
New MCR based on intramolecular Heck reaction under aerobic conditions: a direct access to cytotoxic fused N-heterocycles†
Raju Adepu,
Bagineni Prasad,
Mohd Ashraf Ashfaq,
Nasreen Z. Ehtesham and
Manojit Pal*
Dr Reddy's Institute of Life Sciences, University of Hyderabad Campus, Gachibowli, Hyderabad 500 046, India. Tel: +91 40 6657 1500E-mail: manojitpal@rediffmail.com
First published on 29th September 2014
Abstract
We report a new MCR involving the reaction of isatoic anhydrides, allyl amine and o-bromo arylaldehydes in the presence of Pd(OAc)2, X-Phos and air to afford various isoquinolino[1,2-b]quinazolinones as new cytotoxic agents. The strategy was extended successfully towards the synthesis of a methyl analogue of 7,8-dehydrorutaecarpine.
Multi component reactions (MCRs) catalyzed by transition metals have emerged as powerful tools in modern organic synthesis as they allow quick introduction of complexity into a particular scaffold in a single-step.1 Pd-catalyzed reactions on the other hand allow creation of new C–Z (Z = C or N or O) bonds under milder conditions.2 Thus development of new MCRs based on Pd catalyzed reactions2b is of high demand as these methodologies can easily construct pre-designed complex frameworks relevant to natural products access of which may be difficult or cumbersome via conventional multi-step methods.
Natural products containing N-heterocycles are attractive scaffolds in medicinal/pharmaceutical chemistry as well as in the early stage of drug discovery due to their wide range of remarkable pharmacological properties. For example, indolopyridoquinazolinone alkaloid rutaecarpine (A, Fig. 1) isolated from rutaceae (a tropical family of trees and shrubs) has been explored and evaluated as a potential agent in various therapeutic areas.3 Among the various naturally occurring analogues of rutaecarpine, the 7,8-dehydrorutaecarpines (B, Fig. 1) attracted our particular attention due to their potential in the treatment of Alzheimer's disease (AD).4 At the same time, we also became interested in the structurally similar another framework C i.e. isoquinolino[1,2-b]quinazolinone (Fig. 1) as compounds based on C though seemed to have drug-like properties are rather uncommon in the literature.5a Additionally, in view of the well known anticancer properties5b of isoquinolines and quinazolinones individually, we anticipated that their combined form i.e. C might show anti-proliferative effects against cancer cells. The reported synthesis of B and its analogues are few and involved a lengthy and multistep process.4,6–8 In some cases B has been isolated as a side product in low or poor yield. Notably, the reported synthesis of isoquinolino[1,2-b]quinazolinones are also few.9 We therefore decided to develop a new and general route to construct the framework C extendable to the framework of B. In a retro synthetic analysis we envisaged that ring opening of isatoic anhydride with allyl amine could provide the quinazolin-4-one precursor in situ which on condensation with o-halo benzaldehyde followed by intramolecular Heck reaction could construct the framework C (Fig. 2). Indeed, this strategy worked well as the MCR of isatoic anhydrides (1), allyl amine (2) and o-bromo arylaldehydes (3) in the presence of Pd(OAc)2, ligand X-Phos and air afforded the desired isoquinolino[1,2-b]quinazolinones (4) smoothly (Scheme 1). While combination of MCR with a Pd-catalyzed reaction e.g. Ugi/Heck,10a,b Ugi/C–H activation,10c Ugi–Smiles/Heck10d or Biginelli–Heck10e is known in majority of these cases the individual steps were performed separately in a single pot with the addition of Pd-catalyst after completion of the initial step.10f In contrast the present methodology does not require the monitoring or completion of any initial step as the Pd-catalyst can be added in the beginning of the reaction. Thus the strategy presented here is not common in the literature.
 | ||
| Fig. 1 Rutaecarpine (A), 7,8-dehydrorutaecarpines (B) and isoquinolino[1,2-b]quinazolinone (C). | ||
 | ||
| Fig. 2 Retro synthetic analysis for the construction of framework C. | ||
 | ||
| Scheme 1 New MCR based on intramolecular Heck reaction under aerobic condition leading to 4. | ||
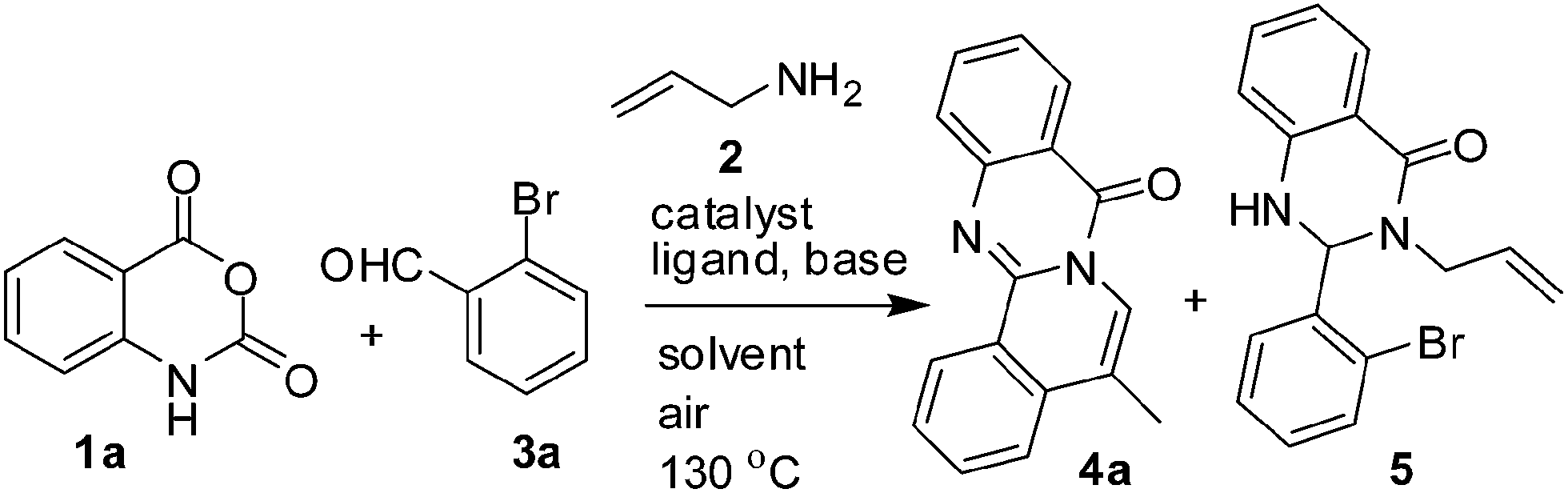
Herein we present our preliminary results of this study.11 Initially, the reaction of isatoic anhydride (1a), allyl amine (2) and 2-bromo benzaldehyde (3a) was examined under various conditions (Table 1). We preferred 3a over the corresponding 2-iodo derivative as 3a is less expensive. The reaction was performed in the presence of Pd(OAc)2 and DIPEA in DMF when the desired product 4a was isolated in poor yield (entry 1, Table 1). A 2,3-dihydroquinazolin-4(1H)-one derivative 5 (thought to be an reaction intermediate) was obtained as a major product in this case. We then examined the effect of ligands such as PPh3, X-Phos and bipyridine (entries 2–4, Table 1). While yield of 4a was improved in all these cases the best result was obtained when X-Phos was used (entry 3, Table 1). To improve the yield of 4a further, the catalyst Pd(OAc)2 was replaced with other Pd-catalysts e.g. Pd2(dba)3, Pd(PPh3)2Cl2 and Pd(PPh3)4 (entry 5–7, Table 1). However, the yield of 4a was not improved. The use of Cu(OAc)2 as an additive also did not improve the yield (entry 8, Table 1). Similar observation was noted when the base DIPEA was replaced with Et3N (entry 9, Table 1) or inorganic bases like K2CO3 and Cs2CO3 (entries 10–11, Table 1). The use of other solvents like DMA, 1,4-dioxane and MeCN in place of DMF afforded lower yield of 4a compared to that of DMF (entry 3 vs. entry 12–14, Table 1). We also varied the quantity of Pd-catalyst used. While the use of a lower quantity of Pd-catalyst decreased the product yield (entry 15, Table 1), the MCR did not provide 4a in the absence of Pd(OAc)2 (entry 16, Table 1) confirming the key role played by the catalyst. Overall, the combination of Pd(OAc)2, X-Phos and DIPEA in DMF was found to be optimum. The compound 4a was characterized by spectral data (see ESI†). The 1HNMR of 4a showed the appearance of Me protons at δ 2.53 along with an allylic coupling i.e. J = 1.2 Hz between the Me protons and the aromatic C-6 proton. The proximity of Me with aromatic C-4 and C-6 protons was confirmed by NOE study (Fig. 3).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Ligand/additive | Base/solvent | % yieldb | |
| 4a | 5 | ||||
| a Reactions were carried out using 1a (1 mmol), 2 (1 mmol), 3a (1 mmol), catalyst (5 mol%), ligand (10 mol%) and base (3 mmol) in a solvent (2 mL) at rt for 5 min and then slowly at 130 °C for 18 h in the presence of air under anhydrous conditions.b Isolated yield.c 1 equiv. of Cu(OAc)2 was used.d Reaction was performed at 100 °C.e Reaction was performed at 80 °C.f 2.5 mol% catalyst and 5 mol% of ligand was used.g Reaction was performed without catalyst and ligand.h N-allyl-2-aminobenzamide was isolated in >70% yield. | |||||
| 1 | Pd(OAc)2 | — | DIPEA/DMF | 30 | 57 |
| 2 | Pd(OAc)2 | PPh3 | DIPEA/DMF | 55 | 21 |
| 3 | Pd(OAc)2 | X-Phos | DIPEA/DMF | 78 | — |
| 4 | Pd(OAc)2 | 2,2′-bipyridine | DIPEA/DMF | 64 | 12 |
| 5 | Pd(dba)3 | — | DIPEA/DMF | 34 | 51 |
| 6 | Pd(PPh3)2Cl2 | — | DIPEA/DMF | 34 | 49 |
| 7 | Pd(PPh3)4 | — | DIPEA/DMF | 31 | 49 |
| 8c | Pd(OAc)2 | Cu(OAc)2 | DIPEA/DMF | 42 | 33 |
| 9 | Pd(OAc)2 | X-Phos | Et3N/DMF | 45 | 29 |
| 10 | Pd(OAc)2 | X-Phos | K2CO3/DMF | 32 | 45 |
| 11 | Pd(OAc)2 | X-Phos | Cs2CO3/DMF | 35 | 44 |
| 12 | Pd(OAc)2 | X-Phos | DIPEA/DMA | 73 | — |
| 13d | Pd(OAc)2 | X-Phos | DIPEA/1,4-dioxane | 45 | 34 |
| 14e | Pd(OAc)2 | X-Phos | DIPEA/MeCN | 22 | 25 |
| 15f | Pd(OAc)2 | X-Phos | DIPEA/DMF | 52 | 30 |
| 16g | — | — | DIPEA/DMF | — | 20h |
 | ||
| Fig. 3 Allylic coupling and NOE correlations observed for compound 4a. | ||
To assess the scope and generality of the newly developed MCR, a number of isatoic anhydrides (1a–e) and a range of 2-bromo arylaldehydes (3a–j) were employed (Table 2). Both electron donating e.g. OH, OMe and OiPr (entry 4–5 & 7–10, Table 2) and electron withdrawing e.g. NO2 group (entry 3 & 13, Table 2) present on the benzene ring of 3 were well tolerated. However, the O-acetyl group was not tolerated under the conditions employed and de-acetylation was observed (entry 6, Table 2). While isatoic anhydrides containing Cl or OMe participated well in the MCR (entries 11–17, Table 2) the presence of an electron withdrawing NO2 group was not favorable (entry 18, Table 2). Since all the reactions were performed in the presence of air hence the methodology does not require the use of any closed vessel or inert atmosphere thereby reducing the risk of pressure development in scale up synthesis. Nevertheless the present MCR appeared to be a general method for accessing isoquinolino[1,2-b]quinazolinones. To expand the scope of this methodology further we extended this strategy successfully towards the synthesis of a natural product analog i.e. 8-methyl-7,8-dehydrorutaecarpine (6). Thus, 6 was prepared via the reaction of 1a, 2 and 3-bromo-1H-indole-2-carbaldehyde (3k)12 in a single synthetic operation (Scheme 2) indicating the potential of this methodology over the conventional multi-step sequences.
|
||||
|---|---|---|---|---|
| Entry | Isatoic anhydride R1, R2 (1) | Aldehyde R3, R4, R5 (3) | Product R1, R2, R3, R4, R5 (4) | % yieldb |
| a All the reactions were carried out using 1 (1 mmol), 2 (1 mmol), 3 (1 mmol), Pd(OAc)2 (5 mol%), X-Phos (10 mol%) and DIPEA (3 mmol) in DMF (2 mL) at rt for 5 min and then slowly at 130 °C for 18 h in the presence of air under anhydrous conditions.b Isolated yield.c Deacetylated product was obtained. | ||||
| 1 | H, H, 1a | H, H, H, 3a | H, H, H, H, H, 4a | 78 |
| 2 | 1a | H, H, F, 3b | H, H, H, H, F, 4b | 69 |
| 3 | 1a | H, H, NO2, 3c | H, H, H, H, NO2, 4c | 65 |
| 4 | 1a | OH, OMe, H, 3d | H, H, OH, OMe, H, 4d | 70 |
| 5 | 1a | OMe, OMe, H, 3e | H, H, OMe, OMe, H, 4e | 71 |
| 6 | 1a | H, OCOCH3, OMe, 3f | H, H, H, OH, OMe, 4f | 62c |
| 7 | 1a | H, OH, OMe, 3g | H, H, H, OH, OMe, 4f | 65 |
| 8 | 1a | H, OMe, OMe, 3h | H, H, H, OMe, OMe, 4g | 70 |
| 9 | 1a | H, OiPr, OMe, 3i | H, H, H, OiPr, OMe, 4h | 73 |
| 10 | 1a | H, OMe, OiPr, 3j | H, H, H, OMe, OiPr, 4i | 72 |
| 11 | Cl, H, 1b | 3a | Cl, H, H, H, H, 4j | 79 |
| 12 | 1b | 3b | Cl, H, H, H, F, 4k | 64 |
| 13 | 1b | 3c | Cl, H, H, H, NO2, 4l | 64 |
| 14 | H, Cl, 1c | 3a | H, Cl, H, H, H, 4m | 75 |
| 15 | 1c | 3b | H, Cl, H, H, F, 4n | 62 |
| 16 | OMe, OMe, 1d | 3a | OMe, OMe, H, H, H, 4o | 66 |
| 17 | 1d | 3b | OMe, OMe, H, H, F, 4p | 60 |
| 18 | NO2, H, 1e | 3a | NO2, H, H, H, H, 4q | 45 |

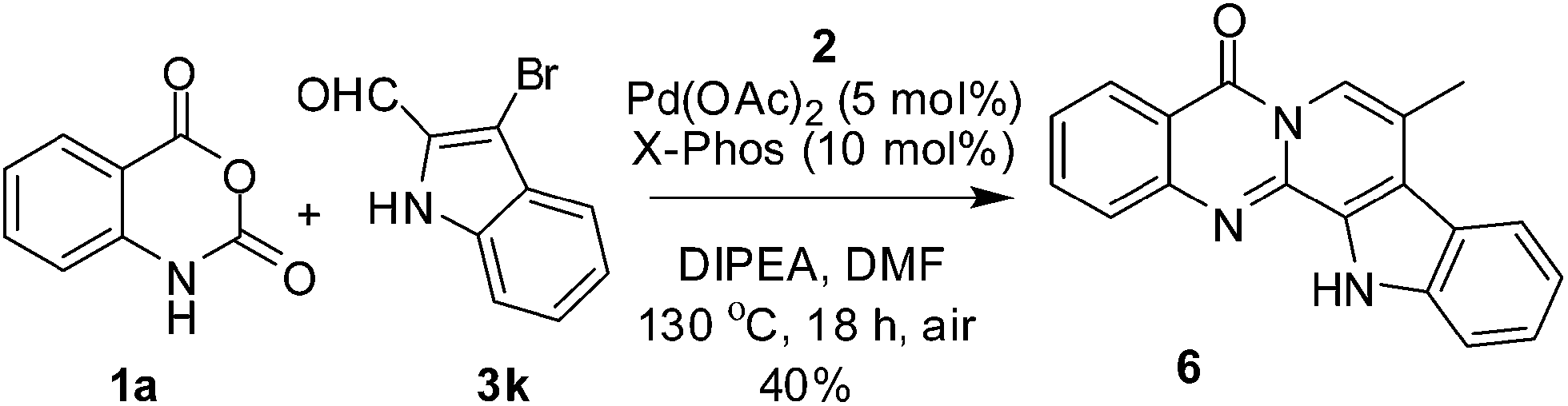 | ||
| Scheme 2 One-step synthesis of a natural product analog 8-methyl 7,8-dehydrorutaecarpine (6). | ||
Based on our observations summarized in Table 1, the reaction seemed to proceed (Scheme 3) via a rapid in situ generation of E-1 (ref. 13) (from 1 and 2) followed by condensation with 3 to give E-2 (cf. compound 5, Table 1). The results of entry 16 of Table 1 indicate that the ligand may have a role in this step. The E-2 then underwent intramolecular Heck reaction via an exo trig fashion in the presence of Pd(0) species generated from Pd(II) in situ.14 The exo trig cyclization leading to E-4 was favored over the endo trig pathway perhaps due to the unfavorable ring strain encountered by the 7-membered ring that would formed in the second case. A typical β-hydride elimination from E-4 followed by the base mediated isomerization afforded E-6 which on aerial oxidation gave 4. To gain further evidence we performed two reactions separately. The N-allyl-2-aminobenzamide 7, prepared from 1a and 2 was treated with 3a under the condition of entry 3 of Table 1 for 18 h when 4a was isolated in 77% yield (Scheme 4). Similarly, the compound 5 afforded 4a in 82% yield when treated with the Pd catalyst maintaining the condition of entry 3 of Table 1 for 10 h (Scheme 4). These observations suggested that the reaction proceeded via E-1 followed by E-2.15
 | ||
| Scheme 3 Proposed reaction mechanism leading to compound 4. | ||
 | ||
| Scheme 4 The reaction of N-allyl-2-aminobenzamide 7 with aldehyde 3a and conversion of 5 to 4a via intramolecular Heck reaction. | ||
Compounds 4a–q were tested against the breast cancer cell e.g. MCF-7 and non-cancer cells e.g. HEK 293T in vitro at 10 μM using the sulphorhodamine B (SRB) assay16a,b with gemcitabine16c as a reference compound. Among the active compounds (Fig 4), 4a, 4e and 4h showed 66, 43 and 47% inhibition (better or comparable to gemcitabine's 49% inhibition) and 4a being the best. However, none of them showed significant inhibition of HEK 293T cells indicating their selectivity towards cancer cells. The 4a therefore is of further interest as a potential agent against breast cancer.
 | ||
| Fig. 4 % inhibition of MCF-7 cells by 4 at 10 μM after 72 h of compound treatment (Gem = Gemcitabine). | ||
In conclusion, a Pd-based new MCR has been developed for the direct access to novel isoquinolino[1,2-b]quinazolinones as potential anticancer agents. The MCR involved the reaction of commercially available isatoic anhydrides, allyl amine and o-bromo arylaldehydes in the presence of Pd(OAc)2, X-Phos and air. This straightforward and operationally simple.
Methodology does not require the monitoring or completion of the initial step before adding the Pd-catalyst. Some of the compounds synthesized showed promising inhibition of breast cancer cell growth and one of them is undergoing further pharmacological evaluation. The strategy was extended successfully towards the synthesis of 8-methyl-7,8-dehydrorutaecarpine avoiding a multi-step procedure. The methodology would find application in constructing library of molecules based on complex and fused N-heterocycles useful for medicinal/pharmaceutical chemistry as well as in the early stage of drug discovery.
RA, BP thank CSIR, for research fellowships. Authors thank DRILS and CSIR [Grant 02(0127)/13/EMR-II] for support.
Notes and references
- For in depth reviews, see:
(a) G. Balme, D. Bouyssi, N. Monteiro, Metal-Catalyzed Multicomponent Reactions in Multicomponent Reactions, ed. J. Zhuand H. Bienayme, Wiley-VCH, Weinheim, Germany, 2005, pp. 224–276 Search PubMed
; (b) D. M. D'Souza and T. J. J. Müller, Multi-component syntheses of heterocycles by transition-metal catalysis, Chem. Soc. Rev., 2007, 37, 1095 RSC
-
(a) For a historical perspective, see: S. C. C. Johansson, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed. Engl., 2012, 51, 5062 CrossRef PubMed
- S. H. Lee, J.-K. Son, B. S. Jeong, T.-C. Jeong, H. W. Chang, E.-S. Lee and Y. Jahng, Molecules, 2008, 13, 272 CrossRef CAS PubMed
- Y. He, P. F. Yao, S. B. Chen, Z. H. Huang, S. L. Huang, J. H. Tan, D. Li, L. Q. Gu and Z. S. Huang, Eur. J. Med. Chem., 2013, 63, 299 CrossRef CAS PubMed
-
(a) For recent examples, see: H. Lu, Q. Yang, Y. Zhou, Y. Guo, Z. Deng, Q. Ding and Y. Peng, Org. Biomol. Chem., 2014, 12, 758 RSC
- H. Möhrle, C. Kamper and R. Schmid, Arch. Pharm., 1980, 313, 990 CrossRef
-
(a) J. Bergman and S. Bergman, J. Org. Chem., 1985, 50, 1246 CrossRef CAS
- M.-C. Tseng, H.-T. Cheng, M.-J. Shen and Y.-H. Chu, Org. Lett., 2011, 13, 4434 CrossRef CAS PubMed
- For previous synthesis of isoquinolino[1,2-b]quinazolinones, see:
(a) A. Maity, S. Mondal, R. Paira, A. Hazra, S. Naskar, K. B. Sahu, P. Saha, S. Banerjee and N. B. Mondal, Tetrahedron Lett., 2011, 52, 3033 CrossRef CAS
- For selected examples, see:
(a) Z. Ma, Z. Xiang, T. Luo, K. Lu, Z. Xu, J. Chen and Z. Yang, J. Comb. Chem., 2006, 8, 696 CrossRef CAS PubMed
- For our earlier efforts on MCR, see:
(a) K. S. Kumar, P. M. Kumar, K. A. Kumar, M. Sreenivasulu, A. A. Jafar, D. Rambabu, G. R. Krishna, C. M. Reddy, R. Kapavarapu, K. Shivakumar, K. K. Priya, K. V. L. Parsa and M. Pal, Chem. Commun., 2011, 47, 5010 RSC
- The indole 3k is commercially available (CAS: 906440-21-9). However, it can also be prepared according to the reported methods, see:
(a) L. Wang, X. Xie and Y. Liu, Angew. Chem., Int. Ed., 2013, 52, 13302 CrossRef CAS PubMed
- The effervescence of CO2 was observed in the beginning of the reaction during rapid generation of E-1. Notably, in spite of low bp of 2 (55–58 °C) the reaction proceeded well at 130 °C as the highly reactive 1 reacted with 2 within initial 15 min when the temp was increased gradually. This perhaps also explains the preferred formation of E-1 over the imine from 2 and 3. Alternatively, the equilibrium of imine formation shifts towards left as more of 2 is consumed by 1.
-
(a) The intramolecular Heck reaction proceeds via (i) the dissociation of a ligand bound to the Pd(0) complex E-3, (ii) coordination of Pd with the alkene moiety followed by (iii) insertion of alkene into the C–Pd bond to give E-4. For a scholarly review, see: A. Jutand, in The Mizoroki–Heck Reaction, ed. M. Oestreich, John Wiley & Sons, Ltd, 2009. ISBN: 978-0-470-03394-4;
(b) The reason for the aerobic Heck coupling working better with X-Phos was probably due to the ability of this ligand to avoid a rapid oxidation of Pd(0) in the presence air;
(c) Notably, no Pd(0) catalyzed deallylation F. Garro-Helion, A. Merzouk and F. Guibe, J. Org. Chem., 1993, 58, 6109 CrossRef CAS
- Alternatively, while the reaction may proceed through an imine (via the reaction of 2 with 3) which on reaction with 1 can afford E-2 (see: B. K. Banik, V. S. Raju, M. S. Manhas and A. K. Bose, Heterocycles, 1998, 47, 639 CrossRef CAS
-
(a) L. V. Rubinstein, R. H. Shoemaker, K. D. Paull, R. M. Simon, S. Tosini, P. Skehan, D. A. Scudiero, A. Monks and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1113 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra10702k |
| This journal is © The Royal Society of Chemistry 2014 |