
Antitumor efficacy of a PLGA composite nanofiber embedded with doxorubicin@MSNs and hydroxycamptothecin@HANPs†
Mengxia Chen‡
a,
Wei Feng‡a,
Si Lina,
Chuanglong He *a,
Yu Gaob and
Hongsheng Wang*a
*a,
Yu Gaob and
Hongsheng Wang*a
aCollege of Chemistry, Chemical Engineering and Biotechnology, Donghua University, 2999 North Renmin Road, Shanghai 201620, China. E-mail: whs@dhu.edu.cn; hcl@dhu.edu.cn; Fax: +86 21 6779 2742; Tel: +86 21 6779 2742
bResearch Institute of Donghua University, Donghua University, Shanghai 201620, China
First published on 7th October 2014
Abstract
A co-delivery system with two or more anticancer drugs has been proposed to minimize the dosage of drug and to achieve the synergistic therapeutic effect in cancer therapy. In this study, we present a dual drug delivery system for the co-release of two anticancer drugs doxorubicin hydrochloride (DOX) and hydroxycamptothecin (CPT). To achieve this goal, DOX and CPT were first separately loaded into mesoporous silica and hydroxyapatite nanocarriers, thus, the two prepared drug loaded nanocarriers were then simultaneously incorporated into poly(lactic-co-glycolic acid) (PLGA) nanofibers by electrospinning. The as-prepared medicated nanofibers were well-characterized by different assays, and the results demonstrated that both of the two drug loaded nanocarriers were successfully incorporated into PLGA nanofibers. The in vitro release study indicated that the loaded DOX and CPT exhibited a sustained and controlled release behavior from the dual drug loaded nanofibers. Furthermore, the dual drug loaded nanofibers displayed a superior capacity of inhibiting HeLa cells in vitro to the single drug loaded PLGA nanofibers. Thus, the synthesized dual drug loaded composite nanofibers may find a promising application for cancer therapy.
Introduction
In recent years, cancer has become a leading cause of death worldwide.1–3 Moreover, local cancer recurrence still is a major clinical problem after surgical treatment for most cancers, which is usually caused by inadequate resection or implantation during surgery.4 In order to cure cancer, numerous anticancer drugs have been used, such as methotrexate,5 vinca alkaloids,6 cisplatin,7 paclitaxel,8 doxorubicin hydrochloride (DOX)4 and hydroxycamptothecin (CPT).9 DOX, a class I anthracycline antibiotic, can effectively kill cancer cells by damaging DNA and its synthesis through mechanisms of intercalation between nucleotides, inhibition of topoisomerase II, and generating oxygen free radicals.10 CPT, a pentacyclic quinoline plant alkaloid, can kill cancer cells efficiently by inhibiting the synthesis of both DNA and RNA.9 In the field of cancer treatment, it is rare to use a single drug due to the toxicity of the drug at high dosage and the heterogeneity of cancer cells and its drug resistance.11,12 To improve the therapeutic efficacy and reduce the other side effects, combination therapy is a promising strategy.13 However, the combination of free drugs is difficult to obtain optimal anticancer effect due to their serious toxic side effects to human bodies, different biochemical activities and pharmacokinetics.13 Therefore, co-delivery system containing two or more different drugs has been proposed to minimize the dosage of drug and to achieve synergistic therapeutic effect in treating cancers.14 Many multi-drug delivery carriers have been developed for co-delivery of different drugs, including nanoparticles,15 liposomes,16 nanofibers,17 polymer–drug conjugates,18 and micelles.19 For example, Zhang and the coauthors have demonstrated that the combination of DOX and CPT could improve the anticancer effect.20In recent years, electrospun nanofibers have received intense attention because of its high specific surface area ratio and high porosity for drug delivery.17 The drug release from electrospun nanofibers can be controlled by the architecture, porosity, and composition of nanofibers.21 Moreover, the medicated nanofibers can be easily set to the targeted area by adjusting their shape and size.22 Thus, the drug-loaded nanofibrous scaffolds has been supposed to have a promising application on preventing local tumor recurrence after surgery.4 Poly(lactic-co-glycolic acid) (PLGA) has been widely used to fabricate nanofibers for drug delivery applications due to its excellent biocompatibility and biodegradability.23 However, the drug encapsulated in electrospun nanofibers result in a burst release behavior due to the drug particles are likely to locate on the fiber surface owing to their high ionic strength in solution and the rapid evaporation of the solvent during electrospinning.24 To overcome this limitation, some nanoscale carriers, including mesoporous silica nanoparticles (MSNs),4,25,26 hydroxyapatite nanoparticles (HANPs),27 and liposomes28 have been incorporated into electrospun nanofibers for potential cancer treatment.23 Mesoporous silica nanoparticles (MSNs) have recently attracted tremendous attention due to their good biocompatibility and drug-loading capacity.29,30 HANPs have also been proposed as a drug carrier because of its high surface activity and strong surface adsorptive capacity.23 To our knowledge, however, no study has been performed to create a dual-drug delivery system that simultaneously contains both MSNs and HANPs nanocarriers for co-release of two different anticancer drugs.
Herein, we fabricated a PLGA-based nanofibrous mats for loading two anticancer drugs, DOX and CPT, which were respectively incorporated into MSNs and HANPs nanocarriers. The fabrication process of the composite nanofibers is schematic represented in Fig. 1. The fabricated electrospun mats were characterized intensively including morphology, structure, drug release and cytotoxicity using various assays.31 These results provided a strong support for the potential application of these dual drug loaded nanofibers in cancer therapy.
 | ||
| Fig. 1 Schematic illustration for the process of fabrication of PLGA/DOX@MSNs & CPT@HANPs electrospun composite nanofibers. | ||
Experimental
Materials
PLGA copolymers with LA/GA ratio of 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 (Mw = 110 kDa) was purchased from Daigang Biomaterials Inc. (Jinan, China). Hydroxyapatite was obtained from Aladdin Chemical Reagent Co., Ltd. (Shanghai, China). Doxorubicin hydrochloride (DOX) and hydroxycamptothecin (CPT) were purchased from Beijing Huafeng United Technology Co., Ltd. Tetraethylorthosilicate (TEOS), and cetyltrimethylammonium bromide (CTAB) were obtained from Sigma-Aldrich (Shanghai) Trading Co., Ltd. (Shanghai, China). Dulbecco's Modified Eagle Medium (DMEM), fetal bovine serum (FBS), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT), trypsin, penicillin (100 U mL−1) and streptomycin (100 μg mL−1) were all obtained from Shanghai Yuanxiang medical equipment Co., Ltd. 4′,6-diamidino-2-phenylindole (DAPI) was purchased from Beyotime Institute of Biotechnology (Jiangsu, China). Alexa-Fluor@488 phalloidin was obtained from Invitrogen Trading Co., Ltd. (Shanghai, China). All other reagents were analytical pure and purchased from Sino-pharm Chemical Reagents Co., Ltd. (Shanghai, China).
:25 (Mw = 110 kDa) was purchased from Daigang Biomaterials Inc. (Jinan, China). Hydroxyapatite was obtained from Aladdin Chemical Reagent Co., Ltd. (Shanghai, China). Doxorubicin hydrochloride (DOX) and hydroxycamptothecin (CPT) were purchased from Beijing Huafeng United Technology Co., Ltd. Tetraethylorthosilicate (TEOS), and cetyltrimethylammonium bromide (CTAB) were obtained from Sigma-Aldrich (Shanghai) Trading Co., Ltd. (Shanghai, China). Dulbecco's Modified Eagle Medium (DMEM), fetal bovine serum (FBS), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT), trypsin, penicillin (100 U mL−1) and streptomycin (100 μg mL−1) were all obtained from Shanghai Yuanxiang medical equipment Co., Ltd. 4′,6-diamidino-2-phenylindole (DAPI) was purchased from Beyotime Institute of Biotechnology (Jiangsu, China). Alexa-Fluor@488 phalloidin was obtained from Invitrogen Trading Co., Ltd. (Shanghai, China). All other reagents were analytical pure and purchased from Sino-pharm Chemical Reagents Co., Ltd. (Shanghai, China).
Preparation of DOX-loaded MSNs
The synthesis of MSNs was as previously described with minor modifications.29 In brief, CTAB (0.36 g) and NH4F (0.6 g) were dissolved in 100 mL of distilled water at 80 °C under vigorous stirring for 1 h. TEOS (1.8 mL) was then added dropwise to the above solution. After 2 h, the product was centrifuged at 10000 rpm for 20 min and washed thoroughly with water and ethanol several times. Thus, obtained nanoparticles were suspended in a mixed solution of 100 mL of ethanol and 2 mL of hydrochloric acid (36–38%) at 80 °C for 12 h to remove the surfactant CTAB. Then, the surfactant-free products were dried under vacuum for further use.
For the preparation of DOX@MSNs, MSNs (0.5 g) was dispersed into DOX aqueous solution with a concentration of 1 mg mL−1. The mixture was stirred under light-sealed conditions for 12 h, the nanoparticles were then vacuumed slowly at room temperature for 3 h. The DOX@MSNs were collected by centrifugation (10000 rpm, 20 min) and washed with PBS (pH 7.4) solution to remove the dissociative DOX. The drug-loaded nanoparticles were dried in the drying oven at 80 °C and stored under light-sealed conditions for future use. To evaluate the loading efficiency of DOX, the supernatant was collected, and the residual DOX content was determined by using high performance liquid chromatography (HPLC). The loading content of DOX in MSNs was calculated by the following equations:

Preparation of CPT-loaded HANPs
For the preparation of CPT@HANPs, CPT (30 mg) was completely dissolved into 15 mL dimethyl sulfoxide (DMSO), and HANPs (0.5 g) was then added into the CPT solution. The mixture was vigorously stirred 12 h under dark condition, and the soaked HANPs were vacuumed for 2 h at room temperature. The CPT@HANPs nanocarriers were separated by centrifugation (10000 rpm, 20 min) and washed three times with DMSO to remove the excess free CPT. The obtained CPT@HANPs nanocarriers were vacuum dried at room temperature overnight to constant weight. The CPT in the supernatant and the washing solutions were collected, and the concentration of CPT was determined by HPLC. The loading content of CPT on the HANPs was also calculated by the above equation.
Preparation of DOX@MSNs & CPT@HANPs-loaded nanofibrous mats
The PLGA/DOX@MSNs & CPT@HANPs nanofibrous mats were fabricated by blend electrospinning of DOX@MSNs, CPT@HANPs and PLGA. The PLGA, PLGA/MSNs, PLGA/HANPs, PLGA/DOX@MSNs and PLGA/CPT@HANPs nanofibrous mats were also prepared for comparison. In a typical procedure, the PLGA solution was prepared by dissolving the PLGA in hexafluoroisopropanol (HFIP) at 20 w/v%. The MSNs & HANPs content in PLGA/DOX@MSNs & CPT@HANPs nanofibrous mats were 2.5 & 2.5, 5 & 2.5, 2.5 & 5 and 5 & 5 wt% with respect to PLGA, thus, the corresponding DOX@MSNs and CPT@HANPs contents in nanofibers can be calculated according to the loading efficiency of the nanocarriers. The spinning solution was then placed into a 10 mL syringe attaching an 18 gauge blunt ended needle. A constant volume flow rate of 2 mL h−1 was maintained using a syringe pump (789100C, Cole-Parmer). The voltage was kept at 14 kV by a static electricity high voltage generator (BGG6-358, BMEI) and the distance between the needle and the collection plate was 15 cm. The electrospun nanofibers were collected on a collection plate covered with aluminum foil. The experiments were carried out at room temperature and relative humidity of 45 ± 5%. The collected nanofibrous mats were vacuum dried overnight to remove the residual organic solvent before further use.Characterization
The structure of nanoparticles and the distribution of nanoparticles in the nanofibers were characterized by transmission electron microscope (TEM, JEM-2100, Japan) at an operating voltage of 200 kV. The morphology of MSNs, HANPs and electrospun mats were observed using a field scanning electron microscope (FESEM, Hitachi S-4800, Japan). The particle size distributions and the polydispersity indexes (PDI) of MSNs were evaluated by dynamic light scattering (DLS) using a BI-200SM multi-angle dynamic/static laser scattering instrument (Brookhaven, USA). The average diameter of nanofibers was obtained from at least 100 measurements on a typical FESEM image using Image J 1.40 G software (NIH, USA). Attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR) spectra were performed by a Nicolet-670 FTIR spectrometer (Thermo Nicolet, USA). X-ray diffraction (XRD) patterns were obtained by a D/MAX-2550 PC diffractometer (Rigaku Inc., Japan) using Cu/Kα radiation with a wavelength of 0.154 nm at 45 kV and 200 mA over the range of 5–60°.Nitrogen adsorption–desorption isotherms were detected with a Micromeritics Tristar II analyzer (Micromeritics, USA). Average pore diameters distributions were calculated from the desorption branches of isotherms by the Barrett–Joyner–Halenda (BJH) method and the specific surface area was calculated according to the Brunauer–Emmett–Teller (BET) method. In order to evaluate the weight loss of the samples in air from the room temperature to 900 °C at a heating rate of 10 °C min−1, the thermogravimetric analysis (TGA) was employed by a thermal analyzer (TG 209 F1, Germany).
The tensile testing of the nanofibrous mats were measured using a universal material tester (H5K-S, Housfield, UK) with a cross-head speed of 10 mm min−1, as described in our previous work.4
In vitro drug release
For drug release study, all the drug-loaded nanofibrous mats were cut into 20 × 20 mm square pieces and their weight were measured accurately. The DOX and CPT release from DOX@MSNs and CPT@HANPs nanocarriers were also investigated for reference. All the samples were dipped into a centrifuge tube filled with 10 mL PBS (pH 7.4). The tube was incubated at 37 °C in a thermostated shaker with the shaking speed of 100 rpm. The release medium (3 mL) was removed at predetermined time point for analysis, and an equal volume of fresh PBS was replaced. The collected release solution was measured using HPLC. The content of DOX or CPT was measured as the average value of three parallel samples.HPLC analyses were performed with a Waters 600 HPLC system connected to a C18 column (Agilent C18, 5 μm, 4.6 mm × 150 mm), operated at room temperature. All samples were filtered by a 0.45 μm filter membrane before testing. The mobile phase consisted in a mixture of deionized water and acetonitrile (30/70, v/v). The detection wavelength of DOX and CPT was 480 nm32 and 360 nm,33 respectively. The detection was performed by a Waters 2489 Detector.
Cytotoxicity assay of the drug-loaded nanofibrous mats
HeLa cells were maintained in DMEM medium supplemented with 10% FBS, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin in 5% CO2 at 37 °C. The medium was changed every two days. Before cell seeding, all the sample materials were sterilized under UV light for 12 h.MTT assay was used to evaluate the cytotoxicity of the medicated nanofibrous mats (PLGA/DOX@MSNs & CPT@HANPs) against HeLa cells. The cytotoxicity of free DOX, free CPT, DOX & CPT, DOX@MSNs and CPT@HANPs with the equivalent amount of DOX or CPT were also assessed for comparison. Briefly, HeLa cells were initially seeded in the 24-well plates (104 cells per well) overnight to allow cells attachment. Then the cells were incubated with MSNs, HANPs, free DOX, free CPT, free DOX & CPT, neat PLGA nanofibers, PLGA/MSNs, PLGA/HANPs, PLGA/DOX@MSNs, PLGA/CPT@HANPs and PLGA/DOX@MSNs & CPT@HANPs nanofibrous mats (DOX at 5.55 μg mL−1 and CPT at 9.6 μg mL−1). After cells were incubated for 24 and 48 h, the fresh culture medium (360 μL) and MTT solution (40 μL, 5 mg mL−1 in PBS) were added into each well for incubation of another 4 h. Then the suspension (400 μL) was removed and DMSO (400 μL) was added to each well to dissolve the precipitate. Finally, the resulting supernatant (100 μL) was carefully transferred to new 96-well plates for MTT assay. Absorbance was measured by using a microplate reader (MK3, Thermo, USA) at the wavelength of 492 nm. The relative cell viability of HeLa cells was calculated by [OD]test/[OD]control × 100%, and the mean value was calculated from six parallel samples.
Confocal laser scanning microscopy (CLSM, Carl Zeiss LSM 700, Germany) was used to observe the morphologies of HeLa cells treated with free DOX, free CPT, free DOX&CPT, DOX@MSNs, CPT@HANPs, PLGA/DOX@MSNs, PLGA/CPT@HANPs and PLGA/DOX@MSNs & CPT@HANPs (the weight of nanoparticles, drug and other nanofibers was obtained from PLGA/DOX@MSNs & CPT@HANPs nanofibrous mats). For CLSM observation, HeLa cells (104 cells per well) were seeded into 24-well plates and incubated for 24 h. After that, the medium was removed and the cells were incubated with MSNs, HANPs, free DOX, free CPT, free DOX & CPT, neat PLGA nanofibers, PLGA/MSNs, PLGA/HANPs, PLGA/DOX@MSNs, PLGA/CPT@HANPs and PLGA/DOX@MSNs & CPT@HANPs nanofibrous mats (DOX at 5.55 μg mL−1 and CPT at 9.6 μg mL−1) at 37 °C for specific time points. Then the HeLa cells were rinsed twice with PBS, fixed with 4% paraformaldehyde for 10 min at 4 °C. Then the cells were washed twice with PBS and permeabilized in 0.1% Triton X-100 in PBS for 5 min, followed by blocking with 1% BSA for 20 min. The fixed cells were stained by using Alexa Fluor@ 488 phalloidin solution (165 nM) for 10 min. In order to label nucleus, the cells were stained by using DAPI solution (100 nM) for 10 min after the cells were washed again with PBS. Finally, all samples were washed with PBS and observed via CLSM.
Statistical analysis
All values were reported as the mean ± standard deviation. Statistical analysis was carried out by the one-way analysis of variance (one-way ANOVA) and Scheffe's post hoc test. The criteria for statistical significance were *P < 0.05 and **P < 0.01.Results and discussion
The morphology and structure of MSNs and HANPs
In this study, two types of nanoparticles including MSNs and HANPs were used as nanocarriers for drug loading. The monodisperse MSNs were successfully synthesized according to our previous study.27 The SEM image of MSNs reveals spherical and monodisperse nanoparticles (Fig. 2A), and TEM image confirms that the prepared MSNs possess wormlike pore structure (Fig. 2B). The hydrodynamic size of MSNs was measured to be 116.5 nm by DLS measurement, with a PDI of 0.18, suggesting uniform particles (Fig. S1A†). Furthermore, the isotherm of MSNs (Fig. S1B†) shows a type IV curve of typical mesoporous material with a specific surface area of 324.2 m2 g−1 and an average pore diameter of 2.5 nm (Fig. S1C†). The XRD pattern of MSNs (Fig. S1D†) appeared a well resolved diffraction peak at around 2θ of 2.0°, which implies a relatively ordered mesoporous structure. These results reveal that the prepared MSNs have a small size suitable for incorporating into polymer nanofibers and a large internal space for drug loading. In addition, the SEM image of commercial HANPs clearly shows a rod-like structure (Fig. 2C), and the TEM image further reveals that the dimensions of the rods are about 40 nm in diameter and 109 nm in length (Fig. 2D). The specific surface area of HANPs (Fig. S2†) was determined to be 47.1 m2 g−1 by the BET method. | ||
| Fig. 2 Morphology and structure of MSNs and HANPs. (A): FESEM image of MSNs; (B): TEM image of MSNs; (C): FESEM image of HANPs; (D): TEM image of HANPs. | ||
Characterization of PLGA/DOX@MSNs & CPT@HANPs nanofibrous mats
The previous study demonstrated that the content of nanoparticles in the polymer is the most important factor in determining the diameter and morphology of the electrospun nanofibers.4 As shown in Fig. 3, the neat PLGA, PLGA/DOX@2.5% MSNs, PLGA/CPT@2.5% HANPs and PLGA/DOX@ 2.5% MSNs & CPT@2.5% HANPs nanofibrous mats were randomly distributed to form the continuous fibrous structure with a smooth surface, and no beads or inorganic nanoparticles were observed on the surface of nanofibers. TEM images indicated that the MSNs or HANPs were successfully embedded in the PLGA nanofiber with uniform distribution within the fibers. The diameter distributions of the PLGA nanofibers were relatively narrow, and the average fiber diameter was increased from 789 to 857 nm after the addition of two kinds of nanocarriers. However, when the content of MSNs and HANPs were totally more than 5%, the fiber diameter distribution became wider and the particles agglomeration can be observed (Fig. S3†). | ||
| Fig. 3 Micrograph and diameter distribution of the nanofibers. (A), (D), (G) and (J) are SEM images (B), (E), (H) and (K) are TEM images; (C), (F), (I) and (L) are diameter distributions. (A): Neat PLGA nanofibers; (D): PLGA/2.5% MSNs composite nanofibers; (G): PLGA/2.5% HANPs composite nanofibers; (J): PLGA/2.5% MSNs & 2.5% HANPs composite nanofibers. | ||
Fig. 4A showed FTIR spectra of as-prepared PLGA/DOX@MSNs & CPT@HANPs nanofibrous mats. For comparison, the FTIR spectra of the DOX, CPT, MSNs, HANPs and neat PLGA nanofibrous mats were also obtained for reference. From the spectrum of MSNs, the peaks near 776 and 1076 cm−1 were the Si–O–Si and Si–OH stretching vibrations of MSNs, respectively.34 For the HANPs, the peaks at 1036 cm−1 can be attributed to the PO43−.23 The characteristic peaks of neat PLGA at 1128 cm−1 assigned to the C–O, at 1186 cm−1 attributed to the C–O–C, at 1755 cm−1 ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and at 2800–2998 cm−1 due to the CH2 stretching vibrations.4 However, for the PLGA/DOX@MSNs & CPT@HANPs composite nanofibers, no characteristic absorption bands of MSNs, HANPs, DOX (the typical absorption bands at 1088, 1285, 1434 and 1621 cm−1) and CPT (the characteristic absorption peaks at 1504, 1588 and 1652 cm−1) were observed, which suggested the DOX@MSNs or/and CPT@HANPs nanocarriers distribute not on the surface but in the inner part of the nanofibers according to the data of TEM. As shown in Fig. 4B, PLGA/DOX@MSNs & CPT@HANPs and neat PLGA nanofibrous mats possess similar XRD patterns without showing any measurable diffraction peaks of MSNs and HANPs, further confirming an effective encapsulation of the DOX@MSNs and CPT@HANPs.
O, and at 2800–2998 cm−1 due to the CH2 stretching vibrations.4 However, for the PLGA/DOX@MSNs & CPT@HANPs composite nanofibers, no characteristic absorption bands of MSNs, HANPs, DOX (the typical absorption bands at 1088, 1285, 1434 and 1621 cm−1) and CPT (the characteristic absorption peaks at 1504, 1588 and 1652 cm−1) were observed, which suggested the DOX@MSNs or/and CPT@HANPs nanocarriers distribute not on the surface but in the inner part of the nanofibers according to the data of TEM. As shown in Fig. 4B, PLGA/DOX@MSNs & CPT@HANPs and neat PLGA nanofibrous mats possess similar XRD patterns without showing any measurable diffraction peaks of MSNs and HANPs, further confirming an effective encapsulation of the DOX@MSNs and CPT@HANPs.
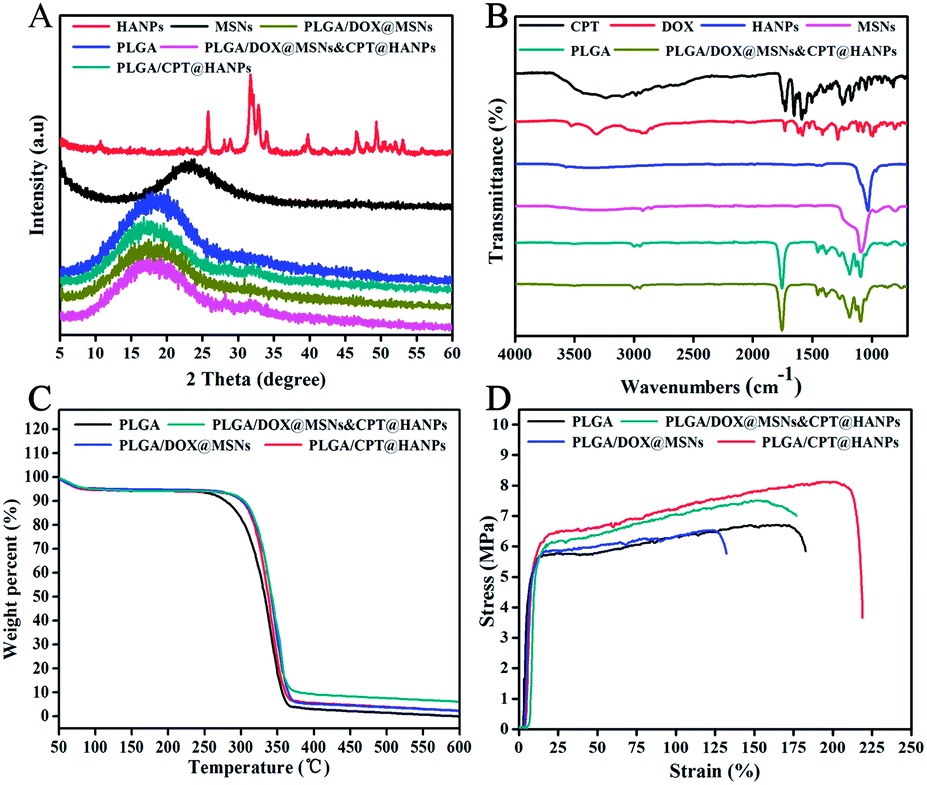 | ||
| Fig. 4 The characterization of the nanofibers. (A): XRD patterns; (B): ATR-FITR spectra; (C):TGA thermograms; (D): typical tensile stress–strain curves. | ||
To determine the thermal properties of the composite nanofibers, TGA curves were measured. As shown in Fig. 4C, it is detected as a temperature-dependent weight reduction. From the TGA curves, there was a moderate weight decrease at less than 100 °C in first step of weight loss, which can be caused by the vaporization of physically adsorbed water in nanofibrous mats. The second step, a large weight loss starting at about 250 °C was due to the decomposition of PLGA and drugs. The curves from PLGA/DOX@MSNs & CPT@HANPs to neat PLGA nanofibrous mats show a consistently higher weight loss as the particles incorporation decreases. For the nanofibrous mats, 4.0, 4.0 and 17.0% of residual MSNs, HANPs and MSNs@HANPs respectively were calculated according to the TGA results, which were almost equal to the initially added weight of inorganic particles. Moreover, the characteristic of thermal stability of the samples were also analyzed from the TGA. The onset temperature of degradation temperature (Tonset) of the neat PLGA sample is about 308.7 °C, which is lower than the composite samples, revealing that the incorporation of nanoparticles can improve the thermal stability of the samples.4
The representative strain–stress curves of the neat PLGA and PLGA composite nanofibers are given in Fig. 4D, and the mechanical properties including the Young's modulus, tensile strength and elongation at break are summarized in the Table 1. Compared with the neat PLGA nanofibers, the breaking strength and Young's modulus of the PLGA/HANPs were improved, which may be due to the efficient loading of HANPs in the PLGA nanofibers.23 It is clear that the breaking and Young's modulus of the PLGA/MSNs decreased when compared with that of the neat PLGA nanofibers, which may be due to the poor interfacial adhesion between the MSNs and the PLGA matrix.4 As a result of the above fact, the breaking strength and Young's modulus of PLGA/MSNs & HANPs composite nanofibers were between that of PLGA/MSNs and PLGA/HANPs composite nanofibers and higher than the neat PLGA nanofibers. This result suggests that the mechanical property of nanofibrous mats can be improved with the addition of MSNs and HANPs.
| Sample | Tensile strength (MPa) | Elongation at break (%) | Young's modulus (MPa) |
|---|---|---|---|
| PLGA | 6.77 ± 1.00 | 178.48 ± 23.89 | 163.97 ± 47.19 |
| PLGA/MSNs | 6.74 ± 0.41 | 126.30 ± 24.27 | 103.89 ± 45.39 |
| PLGA/HANPs | 8.12 ± 1.12 | 164.54 ± 41.38 | 109.90 ± 21.31 |
| PLGA/MSNs & HANPs | 7.87 ± 1.10 | 135.06 ± 34.66 | 165.79 ± 37.49 |
Drug loading and release profiles
To quantify the drug loading content, the collected supernatant solution was measured by HPLC after the loading of DOX and CPT. The amount of the DOX molecules loaded into the mesoporous of MSNs and the CPT loaded onto the surface of HANPs were estimated to be 1.8 and 3.2 mg/100 mg nanoparticles, respectively. So the amount of DOX and CPT loaded into PLGA/DOX@MSN & CPT@HANPs nanofibers were estimated to be 0.045 and 0.08 mg/100 mg nanofibers, respectively.The in vitro release of DOX and CPT from the composite nanofibers was investigated in a simulated physiological environment (PBS, pH = 7.4, 37 °C). Fig. 5A showed the DOX release profiles of DOX@MSNs, PLGA/DOX@MSNs and PLGA/DOX@MSNs & CPT@HANPs nanofibrous mats. As shown in Fig. 4A, all the samples released DOX in a controlled manner. The release of DOX from the DOX@MSNs was faster than that from the other groups due to that DOX@MSNs had open pores and lacked the pore-blocking. DOX released from the PLGA/DOX@MSNs in a very slow fashion, and the cumulative release of DOX was only about 23.5% within 288 h, which may be attributed to the reason that the DOX was firstly released from the mesopores of MSNs to the polymer matrix and subsequently released from the PLGA into the medium. However, the release of DOX from PLGA/DOX@MSNs & CPT@HANPs was about 25.7% in 288 h, a little faster than that of from PLGA/DOX@MSNs, which may because the swelling of the fibers improved with the increase of the nanocarriers content.22
 | ||
| Fig. 5 The cumulative drug release from various samples. (A): The release behavior of DOX; (B): the release behavior of CPT. | ||
Fig. 5B illustrated the cumulative release profiles of CPT from CPT@HANPs, PLGA/CPT@HANPs and PLGA/DOX@MSNs & CPT@HANPs nanofibrous mats. The release of CPT from CPT@HANPs was significantly faster than that of from the nanofibrous mats. CPT was released rapidly in the first 12 h from the CPT@HANPs with 48.8% of the CPT being released, and almost all of the loaded CPT was released after 120 h, which probably due to the lack of strong interaction between CPT molecules and HANPs. In contrast, CPT released from the PLGA/CPT@HANPs nanofibers in a sustained and long-term manner, only 84.1% of the loaded CPT being released after 192 h. This can be explained by the fact that the CPT release from the PLGA/CPT@HANPs nanofibers need two release steps, where the loaded CPT molecules were firstly released from the HANPs nanocarriers and subsequently released from the PLGA matrix into the release medium. It is interesting to note that the release of CPT from PLGA/DOX@MSNs & CPT@HANPs was faster than from PLGA/CPT@HANPs, around 100% of the loaded CPT being released after 192 h, which may because the fiber swelled and the chains of PLGA relaxed in PBS due to the increase of particles content, and in turn resulted in the decrease of interaction between the PLGA and CPT.
Antitumor activity assay in vitro
To investigate the antitumor effect of the PLGA/DOX@MSN & CPT@HANPs composite nanofibers, the cytotoxicity of the nanofibers against HeLa cells was examined in vitro by MTT assay. HeLa cells were treated with different samples at DOX concentration of 5.55 μg mL−1 and CPT concentration of 9.6 μg mL−1 (the relative concentration of MSNs, HANPs, PLGA, PLGA/MSNs and PLGA/HANPs were 0.29, 0.29, 11.30, 12.23 and 11.60 mg mL−1, respectively) for 24 h incubation. Without drug loading, all the bare samples (MSNs, HANPs, neat PLGA nanofibers, PLGA/MSNs nanofibers, PLGA/HANPs nanofibers and PLGA/MSNs & HANPs nanofibers) did not show any cytotoxicity on HeLa cells compared with the control (medium only) (Fig. S4†). As shown in Fig. 6, both the free drug (DOX and CPT) and the drug-loaded nanofibrous mats (PLGA/DOX@MSNs, PLGA/CPT@HANPs and PLGA/DOX@MSNs & CPT/HANPs) effectively inhibited the growth of HeLa cells. Also, the cytotoxicity of free drug and drug-loaded composite nanofibers displayed an apparent treatment time dependent manner. Importantly, the PLGA/DOX@MSNs & CPT@HANPs nanofibrous mats displayed significantly higher cytotoxicity against HeLa cells than the single drug loaded nanofibers (PLGA/DOX@MSNs and PLGA/CPT@HANPs) did, which may result from the synergistic anticancer effect of the released DOX and CPT. This result indicates the combination of DOX and CPT can improve the anticancer effect. | ||
| Fig. 6 Cell viabilities of HeLa cells after treatment with different samples for 24 and 48 h in vitro. | ||
To further confirm the anticancer activity of the composite nanofibers, the morphological changes of HeLa cells treated with different samples at the DOX concentration of 5.55 μg mL−1 and CPT concentration of 9.6 μg mL−1 for 24 and 48 h were observed through CLSM. As shown in Fig. S5,† HeLa cells adopt an extended morphology after treating with neat PLGA nanofibers, bare MSNs and bare HANPs, which were similar to the control, indicating these materials have no cytotoxicity under the conditions of this experiment. From Fig. 7, Fig. S6 and S7,† it can be observed that the HeLa cells treated with free drugs (DOX, CPT, or DOX & CPT), drug-loaded nanocarriers (DOX@MSNs, CPT@HANPs), or drug-loaded nanofibrous mats (PLGA/DOX@MSNs, PLGA/CPT@HANPs or PLGA/DOX@MSNs & CPT@HANPs) for both time points showed apoptotic morphological changes, including cellular shrinkage and cytoplasmic vacuolization. The blue fluorescence of CPT and the red fluorescence of DOX in the nucleus were clearly observed for PLGA/DOX@MSNs & CPT@HANPs nanofibrous mats. The results indicated that DOX and CPT released from the composite nanofibers were cytotoxic on HeLa cells. From these results, we can expect a promising application of PLGA/DOX@MSNs & CPT@HANPs nanofibrous mats in cancer treatment.
 | ||
| Fig. 7 Confocal laser scanning microscopy images of HeLa cells treated with different samples for 24 h. DOX and CPT concentration was 5.55 and 9.6 μg mL−1, respectively. Blue, green and red fluorescence respectively represent the released CPT, Alexa Fluor@488 phalloidin-stained F-actin and the released DOX. Scale bars represent 100 μm. | ||
Although long nanofiber is not suitable to be used by injection, the anticancer drug loaded nanofibrous mats can be easily implanted to the sites where surgical procedures have been made to remove the tumor tissue for preventing local tumor recurrence after surgery. In recent years, considerable efforts have been made to develop such kind of implantable scaffold. Kexin Qiu etc. developed a doxorubicin-loaded electrospun poly(L-lactic acid)/mesoporous silica nanoparticles composite nanofibers for potential postsurgical cancer treatment.4 Touseef Amna etc. prepared a camptothecin loaded poly(ε-caprolactone) nanofibers and tested its antitumor efficacy.35 Fuyin Zheng etc. fabricated a doxorubicin-loaded electrospun poly(lactic-co-glycolic acid)/nano-hydroxyapatite composite nanofibers and examined its antitumor efficacy.23 In the present study, we aimed to develop a new dual anticancer drug loaded composite nanofibers and examined its antitumor efficacy. Our results suggested the PLGA/DOX@MSNs & CPT@HANPs nanofibrous might be used as a potential implantable device for the prevention of cancer recurrence, by surgical implantation into the site or cavity area where a tumor was resected.
Conclusions
In summary, we had successfully fabricated a new dual anticancer drug loaded composite nanofibers, which showed a sustained dual drugs release behavior and a higher antitumor effect than the single drug loaded ones (PLGA/DOX@MSNs and PLGA/CPT@HANPs). The incorporation of DOX@MSNs and CPT@HANPs into the PLGA nanofibers can not only improve the mechanical property and thermal stability of the nanofibers, but also reduce the initial burst release of the drug loaded in nanoparticles, which in turn increase the anticancer efficacy of the drug. According to our data, the dual drug loaded composite nanofibers might have a promising application in preventing local tumor recurrence after surgery.Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (31271028, 51203021), the Natural Science Foundation of Shanghai (12ZR1400300), Shanghai Nano Science Program (11nm0505500), Innovation Program of Shanghai Municipal Education Commission (13ZZ051), Open Foundation of State Key Laboratory for Modification of Chemical Fibers and Polymer Materials (LK1111) and Chinese Universities Scientific Fund (CUSF-DH-D-2014035).Notes and references
- N. A. Fonseca, A. C. Gregorio, A. Valerio-Fernandes, S. Simoes and J. N. Moreira, Cancer Treat. Rev., 2014, 40, 626–635 CrossRef CAS PubMed.
- E. Douglas and D. C. McMillan, Cancer Treat. Rev., 2014, 40, 685–691 CrossRef PubMed.
- Y. Gao, J. Xie, H. Chen, S. Gu, R. Zhao, J. Shao and L. Jia, Biotechnol. Adv., 2014, 32, 761–777 CrossRef CAS PubMed.
- K. Qiu, C. He, W. Feng, W. Wang, X. Zhou, Z. Yin, L. Chen, H. Wang and X. Mo, J. Mater. Chem. B, 2013, 1, 4601 RSC.
- T. Goszczynski, D. Nevozhay, J. Wietrzyk, M. S. Omar and J. Boratynski, Biochim. Biophys. Acta, 2013, 1830, 2526–2530 CrossRef CAS PubMed.
- S. Modok, P. Hyde, H. R. Mellor, T. Roose and R. Callaghan, Eur. J. Cancer, 2006, 42, 2404–2413 CrossRef CAS PubMed.
- R. Das, K. Bhattacharya, S. K. Samanta, B. C. Pal and C. Mandal, Cancer Lett., 2014, 81–90 CrossRef PubMed.
- H. H. Huang, C. L. He, H. S. Wang and X. M. Mo, J. Biomed. Mater. Res., Part A, 2009, 90, 1243–1251 CrossRef PubMed.
- R. P. Hertzberg, M. J. Caranfa and S. M. Hecht, Biochemistry, 1989, 28, 4629–4638 CrossRef CAS.
- G. Das, A. Nicastri, M. L. Coluccio, F. Gentile, P. Candeloro, G. Cojoc, C. Liberale, F. De Angelis and E. Di Fabrizio, Microsc. Res. Tech., 2010, 73, 991–995 CAS.
- I. M. Shaikh, K. B. Tan, A. Chaudhury, Y. Liu, B. J. Tan, B. M. Tan and G. N. Chiu, J. Controlled Release, 2013, 852–861 CrossRef CAS PubMed.
- H. H. Duong and L. Y. Yung, Int. J. Pharm., 2013, 454, 486–495 CrossRef CAS PubMed.
- S. Lv, Z. Tang, M. Li, J. Lin, W. Song, H. Liu, Y. Huang, Y. Zhang and X. Chen, Biomaterials, 2014, 35, 6118–6129 CrossRef CAS PubMed.
- H. Wang, Y. Zhao, Y. Wu, Y. L. Hu, K. Nan, G. Nie and H. Chen, Biomaterials, 2011, 32, 8281–8290 CrossRef CAS PubMed.
- J. Z. Du, C. Q. Mao, Y. Y. Yuan, X. Z. Yang and J. Wang, Biotechnol. Adv., 2014, 32, 789–803 CrossRef CAS PubMed.
- F. Yang, C. Jin, Y. Jiang, J. Li, Y. Di, Q. Ni and D. Fu, Cancer Treat. Rev., 2011, 37, 633–642 CrossRef CAS PubMed.
- S. Chakraborty, I. C. Liao, A. Adler and K. W. Leong, Adv. Drug Delivery Rev., 2009, 61, 1043–1054 CrossRef CAS PubMed.
- B. Felice, M. P. Prabhakaran, A. P. Rodriguez and S. Ramakrishna, Mater. Sci. Eng., C, 2014, 41, 178–195 CrossRef CAS PubMed.
- R. V. Kutty and S. S. Feng, Biomaterials, 2013, 34, 10160–10171 CrossRef CAS PubMed.
- L. Zhang, J. Xia, Q. Zhao, L. Liu and Z. Zhang, Small, 2010, 6, 537–544 CrossRef CAS PubMed.
- M. G. Ignatova, N. E. Manolova, R. A. Toshkova, I. B. Rashkov, E. G. Gardeva, L. S. Yossifova and M. T. Alexandrov, Biomacromolecules, 2010, 11, 1633–1645 CrossRef CAS PubMed.
- B. Song, C. Wu and J. Chang, Acta Biomater., 2012, 8, 1901–1907 CrossRef CAS PubMed.
- F. Zheng, S. Wang, M. Shen, M. Zhu and X. Shi, Polym. Chem., 2013, 4, 933–941 RSC.
- C. L. He, Z. M. Huang and X. J. Han, J. Biomed. Mater. Res., Part A, 2009, 89, 80–95 CrossRef PubMed.
- C. M. Hu and W. G. Cui, Adv. Healthcare Mater., 2012, 1(6), 809–814 CrossRef CAS PubMed.
- C. M. Hu, S. Liu, Y. Zhang, B. Li, H. L. Yang, C. Y. Fan and W. G. Cui, Acta Biomater., 2013, 9(7), 7381–7388 CrossRef CAS PubMed.
- M. V. Jose, V. Thomas, K. T. Johnson, D. R. Dean and E. Nyairo, Acta Biomater., 2009, 5, 305–315 CrossRef CAS PubMed.
- A. Mickova, M. Buzgo, O. Benada, M. Rampichova, Z. Fisar, E. Filova, M. Tesarova, D. Bezdekova, D. Lukas and E. Amler, J. Tissue Eng. Regener. Med., 2012, 6, 35 Search PubMed.
- W. Feng, W. Nie, C. He, X. Zhou, L. Chen, K. Qiu, W. Wang and Z. Yin, ACS Appl. Mater. Interfaces, 2014, 6, 8447–8460 CAS.
- W. Feng, X. J. Zhou, C. L. He, K. X. Qiu, W. Nie, L. Chen, H. S. Wang, X. M. Mo and Y. Z. Zhang, J. Mater. Chem. B, 2013, 1, 5886–5898 RSC.
- Q. J. He, J. M. Zhang, F. Chen, L. M. Guo, Z. Y. Zhu and J. L. Shi, Biomaterials, 2010, 31, 7785–7796 CrossRef CAS PubMed.
- K. Alhareth, C. Vauthier, C. Gueutin, G. Ponchel and F. Moussa, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 887–888, 128–132 CrossRef CAS PubMed.
- S. M. Martins, B. Sarmento, C. Nunes, M. Lucio, S. Reis and D. C. Ferreira, Eur. J. Pharm. Biopharm., 2013, 85, 488–502 CrossRef CAS PubMed.
- E. J. Lee, S. H. Teng, T. S. Jang, P. Wang, S. W. Yook, H. E. Kim and Y. H. Koh, Acta Biomater., 2010, 6, 3557–3565 CrossRef CAS PubMed.
- T. Amna, N. A. M. Barakat, M. S. Hassan, M. S. Khil and H. Y. Kim, Colloids Surf., A, 2013, 431, 1–8 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experimental results. See DOI: 10.1039/c4ra09122a |
| ‡ These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2014 |