
Smart functional polymers – a new route towards creating a sustainable environment
Abstract
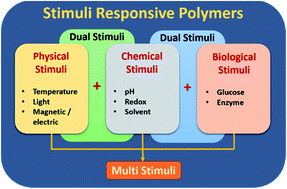
Smart functional polymers have gained a huge amount of interest in recent times due to their innumerable applications in areas including sensors, actuators, switchable wettability, bio-medical and environmental applications. Numerous intensive research studies have been carried out to develop smart functional polymers using stimuli responsive polymeric moieties. This review article encapsulates recent developments in the area of smart functional polymers with a focus on various (physical, chemical and biological) stimuli responsive systems and their applications. Furthermore, this review also provides useful insights and in-depth analysis on the feasibility of utilizing stimuli responsive polymeric materials/composites in anti-fouling and water harvesting applications which hold tremendous potential to create a sustainable environment.
 Please wait while we load your content...
Please wait while we load your content...

