to form iron hureaulite [(Mn,Fe)5H2(PO4)4·4H2O]. This is the main reason for the requirement of a highly acidic bath composition (pH < 3.0), higher operating temperature (>70 °C) and longer duration of time (>30 min) for manganese phosphating processes.11–15 The Fe2+ ions could not be enriched with the addition of ferrous salts in the phosphating bath since the accelerator present in the bath will oxidize them to Fe3+ ions, which would subsequently combine with the PO43− ions and gets precipitated as FePO4 (sludge) before the onset of the deposition process. Disposal of the solid waste is a major concern in finishing industries. In addition, dissolution of Al in the highly acidic bath requires the addition of fluoride ions to complex the aluminium as AlF63−, which would otherwise poison the coating process.4,18–20 Hence, deposition of manganese phosphate coating enriched with iron and manganese hureaulites on Al by chemical conversion method appears to be a difficult proposition.
In our earlier work, we have used cathodic electrochemical treatment for the deposition of zinc–zinc phosphate composite coatings on steel (using a zinc phosphating bath and, graphite and steel anodes) as well as on aluminium (using dilute phosphoric acid and zinc anode).21–26 The use of such a method for the deposition of zinc phosphate coatings on steel and 316 stainless steel has also been reported by other researchers.27,28 The findings of these studies reveal that it is possible to deposit a variety of metal–metal phosphate composite coatings by a suitable combination of electrolytes having the respective metal ions and/or the anodes.21–28 The cathodic electrochemical treatment could be a viable option to deposit manganese phosphate coatings enriched with iron and manganese hureaulites on Al and it has been attempted in the present study. The study also aims at to evaluate the characteristic properties of the resultant coatings and their corrosion behaviour in 3.5% NaCl.
2. Materials and methods
2.1 Materials, surface preparation, electrolyte, deposition methodology and conditions
Pure Al (99.9%) and mild steel (composition in wt%: C: 0.17; Si: 0.07; Mn: 0.72; P: 0.020; S: 0.010; Cr: 0.05; Ni: 0.04; Fe: balance), both having similar dimensions of 5 cm × 8 cm × 0.2 cm were used as cathode and anode, respectively. The Al sample was polished using SiC coated abrasive paper (800 grit) and etched in 10% NaOH. The mild steel sample was degreased using acetone and pickled in 10% H2SO4. After alkaline etching (Al) and acid pickling (mild steel), the samples were thoroughly rinsed in hot deionised water and dried using a stream of compressed air. An aqueous solution containing 6.9 g L−1 of manganese carbonate, 6 mL L−1 of orthophosphoric acid (85%) and 1.5 mL L−1 of nitric acid (70%) was used as the electrolyte. The initial pH of the electrolyte solution was adjusted to 2.80 and it was maintained at 70 °C using a constant temperature oil bath. The cleaned Al and mild steel samples were respectively connected to the cathode and anode terminal of a DC regulated power supply (K-PAS, Chennai, India). Deposition of manganese phosphate coating on Al was carried out at 2 to 5 mA cm−2 for 10 to 60 min. After deposition, the coated samples were thoroughly rinsed with deionised water and dried using a stream of compressed air.
2.2 Methods of characterization
The colour and uniformity of the coatings deposited under varying experimental conditions were assessed by visual observation while their adhesion was evaluated by pull-off adhesion test using a pressure sensitive adhesive tape. The amount of manganese phosphate coating deposited on Al/steel was determined from the gain in weight after deposition. Triplicate samples were coated to determine the coating weight and the average values were reported. The manganese phosphate coatings deposited on Al/steel were characterized by Fourier transform Infrared (FT-IR) spectroscopy (Agilent Cary 630) and confocal Raman spectroscopy (Nanophoton Raman-11i). The surface morphology of the coatings was assessed by scanning electron microscopy (SEM) (S-3400N Hitachi-Japan) while their chemical composition was determined by energy dispersive X-ray analysis (EDAX) attached with the SEM. The phase content of the coatings was ascertained by X-ray diffraction (XRD) measurements using Cu Kα radiation (Bruker Model D8). The chemical nature of the iron–manganese phosphate composite coated Al samples was evaluated by X-ray photoelectron spectroscopy (XPS) (ESCA+, Omicron Nanotechnology, Germany). It was configured with a combination of high transmission SPHERA small spot hemispherical analyzer and XM 1000 high power monochromator so as to achieve the highest resolution. Analysis was performed on a circular area with 3 mm diameter in ultrahigh vacuum environment (10−10 mbar). The photoelectrons were excited by Al Kα (1486.6 eV) radiation. The survey spectrum was acquired at pass energy of 50 eV and all possible elements that could present were identified from it. The high resolution spectra of Mn 2p, O 1s, P 2p and Fe 2p were acquired at pass energy of 20 eV. The spectral data were analyzed using CasaXPS software. All binding energies in the XPS spectra were corrected using the standard binding energy of C 1s peak at 285 eV. The atomic concentrations of Mn, O, P and Fe were calculated using the integral peak intensities of the respective elements and their sensitivity factors.
2.3 Evaluation of corrosion resistance
The ability of the manganese phosphate coating to improve the corrosion resistance of Al is a function of its porosity, surface coverage and thickness. In the present study, the porosity and surface coverage of the coatings deposited on Al was determined by immersion plating of Cu29 using untreated Al as the control. The plating solution consists of 99.8 g L−1 of CuSO4·5H2O and 99.8 g L−1 of ethylenediamine. The total area of the untreated and coated Al used for Cu plating was 50 cm2. The Cu plating was carried out at 27 °C for 2 h. After Cu plating, the samples were washed thoroughly to remove the excess electrolyte present on their surfaces, dried using a stream of compressed air and photographed.
Quantitative estimation of the amount of Cu plated on untreated and coated Al was also made to enumerate the surface coverage of coatings. Sodium diethyldithiocarbamate (DEDTC) was chosen as the complexing agent following its very high sensitivity towards copper. The absorbance of the Cu–DEDTC complex was determined at 435 nm using a diode array spectrophotometer (Agilent 8453). The amount of Cu plated on untreated and coated Al was estimated after stripping the plated Cu in dilute HNO3. The protocol employed for standardization, calibration and estimation of the amount of Cu plated on untreated and coated Al is described in the ESI 1.†
The corrosion resistance of untreated and coated Al, in 3.5% NaCl, was evaluated by potentiodynamic polarization, current–time transient (CTT) and electrochemical impedance spectroscopy (EIS) studies (Biologic SP-240, France). The untreated/coated Al forms the working electrode, while a saturated calomel electrode (SCE) and a graphite rod served as the reference and auxiliary electrodes, respectively. These three electrodes were placed within a flat cell (Friction and Wear Tech., Chennai, India) in such a way that only 1 cm2 area of the working electrode was exposed to the 3.5% NaCl. Potentiodynamic polarization measurements were carried out in the potential range from −250 mV(SCE) in the cathodic direction to +1000 mV(SCE) in the anodic direction from open circuit potential (OCP) at a scan rate of 100 mV min−1. CTT transients were recorded at −650 mV(SCE), −500 mV(SCE) and −300 mV(SCE) for 900 s. The polarization and CTT studies were repeated at least three times to ensure reproducibility of the test results. Optical micrographs of the untreated and coated Al before corrosion as well as after polarization and CTT studies were acquired to understand the extent of corrosion attack and the nature of corrosion products formed on their surface. EIS studies of untreated/coated Al were performed at their respective OCPs. The impedance spectra were obtained using an excitation voltage of 10 mV rms (root mean square) in the frequency range between 10 kHz and 0.01 Hz. The EIS parameters were determined from the Nyquist plot after fitting the data using ZSimpWin 3.21 software.
3. Results and discussion
3.1 Deposition of manganese phosphate coating on Al by cathodic electrochemical treatment
In the present study, deposition of manganese phosphate coating on Al is explored by cathodic electrochemical treatment using a mild steel anode and an electrolyte solution containing Mn(H2PO4)2, H3PO4 and HNO3. With the onset of current, dissolution of iron occurs at the anode (eqn (1)).| | |
Fe → Fe2+ + 2e− (anode)
| (1) |
Since the electrolytic cell consists of a cathode (Al), an anode (mild steel) and a conducting electrolyte, the Fe2+ ions generated at the anode is likely to be reduced to Fe at the cathode (eqn (2)). Reduction of H+ ions, leading to the evolution of H2 will also occur at the cathode as a competing reaction (eqn (3)). Visual observations indicate that the coating growth is accompanied by a simultaneous hydrogen evolution throughout the duration of deposition.
| | |
Fe2+ + 2e− → Fe (cathode)
| (2) |
| | |
2H+ + 2e− → H2↑ (cathode)
| (3) |
Some of the Fe2+ ions could be oxidized to Fe3+ ions by the NO3− ions present in the electrolyte. Some of the Fe3+ ions could be reduced back to Fe2+ ions at the cathode while the others combine with the PO43− ions to form FePO4. Since the solubility product of FePO4 is very low (Ksp: 1.3 × 10−22 @ 25 °C), it is likely to be precipitated. The accumulation of fine white precipitate as sludge at the bottom of the electrolytic cell further validates this attribute.
The consumption of acidity following the reduction of H+ ions at the cathode (eqn (3)) enables an increase in pH at the cathode–electrolyte interface that favours the conversion of primary to secondary and/or tertiary phosphates, resulting in the deposition of various types of manganese phosphates on Al (eqn (4)–(9)). The reduction of NO3− ions at the cathode helps to accelerate their deposition (eqn (10)).
| | |
Mn(H2PO4)2 → Mn2+ + 2H2PO4− (cathode–electrolyte interface)
| (4) |
| | |
H2PO4− + OH− → HPO42− + H2O (cathode–electrolyte interface)
| (5) |
| | |
Mn2+ + HPO42− + 2.25H2O → MnHPO4·2.25H2O (cathode)
| (6) |
| | |
5Mn2+ + 5Fe2+ + 4H2PO4− + 4H2O + 6e− → (Mn,Fe)5H2(PO4)4·4H2O + 3H2↑ (cathode)
| (7) |
| | |
5Mn(H2PO4)2 + 4H2O ↔ Mn5(PO3(OH))2(PO4)2·4H2O + 6H3PO4 (cathode)
| (8) |
| | |
5MnHPO4 + 4H2O ↔ Mn5(PO3(OH))2(PO4)2·4H2O + H3PO4 (cathode)
| (9) |
| | |
NO3− + 10H+ + 8e− → NH4+ + 3H2O (cathode)
| (10) |
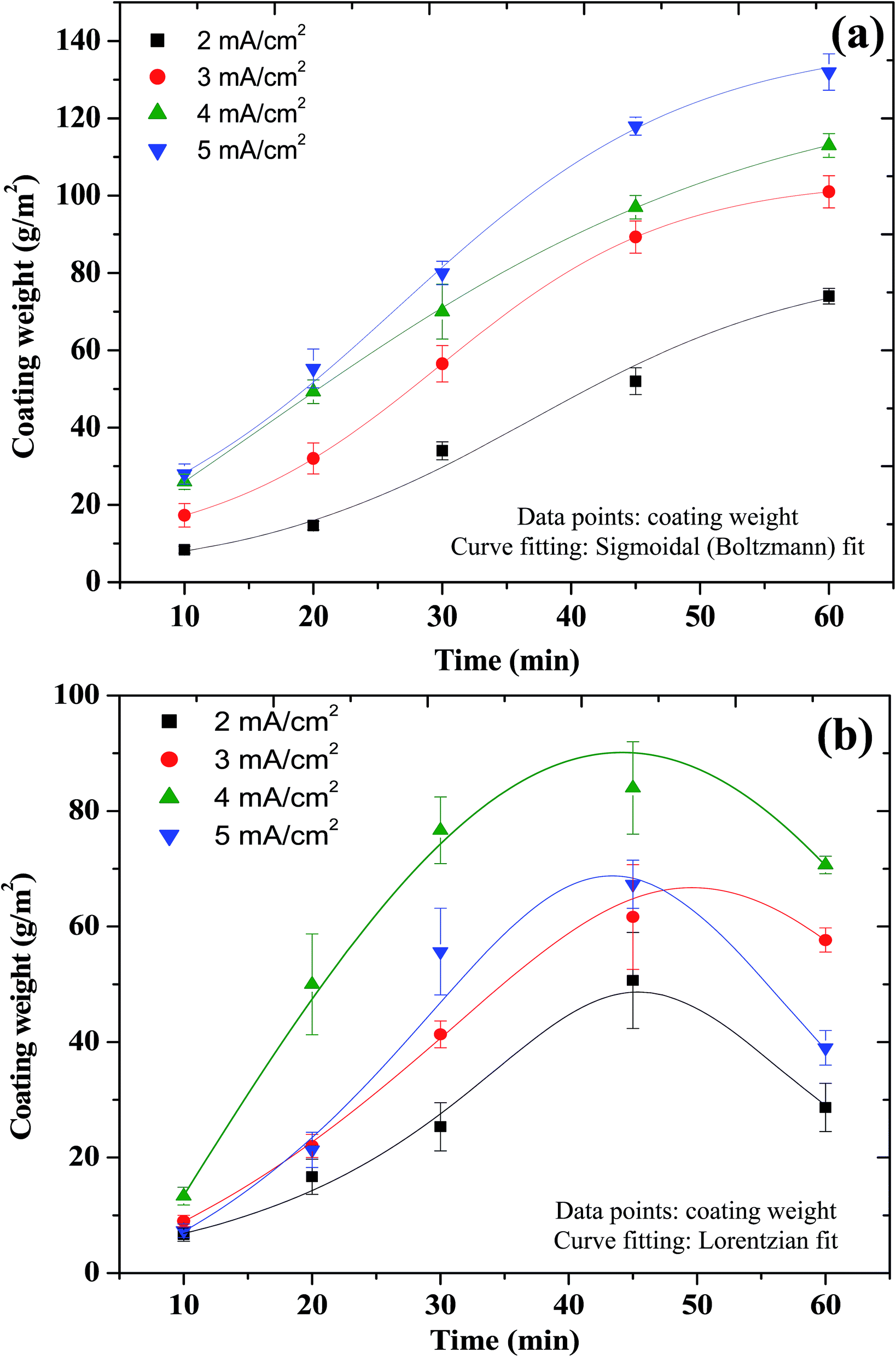
The manganese phosphate coatings deposited on Al at all treatment conditions are uniform and black in colour in contrast to the military green colour of those deposited on ferrous substrates by chemical conversion method.4,5 The adhesion of these coatings when evaluated by a pressure sensitive adhesive tape fails to show any removal of the coated layer from Al. The variation in coating weight obtained at various current densities (2 to 5 mA cm−2) as a function of treatment time (10 to 60 min) is shown in Fig. 1(a). It is evident that the coating weight is increased with an increase in current density as well as treatment time. The trend in variation of coating weight is not linear, rather it follows a sigmoidal (Boltzmann) fit (Fig. 1(a)). The black colour of the coating is believed to be due to the deposition of Fe along with the manganese phosphate. Deposition of Zn during zinc phosphating of Al alloys has been observed earlier.30,31 According to Zhang et al.30 and Sheng et al.,31 the potential difference between Al and Zn promotes the reduction of Zn2+ ions, leading to the deposition of a thin layer of zinc on the surface of the Al alloy. However, once the surface of this zinc layer is covered with a subsequent deposition of zinc phosphate, further reduction of Zn2+ ions becomes negligible.30,31 Hence, in the present study, it would be of much interest to ascertain whether the deposition of Fe on Al occurs only during the initial stage or throughout the duration of deposition.
 |
| | Fig. 1 Variation in coating weight of manganese phosphate coatings deposited on (a) Al cathode; and (b) mild steel anode at 2 to 5 mA cm−2 as a function of treatment time. | |
Considering the various possible chemical and electrochemical reactions that could occur during the deposition process (eqn (1)–(10)), deposition of Fe (eqn (2)) is likely to be the first layer on Al while the interfacial increase in pH following the consumption of H+ ions (eqn (3)), should subsequently enable the deposition of various types of manganese phosphates over the Fe layer. During deposition, the coating weight is increased from 10 to 60 min (Fig. 1(a)). If the initially deposited Fe layer on Al is completely covered by the subsequently deposited manganese phosphates, then further deposition should have been not possible. The continuous evolution of hydrogen visually observed throughout the duration of deposition suggests the availability of metallic sites (Fe) at the surface at any given time. An increase in coating weight from 10 to 60 min is feasible only with a continuous deposition of manganese phosphates and that could happen only with a regular occurrence of an increase in interfacial pH. Hence, it is clear that after deposition of the first layer of Fe on Al, deposition of both Fe as well as manganese phosphates occur simultaneously. It is believed that the deposition of Fe occurs in the form of fixed channels while the manganese phosphates deposit on adjacent areas. Jegannathan et al.21,22,24 and Sankara Narayanan et al.23 have also observed deposition of zinc in the form of fixed channels during cathodic electrochemical deposition of zinc–zinc phosphate composite coatings on steel. This model of coating growth provides ample avenues for the reduction of H+ ions, leading to an interfacial increase in pH, thus favouring the deposition of manganese phosphates along with Fe. During the deposition of manganese phosphate coating by chemical conversion method, the interfacial increase in pH is controlled by the diffusion of H+ ions through the pores of the coating, which limits the amount of coating formation. However, during cathodic electrochemical treatment, the simultaneous deposition of Fe along with manganese phosphates helps to build thicker deposits (130 g m−2 @ 5 mA cm−2 for 60 min). The strong dependence of the coating weight with the applied current density (Fig. 1(a)) further validates this model of coating growth.
Another interesting observation made during the cathodic electrochemical treatment of Al using the electrolyte containing Mn2+, PO43− and NO3− ions is the deposition of coatings on the mild steel anode besides the Al cathode. A rise in interfacial pH is an essential prerequisite for the deposition of phosphate coatings.4,5 Since the reduction of H+ ions with the evolution of hydrogen occurs at the Al cathode, deposition of coatings on the mild steel anode appears to be surprising at the first instance. This can be explained based on the accumulation of Fe2+ ions at the anode–electrolyte interface. When the concentration of Fe2+ ions generated from the anode and their accumulation at the anode–electrolyte interface exceeds a threshold, the displacement of protons away from the interface could cause a rise in the interfacial pH. The availability of Mn2+, Fe2+ and H2PO4− ions at the anode–electrolyte interface along with a rise in the interfacial pH could have favoured the deposition of manganese phosphate coating at the mild steel anode. A similar phenomenon is also observed during the anodic electrochemical treatment of steel in a zinc phosphating bath, resulting in the deposition of zinc phosphate coating on the steel anode.24,32,33 The colour of the manganese phosphate coating deposited on the mild steel anode is dark gray. The variation in coating weight obtained at various current densities (2 to 5 mA cm−2) as a function of treatment time (10 to 60 min) for the coatings deposited on the mild steel anode is given in Fig. 1(b). It is evident that for a given treatment time, the coating weight is increased with an increase in current density from 2 to 4 mA cm−2 whereas the trend is reversed at 5 mA cm−2. Similarly, for a given current density, the coating weight is increased with an increase in treatment time from 10 to 45 min while the trend is reversed beyond 45 min. The reversal in trend observed beyond 4 mA cm−2 as well as after 45 min is due to the spallation of the coating. As a result, the trend in variation of coating weight follows a Lorentzian fit (Fig. 1(b)).
3.2 Characteristics of the manganese phosphate coatings deposited by cathodic electrochemical treatment
The FT-IR and Raman spectrum of manganese phosphate coatings deposited on Al at 4 mA cm−2 for 30 min are shown in Fig. 2(a and b), respectively. The corresponding spectra of the coatings prepared at 2 to 5 mA cm−2 for 30 min are quite similar to the one shown in Fig. 2(a and b). The IR and Raman bands observed at various frequencies along with their corresponding assignments are complied in Table 1. The IR bands observed at 1148, 1077, 1047 and 1028 cm−1 (νas(HPO4)2− and (PO4)3−), at 753 and 704 cm−1 (νs(P–O) of (PO4)3−), at 983 cm−1 (νs(PO4)3−), at 935 cm−1 (ν(P–O) of (HPO4)2−), at 1301 cm−1 (δ(P–O–H) of (HPO4)2−), at 2921, 2851 and 2493 cm−1 (ν(O–H) of H2PO4−) and Raman bands observed at 1075, 1041, 1025 and 1005 cm−1 (ν3HPO42− and PO43−), at 993 cm−1 (ν1(PO43−)), at 945 cm−1 (ν1(P–O) of (HPO42−)), at 598, 582, 543 and 532 cm−1 (ν4HPO42− and PO43− out of plane bending), at 455 and 412 cm−1 (ν2HPO42− and PO43− bending) suggest that type of coating deposited on Al is phosphate. The Raman band observed at 303 cm−1 (δ(M − O)) and the IR bands at 983 and 704 cm−1 (ν(Mn![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and ν(Mn–O–Mn)) indicate that the type of coating is manganese phosphate. The IR bands at 3343, 3309 and 3175 cm−1 (ν(O–H) of H2O) and at 1729 and 1643 cm−1 (δ(H–O–H) of H2O), point out that the manganese phosphate coating is hydrated. The presence of these IR and Raman bands corresponding to the various stretching and bending vibrations of HPO42− and PO43−, H2O, M − O, MnO and Mn–O–Mn (Fig. 2(a and b) and Table 1) has also been observed earlier by other researchers.34–39 The FT-IR and Raman spectrum of the coatings deposited on the mild steel anode are quite similar to the one deposited on the Al cathode (please refer ESI 2, 3 and 4†), suggesting that they are also consists of hydrated manganese phosphate.
O) and ν(Mn–O–Mn)) indicate that the type of coating is manganese phosphate. The IR bands at 3343, 3309 and 3175 cm−1 (ν(O–H) of H2O) and at 1729 and 1643 cm−1 (δ(H–O–H) of H2O), point out that the manganese phosphate coating is hydrated. The presence of these IR and Raman bands corresponding to the various stretching and bending vibrations of HPO42− and PO43−, H2O, M − O, MnO and Mn–O–Mn (Fig. 2(a and b) and Table 1) has also been observed earlier by other researchers.34–39 The FT-IR and Raman spectrum of the coatings deposited on the mild steel anode are quite similar to the one deposited on the Al cathode (please refer ESI 2, 3 and 4†), suggesting that they are also consists of hydrated manganese phosphate.
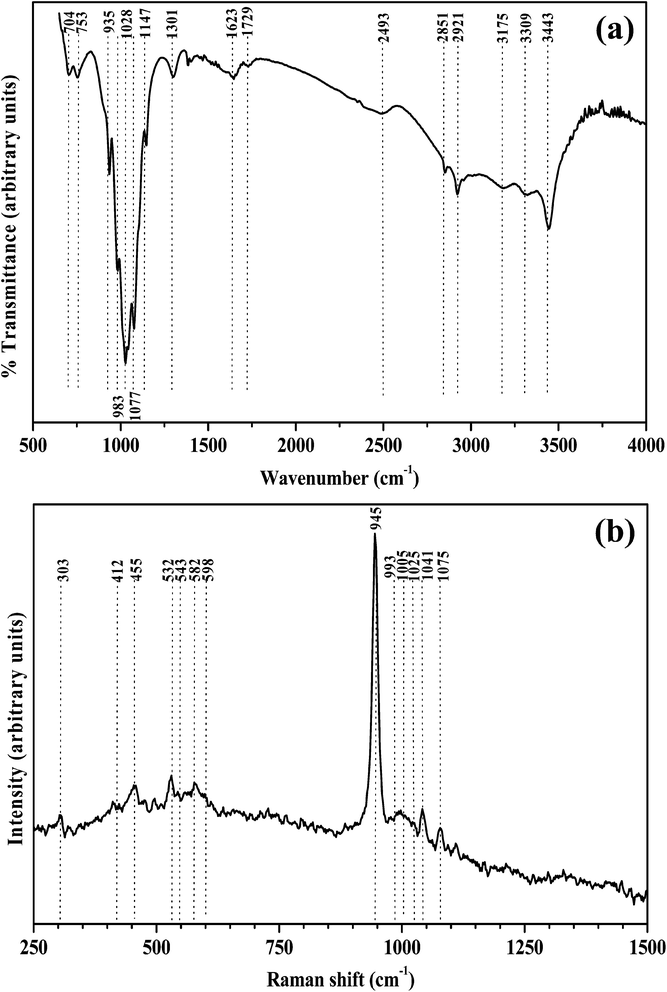 |
| | Fig. 2 (a) FT-IR spectrum; and (b) Raman spectrum of the manganese phosphate coatings deposited on Al at 4 mA cm−2 for 30 min. | |
Table 1 Assignment of IR and Raman bands of the manganese phosphate coatings deposited on Al at 4 mA cm−2 for 30 min
| IR bands (cm−1) |
Assignment |
Raman bands (cm−1) |
Assignment |
| 3443 |
ν(O–H) of H2O (stretching) |
1075 |
ν3HPO42− and PO43− (antisymmetric stretching) |
| 3309 |
ν(O–H) of H2O (stretching) |
1041 |
ν3HPO42− and PO43− (antisymmetric stretching) |
| 3175 |
ν(O–H) of H2O (stretching) |
1025 |
ν3HPO42− and PO43− (antisymmetric stretching) |
| 2921 |
ν(O–H) of H2PO4− (stretching) |
1005 |
ν3HPO42− and PO43− (antisymmetric stretching) |
| 2851 |
ν(O–H) of H2PO4− (stretching) |
993 |
ν1(PO43−) (stretching) |
| 2493 |
ν(O–H) of H2PO4− (stretching) |
945 |
ν1(P–O) of (HPO42−) (stretching) |
| 1729 |
δ(H–O–H) of H2O (bending) |
598 |
ν4HPO42− and PO43− (out of plane bending) |
| 1643 |
δ(H–O–H) of H2O (bending) |
582 |
ν4HPO42− and PO43− (out of plane bending) |
| 1301 |
δ(P–O–H) of (HPO4)2− (bending) |
543 |
ν4HPO42− and PO43− (out of plane bending) |
| 1148 |
ν3HPO42− and PO43− (antisymmetric stretching) |
532 |
ν4HPO42− and PO43− (out of plane bending) |
| 1077 |
ν3HPO42− and PO43− (antisymmetric stretching) |
455 |
ν2HPO42− and PO43− (bending) |
| 1047 |
ν3HPO42− and PO43− (antisymmetric stretching) |
412 |
ν2HPO42− and PO43− (bending) |
| 1028 |
ν3HPO42− and PO43− (antisymmetric stretching) |
303 |
δ(M − O) (bending) |
| 983 |
νs(PO4)3− (symmetric stretching) |
|
|
| 983 |
ν(MnO) and ν(Mn–O–Mn) (stretching) |
|
|
| 935 |
ν(P–O) of HPO42− (stretching) |
|
|
| 753 |
νs(P–O) of (PO4)3− (symmetric stretching) |
|
|
| 704 |
νs(P–O) of (PO4)3− (symmetric stretching) |
|
|
| 704 |
ν(MnO) and ν(Mn–O–Mn) (stretching) |
|
|
The surface morphology of manganese phosphate coatings deposited on Al at 2 and 5 mA cm−2 for 30 min is shown in Fig. 3(a and b), respectively. The morphological features reveal that the coatings are uniform, compact and composed of plate- or sheet-like crystals. Similar morphological features were also observed for manganese phosphate coatings on steel.12,13,15 For coatings prepared at 5 mA cm−2 for 30 min (Fig. 3(b)), in addition to the plate- or sheet-like crystals, formation of spongy crystals are also observed at selected locations (marked as ‘○’ in Fig. 3(b)). The EDAX spectra acquired at selected regions of the manganese phosphate coatings deposited at 2 and 5 mA cm−2 and their corresponding chemical composition are shown in Fig. 4. It is evident that O, P, Mn and Fe are predominant elements for both coatings. A comparison of the chemical composition of the coatings prepared at 2 and 5 mA cm−2 for 30 min (Fig. 4(a and b)) reveals that an increase in current density promotes the Mn, Fe, and P content and decreases the O content. For coatings prepared at 5 mA cm−2, the chemical composition of the bulk crystals appears to be different from those of the spongy crystals (Fig. 4(b and c)); the later one is rich in Fe and O and it has a lower Mn and P content (Fig. 4(c)). The presence of O, P, Mn and Fe in coatings prepared at 2 and 5 mA cm−2 for 30 min (Fig. 4(a and b)) suggests that they could consist of manganese phosphate and manganese iron phosphate as the predominant compounds. The higher Fe and O content observed at the spongy crystals (Fig. 4(c)) suggest some possible codeposition of iron hydroxides along with the various types of manganese phosphates.
 |
| | Fig. 3 Surface morphology of the manganese phosphate coatings deposited on Al at (a) 2 mA cm−2; and (b) 5 mA cm−2 for 30 min. | |
 |
| | Fig. 4 EDAX spectra acquired at selected regions of the manganese phosphate coatings deposited on Al at (a) 2 mA cm−2; and (b and c) 5 mA cm−2 for 30 min and their chemical compositions. | |
The surface morphology of manganese phosphate coating deposited on the mild steel anode at 5 mA cm−2 for 30 min reveal that the coating is uniform and compact (Fig. 5). In spite of a similar plate- or sheet-like morphological feature (Fig. 5(a)) that is highly comparable with the one deposited on the Al cathode (Fig. 3(b)), the presence of pores/voids in the coating (marked as ‘○’ in Fig. 5(b)) is quite evident in coatings deposited on the mild steel anode. The pronounced electrochemical activity at the anode at 5 mA cm−2 could have dislodged the coating, leading to the formation of such pores/voids. The EDAX spectra acquired at selected regions of the manganese phosphate coatings deposited on the mild steel anode at 2 and 5 mA cm−2 for 30 min and their corresponding chemical composition are shown in Fig. 6. The predominance of O, P, Mn and Fe suggests that the coatings deposited on the mild steel anode are composed of manganese phosphate and manganese iron phosphate. A comparison of the chemical composition of the coatings prepared at 2 and 5 mA cm−2 for 30 min (Fig. 6(a and b)) reveals that an increase in current density promotes the Mn, Fe and P content and decreases the O content, a trend quite similar to the one observed for coatings deposited on the Al cathode.
 |
| | Fig. 5 Surface morphology of the manganese phosphate coatings deposited on mild steel at 5 mA cm−2 for 30 min (the pores in the coating are marked as ‘○’ in (b)). | |
 |
| | Fig. 6 EDAX spectra acquired at selected regions of the manganese phosphate coatings deposited on mild steel at (a) 2 mA cm−2; and (b) 5 mA cm−2 for 30 min. and their chemical compositions. | |
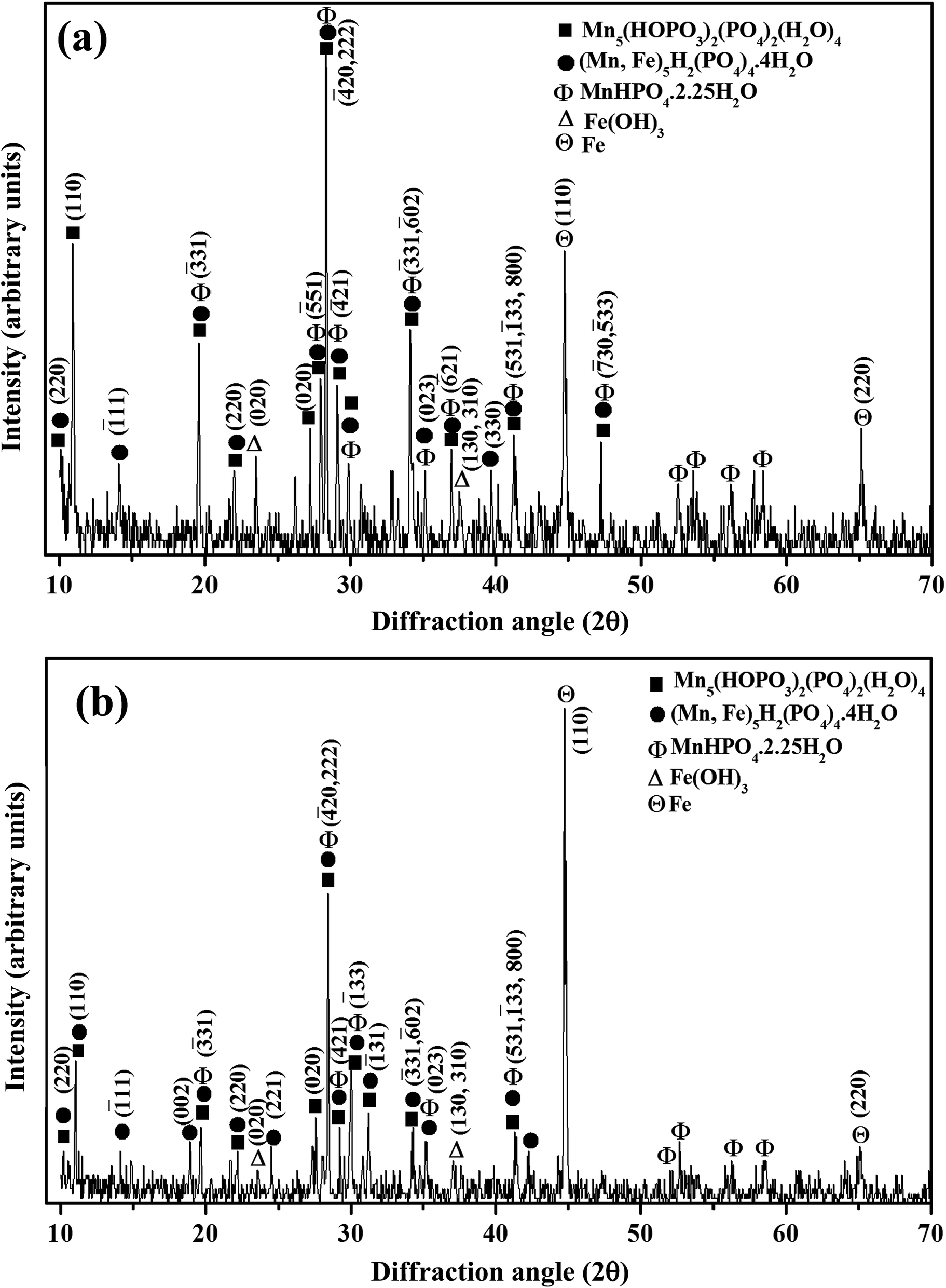
The XRD pattern of the manganese phosphate coating deposited on Al at 4 mA cm−2 for 30 min is shown in Fig. 7(a). It is evident that manganese iron hydrogen phosphate hydrate, referred as iron hureaulite [(Mn,Fe)5H2(PO4)4·4H2O] (JCPDS 16-0383), manganese phosphate hydroxide hydrate, referred as manganese hureaulite [Mn5(PO3(OH)2(PO4)2(H2O)4) (JCPDS 34-0146; 78-1648; and 76-0804) and manganese hydrogen phosphate hydrate [MnHPO4·2.25H2O] (JCPDS 47-0199) are the predominant phases. In addition, the diffraction pattern of this coating also exhibits peaks pertaining to iron [Fe (JCPDS 06-0696 and 85-1410)] and iron hydroxide [Fe(OH)3 (JCPDS 46-1436)]. The presence of iron peaks confirms its codeposition along with other manganese phosphates in the coating. The presence of Fe2+, Fe3+ and NO3− ions in the electrolyte at any given time of the deposition process and the increase in pH at the cathode–electrolyte interface might have promoted the formation of Fe(OH)3, in which a part of it could have codeposited along with the various types of manganese phosphates on Al.
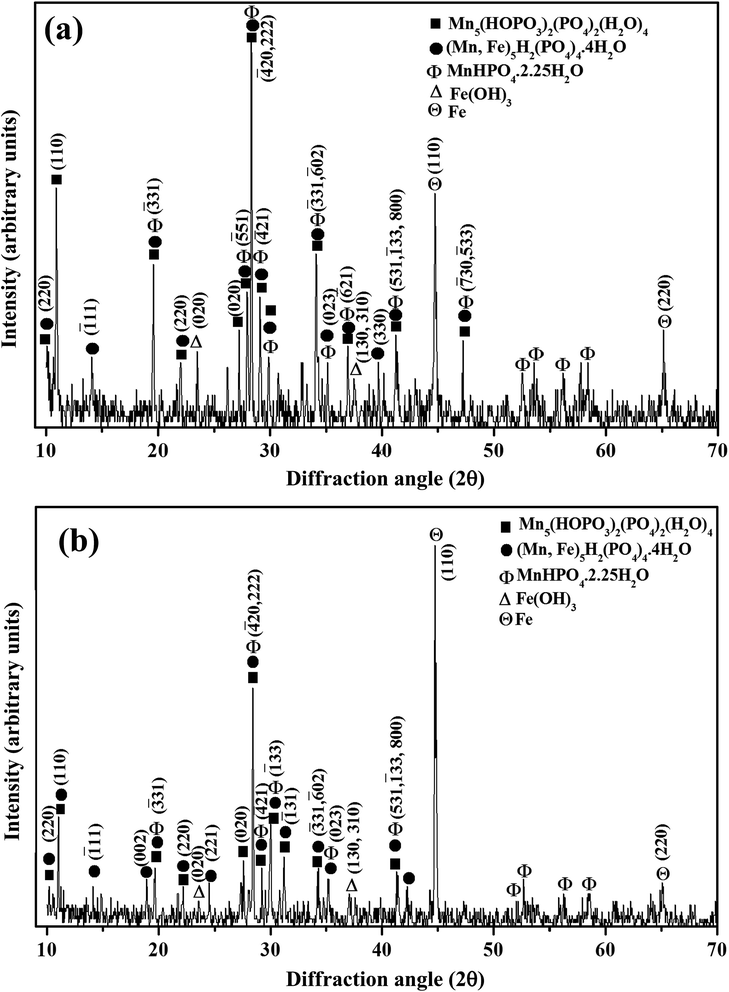 |
| | Fig. 7 XRD pattern of manganese phosphate coatings deposited on (a) Al; and (b) mild steel by cathodic electrochemical treatment at 4 mA cm−2 for 30 min. | |
The XRD pattern of the manganese phosphate coating deposited on mild steel anode at 4 mA cm−2 for 30 min is shown in Fig. 7(b). A comparison of the XRD patterns of coatings deposited on the Al cathode (Fig. 7(a)) and the mild steel anode (Fig. 7(b)) reveals that both of them have similar phase contents. However, the volume fraction of the various types of manganese phosphate phases is relatively less for coatings deposited on mild steel anode (Fig. 7(b)) than those deposited on the Al cathode (Fig. 7(a)). For coatings deposited on the mild steel anode, the peaks pertaining to Fe is likely to be originated from the base metal and the increase in intensity of this peak with current density employed for deposition indicates a decrease in the amount of coating deposited on the anode (Fig. 7(b)). The pronounced electrochemical activity at the anode could have lead to spallation of the manganese phosphate coatings deposited on the mild steel anode. The corresponding patterns of the coatings deposited on Al cathode and mild steel anode at 2 to 5 mA cm−2 for 30 min also consist of similar phase constituents.
Fig. 8(a) shows the XPS survey spectrum taken at the surface of the iron–manganese phosphate coated Al obtained by cathodic electrochemical treatment at 5 mA cm−2 for 30 min, which indicates the presence of Mn, O, P, Fe and C. Since no carbon source is used during the preparation of the coating, the appearance of the C peak in the survey spectra can be ascribed to the adventitious contamination, which could have occurred during the transfer of the sample in air.40 The presence of Mn, O, P and Fe can be attributed to the iron–manganese phosphate composite coating, the existence of which is also pointed out by EDAX analysis (Fig. 4). XRD measurement also confirmed the presence of manganese iron hydrogen phosphate hydrate [(Mn,Fe)5H2(PO4)4·4H2O], manganese phosphate hydroxide hydrate [Mn5(PO3(OH)2(PO4)2(H2O)4) and manganese hydrogen phosphate hydrate [MnHPO4·2.25H2O] as the predominant phases (Fig. 7(a)). The high-resolution XPS spectra of Mn 2p, O 1s, P 2p and Fe 2p are shown in Fig. 8(b–e), respectively. The summary of the XPS analysis indicating the type of elements present along with their core excitation, binding energies, relative atomic percentages and the assignment of chemical groups are compiled in Table 2.
 |
| | Fig. 8 X-ray photoelectron spectra take at the surface of iron–manganese phosphate composite coating deposited on Al by cathodic electrochemical treatment at 5 mA cm−2 for 30 min: (a) survey spectra; (b) high resolution spectra of Mn 2p; (c) high resolution spectra of O 1s; (d) high resolution spectra of P 2p; and (e) high resolution spectra of Fe 2p. | |
Table 2 Summary of the XPS analysis indicating the type of elements present along with their core excitation, binding energies, relative atomic percentages and the possible assignment of chemical groups
| Element |
Core excitation |
Binding energy (eV) |
FWHM (eV) |
Area |
Area ratio |
Content (at%) |
Assignment |
| Mn |
2p3/2 |
641.7 |
4.5 |
1871 |
1.41/2 |
32.12 |
Mn2+ |
| 2p1/2 |
653.7 |
5.0 |
787 |
0.59/2 |
|
Mn2+ |
| O |
1s |
531.6 |
3.4 |
11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 128 128 |
— |
47.52 |
P–O of (PO4)3− and (HPO4)2− |
| P |
2p (1) |
131.8 |
2.6 |
410 |
0.77/2 |
3.13 |
(PO4)3− |
| 2p (2) |
133.6 |
2.0 |
661 |
1.23/2 |
|
(HPO4)2− |
| Fe |
Fe(0) |
706.1 |
4.2 |
109 |
0.34/3 |
17.24 |
Fe0 |
| 2p3/2 |
708.7 |
6.63 |
698 |
1.32/3 |
|
Fe2+ |
| 2p1/2 |
722.0 |
7.88 |
484 |
1.34/3 |
|
Fe3+ |
| Satellite |
713.9 |
2.69 |
62 |
— |
|
Fe3+ |
The high resolution spectrum of Mn 2p indicates two distinct peaks (Fig. 8(b)), Mn 2p3/2 at 641.7 eV and Mn 2p1/2 at 653.7 eV, with a respective area ratio of 1.41/2 and 0.59/2, which can be assigned to Mn2+.41–44 The O 1s peak observed at 531.6 eV (Fig. 8(c)) can be attributed to P–O of (PO4)3− and (HPO4)2−.40,44–50 The peak pertaining to oxygen of metal oxide that commonly appears ∼530 eV could not be identified, which suggests the absence of any metal oxides in the coating. The asymmetric P 2p peak can be deconvoluted into two peaks, one at 131.8 eV while the other at 133.6 eV (Fig. 8(d)). The peaks at 133.6 eV and 131.8 eV, with an area ratio of 1.23/2 and 0.77/2 can be ascribed to (HPO4)2− and (PO4)3−, respectively.40,44–47,51,52 The high resolution spectrum of Fe can be deconvoluted into three main peaks, at 706.1 eV, 708.7 and 722.0 eV, with a satellite peak at 713.9 eV. The peak at 708.7 eV with an area ratio of 1.32/3 is characteristic of Fe2+ while those observed at 722.0 eV with an area ratio of 1.34/3 and the satellite peak at 713.9 eV are characteristic of Fe3+.53–58 The peak at 706.1 eV, with an area ratio of 0.34/3, belongs to metallic iron (Fe0).54–57 Nelson et al.41 have observed the peaks at 641.6 eV (Mn 2p3/2), 531.5 eV (O 1s) and 133.6 eV (P 2p) for Hureaulite. Wang et al.44 have observed the peaks at 642.5 eV (Mn 2p), 531.2 eV (O 1s) and 133.8 eV (P 2p) for zinc–manganese phosphate conversion coatings deposited on Mg–Li alloy. Based on the inferences made from the XPS analysis of the present study and from the findings of Nelson et al.41 and Wang et al.,44 it is clear that the type of coating deposited on Al by cathodic electrochemical treatment is a composite, consisting of phosphates of Mn2+ (only with Mn2+ as well as with Fe2+), metallic Fe and Fe3+, possibly as FeOOH or Fe(OH)3. The chemical nature of the iron–manganese phosphate coatings deposited on Al, evidenced by the XPS analysis (Fig. 8 and Table 2), substantiates the observations made in EDAX analysis (Fig. 4) and XRD measurement (Fig. 7(a)).
The presence of various stretching and bending vibrations of the HPO42− and PO43−, H2O, M − O, MnO and Mn–O–Mn inferred from the FT-IR and Raman spectrum (Fig. 2(a and b) and Table 1), the presence of O, P, Mn and Fe as the predominant elements in the EDAX spectra (Fig. 4 and 6) and, the presence of peaks pertaining to manganese iron hydrogen phosphate hydrate [(Mn,Fe)5H2(PO4)4·4H2O], manganese phosphate hydroxide hydrate [Mn5(PO3(OH)2(PO4)2(H2O)4)], manganese hydrogen phosphate hydrate [MnHPO4·2.25H2O], Fe and Fe(OH)3 in the XRD patterns (Fig. 7(a and b)), confirm the reactions sequences (eqn (1)–(10)) and the mechanism of deposition. XPS analysis confirm the chemical nature of the coatings deposited on Al. Based on the formation mechanism and the nature of the chemical species present, the coatings deposited on Al can be referred as iron–manganese phosphate composite coating in analogy with the zinc–zinc phosphate composite coatings prepared by cathodic electrochemical treatment of steel in a zinc phosphating bath.21–24
Considering the various chemical and electrochemical reactions that occurred during the deposition of manganese phosphate coatings and the characteristic properties of the resultant coatings, a pictorial model is proposed to explain the formation of manganese phosphate coatings on the Al cathode and the mild steel anode (Fig. 9). The coatings deposited on Al are a composite of iron and manganese phosphate while those deposited on mild steel primarily consists of manganese phosphates. The corrosion behaviour of manganese phosphate coatings deposited on mild steel has already been studied earlier by many researchers.12,13,15,16 However, it is important to understand the corrosion behaviour of iron–manganese phosphate composite coating deposited on Al.
 |
| | Fig. 9 Pictorial model depicting the formation of iron–manganese phosphate coatings on Al cathode and manganese phosphate coatings on mild steel anode during cathodic electrochemical treatment. | |
3.3 Corrosion resistance of the iron–manganese phosphate composite coated aluminium
The potentiodynamic polarization curves of the uncoated Al and those coated with iron–manganese phosphate composite by cathodic electrochemical treatment at 2 to 5 mA cm−2 for 30 min, in 3.5% NaCl, are shown in Fig. 10(a). The anodic branch of the polarization curves of coated Al exhibit pronounced changes when compared to the untreated one. Unlike the coated ones, the untreated Al fails to show any distinct passivation and pitting behaviour. The Eb of untreated Al (−736 mV(SCE)) did not vary much from its Ecorr (−803 mV(SCE)). The strong potential dependent dissolution current of several orders of magnitude observed for untreated Al further substantiate the onset of pitting soon after its Ecorr.59 Passivation of Al could be observed if the condition that prevails in the electrolyte medium enables the formation of an aluminium oxide layer on its surface. However, in corrosive mediums such as 3.5% NaCl, the highly aggressive Cl− ions are likely to cause breakdown of the passive film and hinder its repair.60 Several researchers have studied the corrosion behaviour of Al and its alloys in Cl− medium using potentiodynamic polarization, current–time transients, pitting scan technique as well as by mathematical modelling.61–69 It has been established that Cl− ions would cause local thinning of the passive oxide layer, resulting in active dissolution of Al and promote pitting corrosion.63,70,71
 |
| | Fig. 10 (a) Potentiodynamic polarization curves; (b) comparison of the corrosion potential (Ecorr) and (c) comparison of breakdown potential (Eb) of untreated Al and those coated with iron–manganese phosphate composite by cathodic electrochemical treatment at 2 to 5 mA cm−2. | |
Al samples coated with iron–manganese phosphate composites by cathodic electrochemical treatment at 2 to 5 mA cm−2 for 30 min exhibit an anodic shift in Ecorr when compared to the untreated Al (Fig. 10(a)). This is due to the coverage of the Al surface by the iron–manganese phosphate composites, which acts as a barrier layer for the electron transfer process. The extent of anodic shift in Ecorr is relatively lower for Al samples coated at 2 and 3 mA cm−2 when compared to the untreated one while it is relatively higher for those coated at 4 and 5 mA cm−2 (Fig. 10(b)). An increase in current density from 2 to 5 mA cm−2 has increased the weight of the iron–manganese phosphate composite coating deposited on Al (Fig. 1(a)). Hence, it implies that a higher coating weight obtained at 4 and 5 mA cm−2 provides a better surface coverage of Al that enables a higher anodic shift in Ecorr. The ability of the coated Al to limit the charge transfer process is also reflected by the distinct passivation and a higher Eb (Fig. 10(a)). A comparison of the Eb of untreated and coated Al indicates a higher Eb for the later one (Fig. 10(c)). Among the coated ones, the Eb is found to increase with an increase in current density employed for deposition (Fig. 10(c)). In spite of an anodic shift in Ecorr and an increase in Eb, the icorr of coated Al is relatively higher (5.43 to 13.16 μA cm−2) than the untreated one (3.28 μA cm−2). In addition, the icorr of coated Al is found to increase with an increase in current density employed for deposition (from 5.43 to 13.16 μA cm−2 for those coated at 2 to 5 mA cm−2).
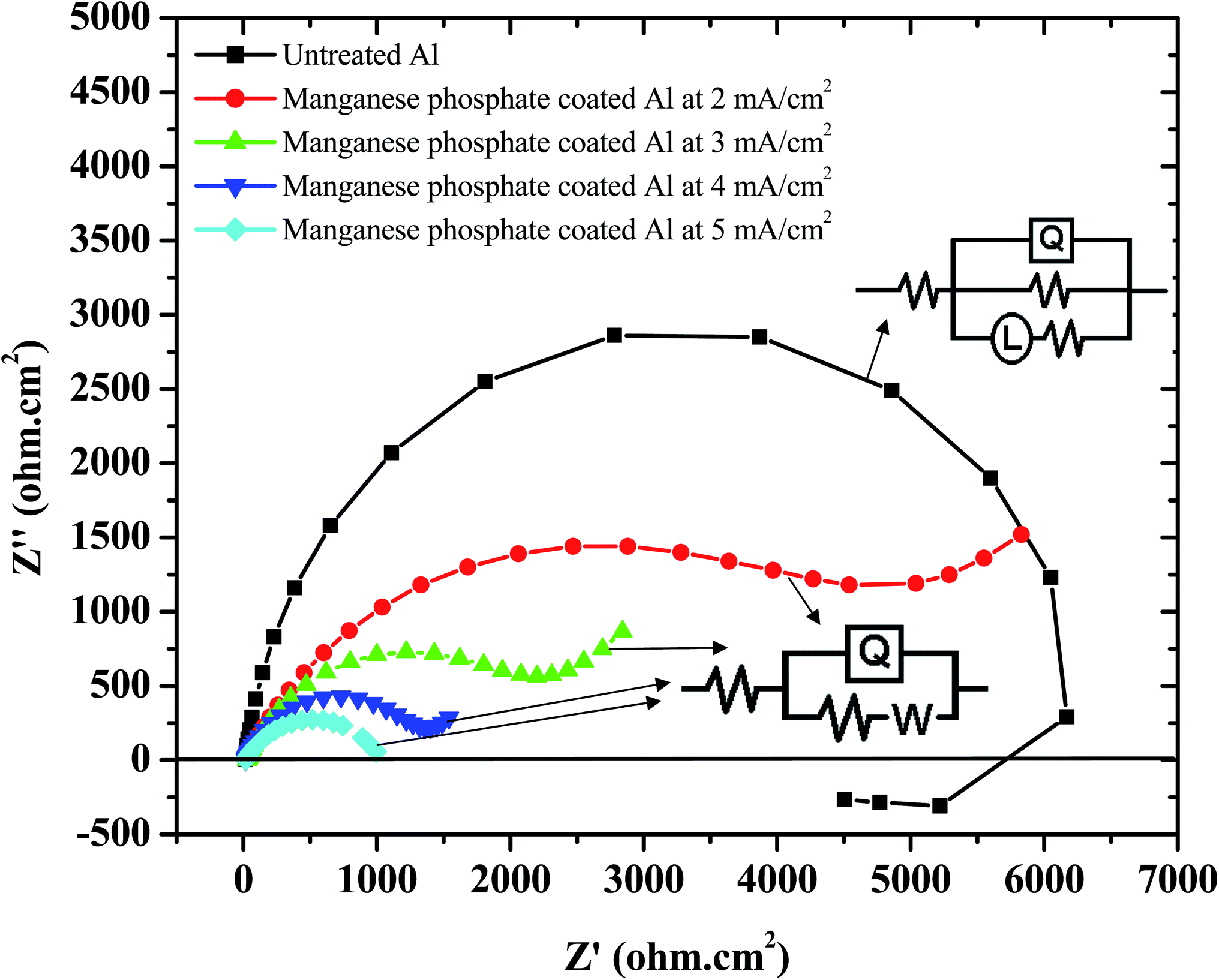
The Nyquist plots of untreated Al and those coated with iron–manganese phosphate composite by cathodic electrochemical treatment at 2 to 5 mA cm−2, in 3.5% NaCl, obtained at their respective OCP's, are shown in Fig. 11. Untreated Al exhibits a semicircle in the high-frequency region followed by an inductive loop at the low frequency region. The occurrence of the inductive loop suggests dissolution of Al in 3.5% NaCl. The Al samples coated with iron–manganese phosphate composite fails to show the inductive loop at the low frequency region. This is due to the coverage of the Al surface by the iron–manganese phosphate composite, which serves as a barrier layer, thus preventing the dissolution of Al in the corrosive medium. The Nyquist plots of the coated Al samples exhibit a semicircle in the high frequency region followed by a typical Warburg impedance behaviour at the low frequency region, suggesting that the corrosion of coated Al is under diffusion control. Hence, two different electrical circuits (shown in the inset of Fig. 11 and marked by arrows) are used to fit the EIS data of untreated and coated Al. The electrochemical parameters derived from the Nyquist plots after fitting are compiled in Table 3. It is evident that the resistance of coated Al samples are relatively lower than that of the untreated one. In addition, among the coated ones, in spite of an increase in coating weight, the resistance is found to decrease with an increase in current density employed for deposition. The inferences made in the EIS studies corroborate well with the observations of the polarization study. It is surprising to note at the first instant that the resistance of coated Al is lower than that of the untreated one and, among the coated ones, those having a higher coating weight exhibit a higher current density/lower resistance. This is due to the chemical nature of the coatings deposited of Al by cathodic electrochemical treatment, which is not pure manganese phosphate, rather, a composite of iron and manganese phosphates. EDAX analysis (Fig. 4) performed on coated Al, XRD pattern (Fig. 7(a)) and XPS analysis (Fig. 8(e)) confirms the presence of Fe in the coating. Hence, the higher icorr and lower resistance observed for the coated Al when compared to the untreated one is due to the dissolution of iron from the iron–manganese phosphate composite coating in 3.5% NaCl.
 |
| | Fig. 11 Nyquist plots of untreated Al and those coated with iron–manganese phosphate composite by cathodic electrochemical treatment at 2 to 5 mA cm−2, in 3.5% NaCl, obtained at their respective OCP's (insets: equivalent electrical circuits used to fit the data of untreated and iron–manganese phosphate coated Al). | |
Table 3 Electrochemical parameters derived from the Nyquist plots of untreated Al and those coated with iron–manganese phosphate composite obtained by cathodic electrochemical treatment at 2 to 5 mA cm−2
| Type of substrate |
Rs (ohm cm2) |
CPE (F cm−2) |
n |
R1 (ohm cm2) |
L (Henri) |
W ohm s−1/2 |
R2 (ohm cm2) |
| Untreated Al |
15.25 |
1.011 × 10−5 |
0.92 |
6581 |
1.959 × 104 |
— |
1.702 × 104 |
| Fe–Mn phosphate coated Al at 2 mA cm−2 |
22.77 |
6.722 × 10−5 |
0.68 |
4658 |
— |
0.001925 |
— |
| Fe–Mn phosphate coated Al at 3 mA cm−2 |
27.81 |
1.271 × 10−4 |
0.72 |
2166 |
— |
0.003605 |
— |
| Fe–Mn phosphate coated Al at 4 mA cm−2 |
17.31 |
9.159 × 10−5 |
0.72 |
1318 |
— |
0.00767 |
— |
| Fe–Mn phosphate coated Al at 5 mA cm−2 |
16.92 |
3.722 × 10−4 |
0.63 |
1014 |
— |
0.8719 |
— |
In order to get a better understanding on the extent of surface coverage of Al by iron–manganese phosphate composite coatings deposited at 2 to 5 mA cm−2 for 30 min, the coated Al samples are subsequently subjected to immersion plating of Cu using a CuSO4-ethylenediamine solution at 27 °C for 2 h. The amount of Cu plated on untreated and coated Al is estimated after stripping the plated Cu in dilute HNO3, complexing it using DEDTC and estimating the absorbance of Cu–DEDTC complex at 435 nm. The photographs of untreated and coated Al after Cu plating along with the amount of Cu plated on them are shown in Fig. 12. It is evident that when compared to the untreated Al and those coated at 2 and 3 mA cm−2, a significant decrease in the extent of Cu plated on Al samples coated at 4 and 5 mA cm−2. Deposition of Cu on the surface of Al by immersion in CuSO4-ethylenediamine is a galvanic plating process driven by the potential difference between Al and Cu. For untreated Al, the entire surface is available for plating and hence a higher amount of Cu (1.8292 mg cm−2) is plated on its surface. For coated Al, the deposition of Cu on Al will be possible only by permeation of the electrolyte through the pores. An increase in current density from 2 to 5 mA cm−2 has increased the amount and compactness of the iron–manganese phosphate composite coating and reduced the porosity of the coating. The pronounced decrease in the amount of Cu plated on Al samples coated at 4 and 5 mA cm−2 points out a better surface coverage.
 |
| | Fig. 12 Amount of copper plated on untreated aluminium and those coated with iron–manganese phosphate composite at 2 to 5 mA cm−2 (inset: photographs of untreated and coated aluminium after immersion in CuSO4-ethylenediamine solution at 27 °C for 2 h). | |
CTT recorded at different potentials for a suitable duration of time provides a good understanding of the tendency of the metal/coated metal to corrode and/or passivate. In addition, recording CTT at potentials near Eb will indicate the susceptibility of the metal/coated metal to pitting corrosion.60,61,64,72 In the present study, CTT were recorded at −650 mV(SCE), −500 mV(SCE) and −300 mV(SCE) for 900 s. CTT recorded at −650 mV(SCE) for 900 s indicates the ability of iron–manganese phosphate composite coated Al in improving the corrosion resistance (Fig. 13) while those recorded at −500 mV(SCE) (Fig. 14(a)) and at −300 mV(SCE) (Fig. 14(b)) for 900 s points out their ability in reducing the susceptibility of Al for pitting corrosion.
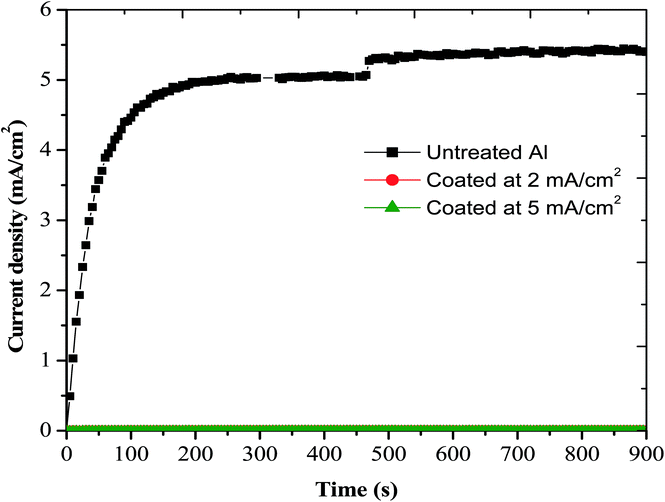 |
| | Fig. 13 Current–time transients of untreated Al and those coated with iron–manganese phosphate composite by cathodic electrochemical treatment at 2 and 5 mA cm−2, in 3.5% NaCl, recorded at −650 mV(SCE) for 900 s. | |
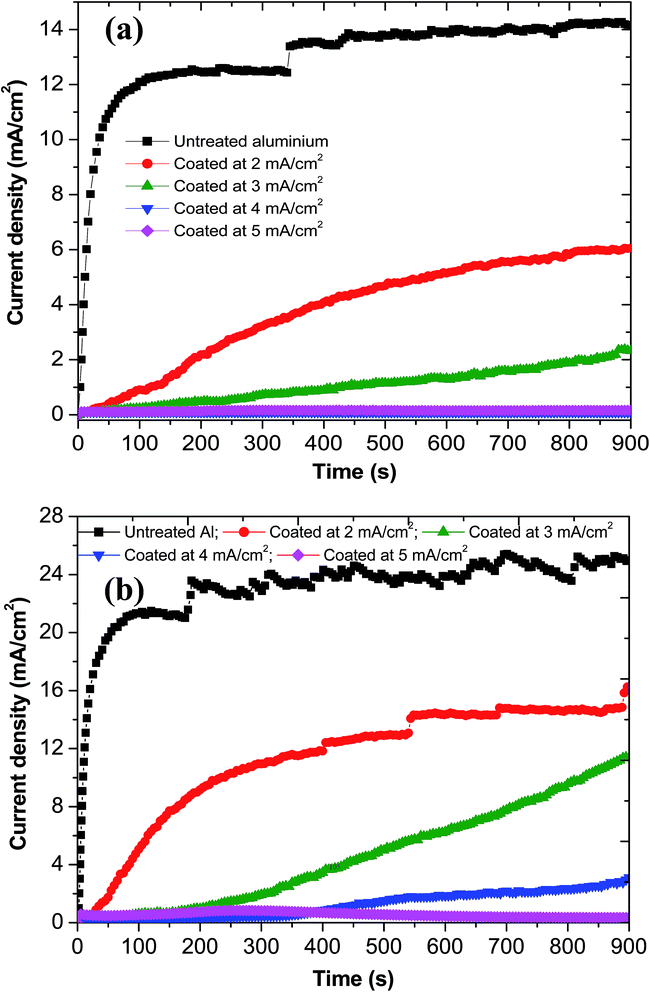 |
| | Fig. 14 Current–time transients of untreated Al and those coated with iron–manganese phosphate composite by cathodic electrochemical treatment at 2 to 5 mA cm−2, in 3.5% NaCl, recorded at (a) −500 mV(SCE) for 900 s; and (b) −300 mV(SCE) for 900 s. | |
It is interesting to note the change in shape of the anodic polarization curves of coated Al at potentials beyond −300 mV(SCE) (Fig. 10(a)). For untreated Al and those coated at 2 mA cm−2, a steady increase in current is observed after their Eb at −736 mV(SCE) and −313 mV(SCE) vs. SCE, respectively. In contrast, those coated at 3, 4 and 5 mA cm−2 exhibit a secondary passivation and a subsequent breakdown at relatively higher potentials. This is believed to be due to the formation of iron oxide on the surface of corroding Al that facilitates the secondary passivation. According to Sherif,72 during corrosion of unalloyed iron in neutral NaCl, the formation and transformation of iron(II) hydroxide into iron oxide by the reaction with the excess of oxygen present in NaCl medium has enabled a larger passivation while a further increase in potential towards more anodic direction has resulted in the breakdown of the passive film. In order to get a better understanding of this phenomenon, optical micrographs of untreated and coated Al after polarization study as well as after the CTT study at −300 mV(SCE) for 900 s are acquired (Fig. 15). The optical micrographs of untreated Al and those coated at 2 to 5 mA cm−2 for 30 min before corrosion are also included in Fig. 15 for an effective comparison. Untreated Al exhibits severe pitting after the polarization and CTT studies (Fig. 15(b and c)). Al samples phosphated at 2 and 3 mA cm−2 also exhibit some pitting while a significant reduction in the extent of pitting is observed for Al samples phosphated at 4 and 5 mA cm−2. The presence of iron corrosion product (regions which appear in brown colour in Fig. 15(k, l, n and o)) is clearly visible on Al samples phosphated at 4 and 5 mA cm−2. Hence, the secondary passivation observed in the polarization curves of these samples is due to the formation of iron oxides on their surface. The extent of iron oxide formation is rather limited on Al samples phosphated at 2 and 3 mA cm−2 (marked as ‘○’ in Fig. 15(e and h)). The extent of iron oxide formation on coated Al is a function of the iron deposited along with manganese phosphates; the higher the current density employed for deposition, the higher the iron content and the higher the iron oxide formation.
 |
| | Fig. 15 Optical micrographs of untreated and iron–manganese phosphate composite coated Al prepared by cathodic electrochemical treatment at 2 to 5 mA cm−2: (a, d, g, j and m) before subjecting them to corrosion; (b, e, h, k and n) after polarization study; and (c, f, i, l and o) after CTT study at −300 mV(SCE) for 900 s: (a–c) untreated Al; (d–f) Al coated at 2 mA cm−2; (g–i) Al coated at 3 mA cm−2; (j–l) Al coated at 4 mA cm−2; and (m–o) Al coated at 5 mA cm−2. | |
4. Conclusions
The findings of this study showed that it is possible to use cathodic electrochemical treatment to deposit manganese phosphate coatings on Al. The coating is uniform, compact and adherent to Al. XRD, FT-IR, Raman and XPS analysis confirm that the coating is a composite of iron and phosphates of manganese. During this treatment, deposition of coatings has also occurred on the mild steel anode besides the Al cathode. The morphological features, chemical nature and phase content of the coatings deposited on mild steel anode are quite similar to those deposited on the Al cathode. Based on the inferences made during deposition and coating characteristics, a pictorial model is proposed to explain the formation of manganese phosphate coatings on Al cathode and mild steel anode. The anodic shift in Ecorr and a higher Eb as well as the absence of an inductive loop indicate the ability of the iron–manganese phosphate composite coating to serve as a barrier layer for the corrosion of Al in 3.5% NaCl. Immersion plating of Cu substantiates the ability of the coated Al to offer a better surface coverage with reduced porosity. CTT studies performed at different impressed potentials confirm the ability of the coated Al to improve the corrosion resistance as well as to reduce the susceptibility of Al for pitting corrosion. Nevertheless, the dissolution of iron from the composite coating leads to a higher icorr and lower charge transfer resistance. The presence of iron in the composite coatings deposited on Al enables a secondary passivation, which is further substantiated by the formation of brown colour iron oxide on their surface after polarization and CTT studies by optical micrographs. Based on the inferences made in the present study, it can be concluded that cathodic electrochemical treatment is a facile approach for the deposition of iron–manganese phosphate composite coatings on Al, which would otherwise be difficult to obtain by the conventional chemical conversion methodology.
Acknowledgements
One of the authors, S. Shanmugam, expresses his sincere thanks to University of Madras for providing financial assistance in the form of Teaching cum Research Fellowship (TRF) to carry out this research work. The authors thank the Director, National Centre for Nano Science and Nanotechnology (NCNSNT), University of Madras for extending facilities, viz., FE-SEM, EDX, XPS and Raman spectroscopy. The support provided by Dr T. M. Sridhar, Dr B. Venkatachalapathy and Dr L. Sujatha, Rajalakshmi Engineering College, Thandalam, Chennai-602105, for performing the electrochemical and optical microstructural studies using Bio-Logic SP-240 work station and Leica DM2700M is gratefully acknowledged.
References
- R. Faria, P. Marques, P. Moura, F. Freire, J. Delgado and A. T. de Almeida, Renewable Sustainable Energy Rev., 2013, 24, 271 CrossRef.
- H. C. Kim and T. J. Wallington, Environ. Sci. Technol., 2013, 47, 6089 CrossRef CAS PubMed.
- A. Mayyas, A. Qattawi, M. Omar and D. Shana, Renewable Sustainable Energy Rev., 2012, 16, 1845 CrossRef.
- K. Ogle and M. Wolpers, in Corrosion: Fundamentals, Testing, and Protection, ASM Handbook, ASM International, Ohio, 2003, vol. 13A, p. 712 Search PubMed.
- T. S. N. Sankara Narayanan, Adv. Mater. Sci., 2005, 9, 130 Search PubMed.
- P. L. Hagans and C. M. Haas, in Surface Engineering, ASM Handbook, ASM International, Ohio, 1994, vol. 5, p. 405 Search PubMed.
- B. L. Jiang and Y. M. Wang, in Surface Engineering of Light Alloys Aluminium, Magnesium and Titanium Alloys, Woodhead Publishing, Series in Metals and Surface Engineering, 2010, ch. 5, p. 110 Search PubMed.
- H. Fadaee and M. Javidi, J. Alloys Compd., 2014, 604, 36 CrossRef CAS.
- M. Franco, W. Sha, S. Malinov and R. Rajendran, Surf. Coat. Technol., 2013, 235, 755 CrossRef CAS.
- A. Farzaneh, M. Mohammadi, M. Ehteshamzadeh and F. Mohammadi, Appl. Surf. Sci., 2013, 276, 697 CrossRef CAS.
- Ph. Hivart, B. Hauw, J. Crapon and J. P. Bricout, Wear, 1998, 219, 195 CrossRef CAS.
- C.-M. Wang, H.-C. Liau and W.-T. Tsai, Surf. Coat. Technol., 2006, 201, 2994 CrossRef CAS.
- Y. Totik, Surf. Coat. Technol., 2006, 200, 2711 CrossRef CAS.
- J. D. B. De Mello, H. L. Costa and R. Binder, Wear, 2007, 263, 842 CrossRef CAS.
- L. Fang, L.-B. Xie, J. Hu, Y. Li and W.-T. Zhang, Phys. Procedia, 2011, 18, 227 CrossRef CAS.
- X.-B. Chen, X. Zhou, T. B. Abbott, M. A. Easton and N. Birbilis, Surf. Coat. Technol., 2013, 217, 147 CrossRef CAS.
- X.-J. Cui, C.-H. Liu, R.-S. Yang, Q.-S. Fu, X.-Z. Lin and M. Gong, Corros. Sci., 2013, 76, 474 CrossRef CAS.
- A. S. Akhtar, K. C. Wong and K. A. R. Mitchell, Appl. Surf. Sci., 2006, 253, 493 CrossRef CAS.
- A. S. Akhtar, K. C. Wong, P. C. Wong and K. A. R. Mitchell, Thin Solid Films, 2007, 515, 7899 CrossRef CAS.
- V. Burokas, A. Martušienė and O. Girčienė, Surf. Coat. Technol., 2007, 202, 239 CrossRef CAS.
- S. Jegannathan, T. S. N. Sankara Narayanan, K. Ravichandran and S. Rajeswari, Surf. Coat. Technol., 2006, 200, 4117 CrossRef CAS.
- S. Jegannathan, T. S. N. Sankara Narayanan, K. Ravichandran and S. Rajeswari, Electrochim. Acta, 2005, 51, 247 CrossRef CAS.
- T. S. N. Sankara Narayanan, S. Jegannathan and K. Ravichandran, Prog. Org. Coat., 2006, 55, 355 CrossRef CAS.
- S. Jegannathan, T. K. Arumugam, T. S. N. Sankara Narayanan and K. Ravichandran, Prog. Org. Coat., 2009, 65, 229 CrossRef CAS.
- C. Kavitha, Ph.D. thesis, University of Madras, 2013.
- C. Kavitha, T. S. N. Sankara Narayanan, K. Ravichandran, I. S. Park and M. Ho Lee, J. Coat. Technol. Res., 2014, 11, 431 CrossRef CAS.
- F. Simescu and H. Idrissi, Corros. Sci., 2009, 51, 833 CrossRef CAS.
- A. A. Oskuie, A. Afshar and H. Hasannejad, Surf. Coat. Technol., 2010, 205, 2302 CrossRef CAS.
- S. Hirsch and C. Rosenstein, Met. Finish., 2002, 100, 421 CrossRef.
- S. Zhang, H. Chen, X. Zhang and M. Zhang, Surf. Coat. Technol., 2008, 202, 1674 CrossRef CAS.
- M. Sheng, C. Wang, Q. Zhong, Y. Wei and Y. Wang, Ultrason. Sonochem., 2010, 17, 21 CrossRef CAS PubMed.
- S. Jegannathan, T. S. N. Sankara Narayanan, K. Ravichandran and S. Rajeswari, Surf. Coat. Technol., 2006, 200, 6014 CrossRef CAS.
- S. Jegannathan, T. S. N. Sankara Narayanan, K. Ravichandran and S. Rajeswari, Prog. Org. Coat., 2006, 57, 392 CrossRef CAS.
- R. L. Frost, Y. Xi, R. Scholz, A. López and F. M. Belotti, Vib. Spectrosc., 2013, 66, 69 CrossRef CAS.
- H. Yin, F. Liu, X. Chen, X. Feng, W. Tan and G. Qiu, Microporous Mesoporous Mater., 2012, 153, 115 CrossRef CAS.
- G. Qiu, Z. Gao, H. Yin, X. Feng, W. Tan and F. Liu, Solid State Sci., 2010, 12, 808 CrossRef CAS.
- E. S. Larreaa, J. L. Mesa, J. L. Pizarro, M. I. Arriortua and T. Rojo, J. Solid State Chem., 2007, 180, 1686 CrossRef.
- B. Boonchom and C. Danvirutai, Ind. Eng. Chem. Res., 2008, 47, 2941 CrossRef CAS.
- C. Danvirutai, B. Boonchom and S. Youngme, J. Alloys Compd., 2008, 457, 75 CrossRef CAS.
- Y. Chen, B. L. Luan, G.-L. Song, Q. Yang, D. M. Kingston and F. Bensebaa, Surf. Coat. Technol., 2012, 210, 156 CrossRef CAS.
- A. J. Nelson, J. G. Reynolds and J. W. Roos, Sci. Total Environ., 2002, 295, 183 CrossRef CAS PubMed.
- C. V. Krishnamohan Sharma, C. C. Chusuei, R. Clerac, T. Moller, K. R. Dunbar and A. Clearfield, Inorg. Chem., 2003, 42, 8300 CrossRef CAS PubMed.
- J. Zong, Q. Peng, J. Yu and X. Liu, J. Power Sources, 2013, 228, 214 CrossRef CAS.
- G. Wang, N. Cao and Y. Wang, RSC Adv., 2014, 4, 59772 RSC.
- L. Wu, L. Zhao, J. Dong, W. Ke and N. Chen, Electrochim. Acta, 2014, 145, 71 CrossRef CAS.
- A. dos Santos, J. R. Araujo, S. M. Landi, A. Kuznetsov, J. M. Granjeiro, L. A. de Sena and C. A. Achete, J. Mater. Sci.: Mater. Med., 2014, 25, 1769 CrossRef CAS PubMed.
- M. Mosiałek, G. Mordarski, P. Nowak, W. Simka, G. Nawrat, M. Hanke, R. P. Socha and J. Michalska, Surf. Coat. Technol., 2011, 206, 51 CrossRef.
- V. Hannelore, H. Noller, M. Ebel and K. Schwarz, J. Chem. Soc., Faraday Trans. 1, 1977, 73, 734 RSC.
- G. Zorn, I. Gotman, E. Y. Gutmanas, R. Adadi, G. Salitra and C. N. Sukenik, Chem. Mater., 2005, 17, 4218 CrossRef CAS.
- G. Zorn, R. Adadi, R. Brener, V. A. Yakovlev, I. Gotman, E. Y. Gutmanas and C. N. Sukenik, Chem. Mater., 2008, 20, 5368 CrossRef CAS.
- T. Ishizaki, R. Kudo, T. Omi, K. Teshima, T. Sonoda, I. Shigematsu and M. Sakamoto, Electrochim. Acta, 2012, 62, 19 CrossRef CAS.
- S. H. Mohamed and M. Raaif, Surf. Coat. Technol., 2010, 205, 525 CrossRef CAS.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. St. C. Smart, Appl. Surf. Sci., 2011, 257, 2717 CrossRef CAS.
- A. A. Hermas, M. Nakayama and K. Ogura, Electrochim. Acta, 2005, 50, 3640 CrossRef CAS.
- A. R. Brooks, C. R. Clayton, K. Doss and Y. C. Lu, J. Electrochem. Soc., 1986, 133, 2459 CrossRef CAS.
- Y. Hao, F. Liu, E.-H. Han, S. Anjum and G. Xu, Corros. Sci., 2013, 69, 77–86 CrossRef CAS.
- B. Lee, C. Kim, Y. Park, T.-G. Kim and B. Park, Electrochem. Solid-State Lett., 2006, 9, E27–E30 CrossRef CAS.
- K. Gopala Krishna, K. Sivaprasad, T. S. N. Sankara Narayanan and K. C. Hari Kumar, Corros. Sci., 2012, 60, 82 CrossRef CAS.
- Z. Szklarska-Smialowska, Corros. Sci., 1999, 41, 1743 CrossRef CAS.
- R. K. Gupta, N. L. Sukiman, M. K. Cavanaugh, B. R. W. Hinton, C. R. Hutchinson and N. Birbilis, Electrochim. Acta, 2012, 66, 245 CrossRef CAS.
- O. Guseva, P. Schmutz, T. Suter and O. von Trzebiatowski, Electrochim. Acta, 2009, 54, 4514 CrossRef CAS.
- J. Ma, J. Wen, Q. Li and Q. Zhang, Int. J. Hydrogen Energy, 2013, 38, 14896 CrossRef CAS.
- A. R. Trueman, Corros. Sci., 2005, 47, 2240 CrossRef CAS.
- K. D. Ralston, D. Fabijanic and N. Birbilis, Electrochim. Acta, 2011, 56, 1729 CrossRef CAS.
- M. Trueba and S. P. Trasatti, Mater. Chem. Phys., 2010, 121, 523 CrossRef CAS.
- M. K. Cavanaugh, N. Birbilis and R. G. Buchheit, Electrochim. Acta, 2012, 59, 336 CrossRef CAS.
- K. D. Ralston, N. Birbilis, M. Weyland and C. R. Hutchinson, Acta Mater., 2010, 58, 5941 CrossRef CAS.
- M. K. Cavanaugh, N. Birbilis, R. G. Buchheit and F. Bovard, Scr. Mater., 2007, 56, 995 CrossRef CAS.
- A. Aballe, M. Bethencourt, F. J. Botana, M. J. Cano and M. Marcos, Corros. Sci., 2001, 43, 1657 CrossRef CAS.
- E. McCafferty, Corros. Sci., 2003, 45, 1421 CrossRef CAS.
- E. M. Sherif, Mater. Chem. Phys., 2011, 129, 961 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra08195a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.