
Ultra-flyweight hydrophobic poly(m-phenylenediamine) aerogel with micro-spherical shell structures as a high-performance selective adsorbent for oil contamination†
Xun Song‡
a,
Siwei Yang‡*b,
Lin Hea,
Shuai Yana and
Fang Liao*a
aChemical Synthesis and Pollution Control Key Laboratory of Sichuan Province, College of Chemistry and Chemical Engineering, China West Normal University, Nanchong 637002, P. R. China. E-mail: liaozhang2003@163.com; Fax: +86 817 2568067; Tel: +86 817 2568067
bState Key Laboratory of Functional Materials for Informatics, Shanghai Institute of Microsystem and Information Technology, Chinese Academy of Science, Shanghai 20050, P. R. China. E-mail: yangsiwei@mail.sim.ac.cn; Fax: +86 021 62511070 420; Tel: +86 021 62511070 420
First published on 18th September 2014
Abstract
In this paper, ultra-flyweight hydrophobic poly(m-phenylenediamine) aerogel (PmPDA) was fabricated via a simple two step approach from a poly(m-phenylenediamine) micro-spherical shell (PmPDMS). The PmPDA was first formed by freeze-drying and then cross-linked via the oxidative polymerization of surface groups following an annealing process. The PmPDA is hydrophilic before the annealing process and becomes hydrophobic (the contact angle is 103.7°) after it is annealed in air. Moreover, the obtained ultra-flyweight PmPDA (ρ = 0.8 mg cm−3) has a large surface area (338 m2 g−1), low thermal conductivity (0.0125 W m−1 K−1 at 25 °C) and excellent mechanical properties. Finally, the regenerated aerogels retain their original shape and ultrahigh absorption capability after more than 10 cycles, making PmPDA an ideal candidate for practical application in the absorption or removal of organics, particularly in environmental protection and pollution control.
1. Introduction
Since aerogels were first reported by Kistler in the 1930s,1 they have attracted the attention of many researchers because of their high porosity, large surface area, electrical conductivity, high water absorptivity, good elasticity, and extremely low bulk density.2–6 The traditional aerogels have mainly focused on silica aerogels,7 and have been utilized or are under investigation for a range of applications such as thermal insulation, adsorption, nuclear waste storage, electrochemical energy storage, cosmic dust collectors and catalysis.8–12 Prakash et al. described a method for preparing silica aerogel films by a simple dipcoating method at ambient pressure without the need for supercritical extraction. It is noteworthy that they added surface groups to the inorganic gel, which made drying shrinkage reversible. In addition, they were able to generate silica aerogel films with 98.5% porosity with this approach.13 However, the preparation of the silica aerogel process is complex,14 which limits its application in numerous fields. On the other hand, with increase in research into carbon materials,15–17 carbon aerogel materials have also been widely researched15,17 with graphene aerogels being one of the most representative examples.7,18–22 Gao et al.15 synthesized carbon aerogels by freeze-drying aqueous solutions of carbon nanotubes and very large graphene oxide (GGO) sheets, followed by the chemical reduction of GGO into graphene with hydrazine vapor. The resulting carbon aerogel had high elasticity, and compression experiments showed a nearly complete recovery after 50–82% compression. Moreover, it possessed super-high absorption capacities and an ultrafast absorption rate for organic solvents and oils. In addition, it is an ultra-flyweight aerogel with a density of less than 1 mg cm−3, which is even smaller than nitrogen. In addition, the carbon aerogel is one of the lightest aerogels in the world. It is noteworthy that it can be easily manufactured on a large-scale due to its tunable size. However, the π–π stacking interactions between graphene sheets can result in the unsuccessful construction of three-dimensional graphene aerogels structures.23 In addition, the potential for obtaining large quantities of graphene by a low-cost production process is difficult.24 However, due to complicated production process, its application is limited to carbon aerogel fields.On the other hand, conducting polymers, such as polyaniline (PANI), polythiophene (PTH), polyparaphenylene (PPP), polypyridine (PPY), and polyaniline derivatives (such as poly(o-phenylenediamine) (PoPD), poly(p-phenylenediamine) (PpPD), and poly(m-phenylenediamine) (PmPD)), have been the subject of considerable attention for potential applications in catalysis, optoelectronic materials, fuel cells, fluorescent probes, sensors, organic batteries, and diodes due to their unique electrical, optical and photoelectric properties.25–43 Among numerous conducting polymers, PANI has attracted persistent attention due to its low cost, environmental compatibility, excellent electrical conductivity, large pseudo-capacitance, and fast doping/dedoping rate during charge/discharge process, which is the most typical conducting polymer.44 Sun et al. synthesized magnetically responsive Fe3O4@PANI@Au nanocomposites via an ultrasound-assisted in situ surface polymerization method. They also proved that negatively charged Au nanoparticles with a diameter of about 4 nm were effectively assembled onto the positively charged surface of the as-synthesized Fe3O4@polyaniline core/shell microspheres via electrostatic attraction.45 Sun et al. reported the simple synthesis of submicrometer scale conjugation polymer PoPD colloids by the direct mixing of aqueous PoPD and potassium bichromate solutions at room temperature.45 Our previous research demonstrated poly(p-phenylenediamine) nanofibers with conjugated structures and showed that they could be used as a highly selective fluorescent probe for thiols, L-cysteine and caffeine.25,31,32 We also revealed the synthesis of Ag nanoparticles-decorated poly (m-phenylenediamine) hollow spheres and their application for hydrogen peroxide detection.35 However, to the best of our best knowledge, the synthesis of poly(m-phenylenediamine) aerogel has not been reported clearly.
Herein, we report an ultra-flyweight hydrophobic poly(m-phenylenediamine) aerogel (PmPDA), which was fabricated via a simple two step approach from a poly(m-phenylenediamine) micro-spherical shell (PmPDMS). The freeze-drying–annealing cross-linking process was easy to carry out and was low cost. PmPDA was first formed by freeze-drying and then cross-linked together via the oxidative polymerization of surface groups following the annealing process. It was interesting to note that the PmPDA was hydrophilic before annealing and was hydrophobic after annealling in air. The obtained ultra-flyweight (ρ = 0.8 mg cm−3) PmPDA had a large surface area (BET = 338 m2 g−1), low thermal conductivity (0.0125 W m−1 K−1 at 25 °C) and excellent mechanical properties. The formation mechanism of PmPDA was demonstrated by X-ray photoelectron spectrum (XPS) results. Furthermore, the PmPDA could be used as a high-performance selective adsorbent for oil contamination.
2. Experimental
2.1 Materials
All the chemicals (m-phenylenediamine (mPD), (NH4)2S2O8 (APS), Sudan Red, and methylbenzene) were purchased from Aladdin (Shanghai, China) and were used as received without further purification. Water used in all the experiments was purified through a Millipore system.2.2 Characterization
The structure and morphology were investigated by scanning electron microscope (SEM) (FEI Sirion-F250) at an accelerating voltage of 20 kV, and transmission electron microscope (TEM) (FEI Tecnai G2 20 STWIN) at an accelerating voltage of 200 kV. Raman spectra were recorded on a Thermo Fisher DXR using an Ar+ laser (wavelength 532 nm, 2 mW) with a 1 μm laser spot equipped with optical microscopy. Fourier transform infrared (FTIR) was performed on a Nicolet 6700 (resolution 0.4 cm−1) infrared spectrometer and samples were dispersed in potassium bromide and compressed into pellets. XPS was derived from measurements conducted on an X-ray photoelectron spectrometer using an Mg-Kα radiation excitation source (AXIS ULTRA DLD, Kratos). The phase compositions of the as-prepared products were determined by X-ray diffraction analysis (XRD, Rigaku D/max-2500) using graphite-monochromised Cu-Kα radiation. Specific surface area, pore size, and pore volume were determined by a Brunauer–Emmett–Teller (BET) apparatus, V-Sorb 2800P (Gold APP Instruments Corporation, China). Surface area was determined by evaluating the isotherm of the physical adsorption of nitrogen, and specific surface area was determined by the BET method. The surface area of the mesopores was calculated by applying the Barrett–Joyner–Halenda adsorption (BJH) approach.2.3 Synthesis of PmPDMS
The typical preparation process of PmPDMS is as follows: 4.5 g mPD was dissolved in 30 mL deionized water. Then, the mPD solution was cooled in an ice-water bath for 2 h in the dark before the oxidative polymerization. Then, 18 mL of 1.67 M APS aqueous solution was added gradually to the mPD solution with magnetic stirring. Afterward, the resulting solution was stirred for another 5 min to ensure complete mixing and then the reaction was allowed to proceed without agitation for 24 h at 4 °C. Finally, the product was centrifuged and washed with deionized water until the filtrate became colorless and then dried in a vacuum at 60 °C for 24 h.2.4 Synthesis of PmPDA
The typical preparation process of PmPDA is as follows: first, 4 mg PmPDMS was dispersed in 5 mL deionized water and then the resulting mixture was sonicated for about 10 h to yield a homogenous suspension. Then, the suspension was poured into a cylindrical crucible and freeze-dried for 2 days. The as-prepared PmPDA foam was cross-linked by annealing at 160 °C in air for 24 h, followed by vacuum-drying at 80 °C for 24 h.3. Results and discussions
3.1 Characterization of PmPDA
Fig. 1 shows the typical SEM and TEM images of PmPDA. Clearly, PmPDA with a porous structure is deposited from the homogeneous micro-spherical shell (PmPDMS) with a size distribution of 80–120 nm (Fig. 1a, b and 2a). As seen from typical high magnification SEM and TEM images (Fig. 1c and d) of a single PmPDMS, the edges of the PmPDMS can be clearly recognised, and the wall thickness of a single particle is about 10 nm. The high surface area and porous structure will be conducive to rapid dye absorption. The electron diffraction data (Fig. 1d inset) shows a crystalline structure of PmPDMS, which indicates that the products are amorphous. | ||
| Fig. 1 (a, b and c) FESEM images of PmPDA under different magnification, (d) TEM image of PmPDA. Inset: electron diffraction data of PmPDMS in PmPDA. | ||
 | ||
| Fig. 2 (a) Corresponding size distribution histograms of PmPDMS in PmPDA, (b) Raman spectra of PmPDA and PmPDMS, (c) FTIR spectra and (d) XRD spectrum of PmPDA and PmPDMS. | ||
The Raman spectra of PmPDA and PmPDMS are shown in Fig. 2b. There are obvious difference in the Raman spectra of PmPDA and PmPDMS. The peak at 1170 cm−1 is due to the C–H in-plane bending mode, which indicates the presence of a quinoid ring. The weak band at 1200 cm−1 is assigned to the C–N stretching mode of polaronic units.36,38–40 The peaks located between 1420 cm−1 and 1370 cm−1 are assigned to the C–N+ stretching modes of the delocalized polaronic charge carriers.39 The peaks at 1520, 1549, and 1494 cm−1 correspond to the N–H bending deformation mode, and the C–C deformation of benzenoid rings and quinoid rings, respectively.38 Moreover, the peak at 1622 cm−1 is assigned to the benzene ring.40
The FT-IR spectrum of PmPDA is shown in Fig. 2b. The peaks at 761 and 565 cm−1 are characteristic of the C–H out-of-plane bending vibrations of benzene nuclei in the phenazine skeleton.38,39 The peaks at 905 and 832 cm−1 correspond to the out-of-plane deformation of C–H on a 1, 2, 3, 5-tetrasubstituded benzene ring. The peaks at 1242 and 1371 cm−1 are associated with the C–N stretching in the benzenoid and quinoid imine units.37 The strong peaks at 1600 and 1545 cm−1 are assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CC stretching vibrations in the phenazine structure, respectively. Moreover, the peaks at 3325, 3244 and 3206 cm−1 correspond to the N–H stretching mode, and imply the presence of secondary amino groups, respectively.43 The XRD patterns of PmPDA and PmPDMS are shown in Fig. 2c. Both PmPDA and PmPDMS show a broad band centered at 2θ = 18–30°, indicating that the PmPDA and PmPDMS are amorphous.
N and CC stretching vibrations in the phenazine structure, respectively. Moreover, the peaks at 3325, 3244 and 3206 cm−1 correspond to the N–H stretching mode, and imply the presence of secondary amino groups, respectively.43 The XRD patterns of PmPDA and PmPDMS are shown in Fig. 2c. Both PmPDA and PmPDMS show a broad band centered at 2θ = 18–30°, indicating that the PmPDA and PmPDMS are amorphous.
Fig. 3a shows the photos of the PmPDA cylinder before and after the annealing process. It is clear that before the annealing process, the PmPDA can be easily destroyed (Fig. 3b). This indicates the PmPDA has a non-compact structure before the annealing process. Moreover, it can be wetted easily by water, which illustrates that the PmPDA is hydrophilic before the annealing process. In contrast, the PmPDA cylinder shows good mechanical properties, and can float on water after the annealing process. This indicates that the PmPDA have a stable structure and are hydrophobic (the contact angle is 103.7°) after being annealed in air. Moreover, the apparent density of PmPDA is 0.8 mg cm−3, which is significantly smaller than air (1.25 mg cm−3, 25 °C). Fig. 3c shows a 5 cm3 PmPDA cylinder standing on flowers.
 | ||
| Fig. 3 (a) Digital photo of the preparation process of PmPDA, (b) digital photo of PmPDA under pressure before (left) and after (right) annealing, and (c) a 5 cm3 PmPDA cylinder standing on a flower. | ||
3.2 Formation mechanism of PmPDA
To study the formation mechanism of PmPDA, the XPS data of PmPDMS and PmPDA were investigated (Fig. 4). Fig. 5a shows the XPS survey spectrum of PmPMS, where N (399 eV) and C (284 eV) could be detected. The N/C atomic ratio is 32.7%, which is close to the monomer (mPD, N/C = 33.3%). | ||
| Fig. 4 XPS spectra of PmPDA (a) before annealing, (b) after annealing; C 1s core-level and corresponding deconvoluted spectra for PmPDA (c) before annealing, (d) after annealing, respectively; N 1s core-level and corresponding deconvoluted spectra for PmPDA (e) before annealing, (f) after annealing, respectively. | ||
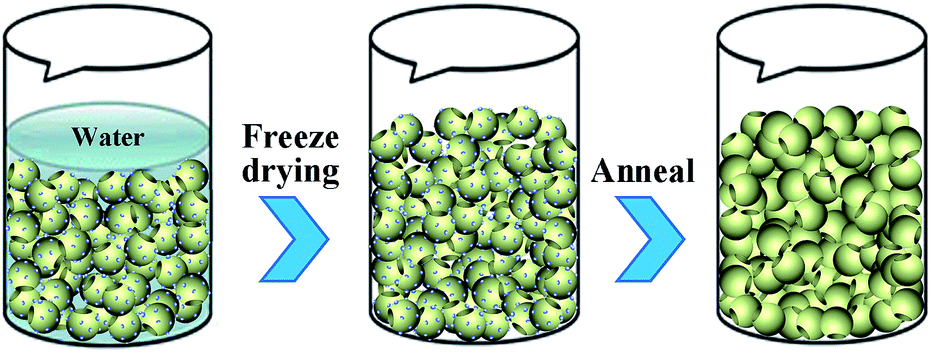 | ||
| Fig. 5 Schematic diagram of the morphology forming mechanism. The blue ball represents the surface groups in PmPDMS. | ||
The core-level XPS spectrum of C 1s and the corresponding deconvoluted spectra for the PmPDMS are shown in Fig. 4c. The peak with a binding energy centered at 284.73 eV could be attributed to the C–C bond. The peaks with binding energies of 286.09 and 288.77 eV can be assigned to the C–N and CN bonds, respectively.37 The high-resolution N 1s spectrum (Fig. 4e) reveals the presence of amidogen N (398.45 eV) and C–NC (phenothiazine structure, 400.50 eV).36,37 The proportion of amidogen N in the total N element is about 36.5%. While for PmPDA, the intensity of the N 1s peak becomes weaker, and the N/C atomic ratio is 24.1% (Fig. 4b), suggesting the loss of N. The proportion of amidogen N in total N element is about 17.6% (Fig. 4f), which is considerably lower than PmPDMS. The reduction in amidogen N indicates that most amidogen N were cross-linked in the annealing process. Moreover, the reduction in amidogen N is also the key factor for the hydrophobicity of this aerogel. When there are numerous hydrophilic groups (mostly amido) in PmPDMS before the annealing process, the products have good hydrophilicity. However, most amidogen N were cross-linked after the annealing process, hence the PmPDA was hydrophobic and could easily float on water.
The formation mechanism of PmPDA is shown in Fig. 5 and is as follows: first, hydrophilic PmPDMS with a non-compact structure is formed in a special shape via a freeze drying process. The PmPDMS piles up and there are no chemical bonds between these particles. Furthermore, the PmPDMS includes interactive surface groups (amido) at the surface. Subsequently, the PmPDMSs are cross-linked together via the oxidative polymerization of surface groups in the annealing process. Hydrophilic PmPDA was formed and had good mechanical properties.
3.3 Mechanical properties and absorption of oil contamination
The obtained PmPDA is ultra-flyweight and has excellent mechanical properties. The apparent density of PmPDA is 0.8 mg cm−3, which is significantly smaller than air (1.25 mg cm−3, 25 °C). The low density of PmPDA indicates that the PmPDA has a large surface area. Fig. 6 shows the N2 adsorption/desorption isotherms and corresponding pore size distribution curve for PmPDA. The isotherms exhibit the characteristics of type IV isotherms with a hysteresis loop (Fig. 6a).46 It can be seen that the isotherms showed little uptake at low relative pressures (P/P0 < 0.1), indicating the presence of micropores. Significant hysteresis of the isotherms at high relative pressures (P/P0 > 0.1) can be seen, which indicated that PmPDA was rich in mesopores. Moreover, there was an upward trend at high relative pressures (P/P0 > 0.90), which may originate from the presence of macropores. PmPDA exhibited a highly developed, hierarchical porosity framework of micropores together with macropores and mesopores, which matched the SEM observations (Fig. 6a). The pore size distribution data for the aerogel ranged from 2 nm to 800 nm (Fig. 6b), and the size of most of the pores were between 2 nm and 15 nm. The average pore size was 3.21 nm. The specific surface area was determined by the BET model to be 338 m2 g−1, whereas the pore volume was determined from the BJH model to be 0.72 cm3 g−1. | ||
| Fig. 6 (a) N2 adsorption/desorption curve and (b) pore size distribution based on the BJH model of the PmPDA. | ||
The obtained aerogel also had good mechanical properties: 4 mg of PmPDA cylinder could support a static load of at least 100 g, and exhibited good resilience when released from compression. As shown in Fig. 7a–c, when the compression was released, the sample almost recovered its original shape, indicating that the sample was strong and resilient. Moreover, the PmPDA could be used as a spring due to its good recovery performance. Fig. 7d shows the height change of the PmPDA cylinder after 50 compressing cycles. It is clear that there is no significant change in the height of the PmPDA cylinder, even after it was compressed 50 times.
 | ||
| Fig. 7 (a–c) Digital photos of PmPDA under 100 g added mass, (d) height change of PmPDA cylinder under 50 compressing cycles. | ||
The good mechanical properties and large surface area of ultra-flyweight hydrophilic PmPDA indicate that this aerogel posses high absorption capacities and an ultrafast absorption rate for organic solvents and oils. As shown in Fig. 8a–d, 0.5 g methylbenzene (stained with Sudan Red) on water is absorbed completely by 4 mg PmPDA within 5 s, indicating an average absorption rate of 25 g g−1 s−1. The high absorption capacities and adsorption rate are remarkable. The PmPDA has good regioselectivity for organics, which can be easily ignited after the adsorption process (Fig. 8e, Video 1 in the ESI†) due to the hydrophobicity of PmPDA. Moreover, this aerogel can be easily picked up by hand after burning (see Video 1 in the ESI†), which indicates that PmPDA has a low thermal conductivity. The low thermal conductivity (0.0125 W m−1 K−1 at 25 °C) can be attributed to the three-dimensional porous structure of PmPDA.
 | ||
| Fig. 8 Digital photos of the absorption process of toluene (stained with Sudan Black B) on water by PmPDA within 5 s, (a) 0 s, (b) 2 s, (c) 4 s, (d) 5 s; and (e) digital photos of ignited PmPDA after the absorption process. (f) The restored weight of PmPDA after removing methylbenzene by completely burning it off from the aerogels. | ||
Table 1 shows the absorption capacities (Q is the ratio of the final weight after full absorption as a percentage of the initial weight of PmPDA) of PmPDA to various solvents and oils. The absorption capacities range from 837–1986 g g−1 for various solvents. The large absorption capacities indicate that this aerogel has excellent adsorption capacity.
| Solvents | Q/% |
|---|---|
| Methylbenzene | 1002 |
| Benzene | 968 |
| CCl4 | 1986 |
| CHCl3 | 1769 |
| Diethyl ether | 894 |
| n-Hexane | 837 |
| Cyclohexane | 951 |
| Ethyl acetate | 1139 |
| Nitrobenzene | 1420 |
| Chlorobenzene | 1288 |
Moreover, because of the fine elasticity and low thermal conductivity of PmPDA, the absorbed liquids can be completely burned off from the aerogels in air without destroying the porous structures. The regenerated aerogels still retain their original shape and ultrahigh absorption capability after more than 10 cycles (Fig. 8f), making PmPDA an ideal candidate for practical applications in the absorption/removal of organics, particularly in environmental protection and pollution control.
4. Conclusions
In summary, ultra-flyweight hydrophobic PmPDA was fabricated via a simple two step approach from PmPDMS. The PmPDA was first formed by freeze-drying and then cross-linked together via oxidative polymerization of its surface groups following the annealing process. The obtained PmPDA had a large surface area (338 m2 g−1), low thermal conductivity (0.0125 W m−1 K−1 at 25 °C) and excellent mechanical properties with a density smaller than air (ρ = 0.8 mg cm−3). The formation mechanism of PmPDA and the switch from hydrophilic to hydrophobic is clearly demonstrated by XPS. Moreover, the PmPDA can be used as a high-performance selective adsorbent for oil contamination. These findings should be helpful for the fabrication of polymer-based aerogels and provide insights into the potential use of polymer-based aerogels.Acknowledgements
This work was supported by China West Normal University Fund (12B017) and China West Normal University Students Research Fund (42713070).References
- S. S. Kistler, Nature, 1931, 127, 74 CrossRef.
- H. Hu, Z. B. Zhao, W. B. Wan and J. S. Qiu, Mater. Interfaces, 2014, 6, 3242 CrossRef CAS PubMed.
- X. L. Wu, T. Wen, H. L. Guo, S. B. Yang, X. K. Wang and A. W. Xu, NANO, 2013, 4, 3589 Search PubMed.
- A. C. Pierre and G. M. Pajonk, Chem. Rev., 2002, 102, 4243 CrossRef CAS PubMed.
- J. Z. Feng, C. R. Zhang, J. Feng, Y. G. Jiang and N. Zhao, Mater. Interfaces, 2011, 3, 4796 CrossRef CAS PubMed.
- Y. J. Wang, G. H. Zhao, S. N. Chai, H. Y. Zhao and Y. B. Wang, Mater. Interfaces, 2013, 5, 842 CrossRef CAS PubMed.
- Y. R. Lin, G. J. Ehlert, C. Bukowsky and H. A. Sodano, Mater. Interfaces, 2011, 3, 2200 CrossRef CAS PubMed.
- D. Sanli and C. Erkey, Mater. Interfaces, 2013, 5, 11708 CrossRef CAS PubMed.
- R. Baetens, B. P. Jelle and A. Gustavsen, Energ. Build., 2011, 43, 761 CrossRef PubMed.
- A. C. Pierre and G. M. Pajonk, Chem. Rev., 2002, 102, 4243 CrossRef CAS PubMed.
- J. P. Randall, M. A. B. Meador and S. C. Jana, Mater. Interfaces, 2011, 3, 613 CrossRef CAS PubMed.
- M. Tabata, I. Adachi, H. Kawai, T. Sumiyoshi and H. N. I. Yokogawa, Nucl. Instrum. Methods Phys. Res., Sect. A, 2012, 668, 64 CrossRef CAS PubMed.
- S. S. Prakash, C. Jeffrey, A. J. Hurd and S. M. Rao, Nature, 1995, 374, 440 CrossRef.
- G. Hayase, K. Kanamori, K. Abe, H. Yano, A. Maeno, H. Kaji and K. Nakanishi, Mater. Interfaces, 2014, 6, 9466 CrossRef CAS PubMed.
- H. Y. Sun, Z. Xu and C. Gao, Adv. Mater., 2013, 25, 2554 CrossRef CAS PubMed.
- Z. Y. Sui, Y. Cui, J. H. Zhu and B. H. Han, Mater. Interfaces, 2013, 5, 9172 CrossRef CAS PubMed.
- H. P. Cong, X. C. Ren, P. Wang and S. H. Yu, NANO, 2012, 6, 2693 CAS.
- L. Xiao, D. Q. Wu, S. Han, Y. S. Huang, S. Li, M. Z. He, F. Zhang and X. L. Feng, Mater. Interfaces, 2013, 5, 3764 CrossRef CAS PubMed.
- Z. S. Wu, S. B. Yang, Y. Sun, K. Parvez, X. L. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082 CrossRef CAS PubMed.
- M. A. Worsley, P. J. Pauzauskie, T. Y. Olson, J. Biener, J. H. Satcher and T. F. Baumann, J. Am. Chem. Soc., 2010, 132, 14067 CrossRef CAS PubMed.
- G. Rother, L. Vlcek, M. S. Gruszkiewicz, A. A. Chialvo, L. M. Anovitz, L. Bañuelos, D. Wallacher, N. Grimm and D. R. Cole, J. Phys. Chem. C, 2014, 4, 546 Search PubMed.
- G. Rother, E. G. Krukowski, D. Wallacher, N. Grimm, R. J. Bodnar and D. R. Cole, J. Phys. Chem. C, 2012, 116, 91 Search PubMed.
- C. Y. Hou, Q. H. Zhang, Y. G. Li and H. Z. Wang, Carbon, 2012, 50, 1959 CrossRef CAS PubMed.
- S. H. Lee, H. W. Kim, J. O. Hwang, W. J. Lee, J. Kwon, C. W. Bielawski, R. S. Ruoff and S. O. Kim, Angew. Chem., Int. Ed., 2010, 49, 10084 CrossRef CAS PubMed.
- T. T. Zhang, S. W. Yang, J. Sun, X. B. Li, L. He, S. Yan, X. Y. Kang, C. S. Hu and F. Liao, Synth. Met., 2013, 181, 86 CrossRef CAS PubMed.
- S. W. Yang, D. Liu, F. Liao, T. T. Guo, Z. P. Wu and T. T. Zhang, Synth. Met., 2012, 162, 2329 CrossRef CAS PubMed.
- S. W. Yang and F. Liao, NANO, 2011, 6, 597 CrossRef CAS.
- P. Xiong, H. J. Huang and X. Wang, J. Power Sources, 2014, 245, 938 Search PubMed.
- A. Madhankumar and N. Rajendran, Prog. Org. Coat., 2013, 76, 1445 CrossRef CAS PubMed.
- Z. F. Wang, F. Liao, S. W. Yang and T. T. Guo, Mater. Lett., 2012, 67, 121 CrossRef CAS PubMed.
- S. W. Yang and F. Liao, Synth. Met., 2012, 162, 1343 CrossRef CAS PubMed.
- S. W. Yang, S. Q. S. Huang, D. Liu and F. Liao, Synth. Met., 2012, 162, 2228 CrossRef CAS PubMed.
- X. Wang, H. D. Li and P. Liu, Electrochim. Acta, 2014, 125, 630 CrossRef CAS PubMed.
- F. Liao, S. W. Yang, X. B. Li, S. Yan, C. S. Hu, L. He, X. Y. Kang, X. Song and T. Y. Ren, Synth. Met., 2014, 190, 79 CrossRef CAS PubMed.
- Z. P. Wu, S. W. Yang, Z. Chen, T. T. Zhang, T. T. Guo, Z. F. Wang and F. Liao, Electrochim. Acta, 2013, 98, 104 CrossRef CAS PubMed.
- T. T. Guo, F. Liao, Z. F. Wang and S. W. Yang, J. Mater. Res., 2012, 3, 25 Search PubMed.
- S. Yang, J. Sun, X. Li, W. Zhou, Z. Wang, P. He, G. Ding, X. Xie, Z. Kang and M. Jiang, J. Mater. Chem. A, 2014, 2, 8660 CAS.
- U. Olgun and M. Gulfen, React. Funct. Polym., 2014, 77, 23 CrossRef CAS PubMed.
- F. Liao, S. W. Yang, X. B. Li, L. Yang, Z. Xie, C. S. Hu, S. Yan, T. Y. Ren and Z. Liu, Synth. Met., 2014, 189, 126 CrossRef CAS PubMed.
- F. Liao, S. W. Yang, X. B. Li, S. Yan, C. S. Hu, L. He, X. Y. Kang, X. Song and T. Y. Ren, Synth. Met., 2014, 190, 79 CrossRef CAS PubMed.
- X. F. Wang, Y. H. Shen, A. J. Xie, L. G. Qiu, S. K. Li and Y. Wang, J. Mater. Chem., 2011, 21, 9641 RSC.
- S. W. Yang, C. S. Hu, D. Liu, T. T. Zhang, T. T. Guo and F. Liao, J. Cluster Sci., 2014, 25, 337 CrossRef CAS.
- F. Liao, S. W. Yang, X. B. Li, L. Yang, Z. Xie, C. S. Hu, L. He, X. Y. Kang, X. Song and T. Y. Ren, Synth. Met., 2014, 189, 135 CrossRef CAS PubMed.
- S. H. Xuan, Y. X. J. Wang, J. J. C. Yu and K. C. F. Leung, Langmuir, 2009, 25, 11835 CrossRef CAS PubMed.
- J. Tian, H. Li, Y. Luo, L. Wang, Y. Zhang and X. P. Sun, Langmuir, 2010, 27, 874 CrossRef PubMed.
- X. B. Li, S. W. Yang, J. Sun, P. He, X. G. Xu and G. Q. Ding, Carbon, 2014, 78, 38 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09080b |
| ‡ These authors are co-first authors. |
| This journal is © The Royal Society of Chemistry 2014 |