
Curvature effect of SiC nanotubes and sheets for CO2 capture and reduction†
P. Zhangab,
X. L. Hou*a,
J. L. Mia,
Q. Jiangc,
H. Aslanb and
M. D. Dong*b
aInstitute for Advanced Materials, School of Materials Science and Engineering, Jiangsu University, Zhenjiang 212013, China. E-mail: houxiuli@ujs.edu.cn
bCenter for DNA Nanotechnology (CDNA), interdisciplinary Nanoscience Center (iNANO), Aarhus University, DK-8000 Aarhus, Denmark. E-mail: dong@inano.au.dk
cKey Laboratory of Automobile Materials, Ministry of Education, Department of Materials Science and Engineering, Jilin University, Changchun 130022, China
First published on 22nd September 2014
Abstract
The environmental crisis due to greenhouse gas CO2 emissions is motivating researchers to discover new materials and efficient technologies for CO2 capture and conversion. In this work, density functional theory (DFT) has been employed to investigate the surface curvature dependence of the adsorption and (electro) reduction of CO2 on SiC nanomaterials, including single layer SiC sheets and nanotubes. The DFT calculations show that both the adsorption energy and reduction free energy decrease with the decrease of the curvature of SiC nanotubes. SiC nanotubes with suitable curvature can capture and reduce CO2 effectively. However a single layer SiC sheet (without curvature) cannot adsorb CO2 at all. These findings are particularly relevant to generate fuels with a carbon-neutral footprint.
Introduction
The global climate change has become a significant challenge due to the magnitude of greenhouse gas emission. CO2 is believed to be partly responsible due to the combustion of fossil fuels.1,2 CO2 chemistry has become a very attractive area of research, not only because of environmental concerns, but also due to the potential use of CO2 as an alternative and economical feedstock.3 The recovery of CO2 for its hydrogenation to formic acid, alcohols or other hydrocarbon compounds is an important approach to recycle released CO2. However, it is a difficult task due to the challenges associated with the chemical inertness of CO2.The combination of CO2 capture and conversion is an attractive strategy for efficiently reducing CO2 emissions. One example of CO2 conversion to a useful hydrocarbon is hydrogenation of CO2 to formic acid, an important chemical fuel in fuel cells.4,5 An ideal CO2 sequestration material should have large surface area and strong adsorption ability. Several CO2 adsorbents have been proposed previously including zeolite, carbon, boron nitride, alumina and metal–organic frameworks (MOFs).6–16 Recently, it has been shown that CO2 can be adsorbed strongly on boron antisite in boron-rich boron nitride (BN) nanotubes.17 In addition, the process of CO2 capture/release can be simply controlled by switching on/off the charges carried by BN nanomaterials.18 A lot of attention has also been focused on the hydrogenation of CO2 on metals, carbides, and metal/oxide catalysts.19–30 Cu electrodes, of all experimentally examined metals, have shown a unique ability to produce hydrocarbon products at reasonable currents and efficiencies, but with a still relatively large over-potential of approximately 1 V.31,32 Carbon monoxide dehydrogenase (CODH) enzyme has been shown to have better catalytic activity than Cu.25 With a Pt working electrode in acidic pyridine solutions, CO2 can be reduced by H atoms bound to the Pt surface that are transferred to CO2 in a proton-coupled hydride transfer mechanism activated by pyridinium at low over-potentials (−0.58 V vs. SCE).33,34 It is essential that the materials can capture and convert CO2 at atmospheric pressure and room temperature with only heat from surrounding environments to avoid the generation of new CO2.8 Thus far, few materials can satisfy these requirements.8
SiC with the atomic ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 can be potentially provide a lot of active sites for CO2 adsorption. In this study, CO2 adsorption and reduction on two types of SiC nanomaterials (single layer sheet and nanotubes) are studied based on density functional theory (DFT). The theoretical investigations show that CO2 adsorption and reduction on SiC nanomaterials display a curvature effect. SiC nanotubes with suitable curvatures can capture and reduce CO2 effectively, while single layer SiC sheet cannot.
:1 can be potentially provide a lot of active sites for CO2 adsorption. In this study, CO2 adsorption and reduction on two types of SiC nanomaterials (single layer sheet and nanotubes) are studied based on density functional theory (DFT). The theoretical investigations show that CO2 adsorption and reduction on SiC nanomaterials display a curvature effect. SiC nanotubes with suitable curvatures can capture and reduce CO2 effectively, while single layer SiC sheet cannot.
Computational methods
All calculations are based on spin-polarized DFT framework as implemented in DMol3 code.35,36 The generalized gradient approximation (GGA) with Perdew–Burke–Ernzerhof functional (PBE) is employed to describe the exchange-correlation potential.37 All electron relativistic core treatment method is implemented for relativistic effects, which explicitly includes all electrons and introduces some relativistic effects into the core. The double numerical plus polarization (DNP) basis set is adopted for the spin-polarized DFT calculations.35 A smearing of 0.005 Ha (1 Ha = 27.21 eV) to the orbital occupation is applied to achieve accurate electronic convergence. To ensure high-quality results, the real-space global orbital cutoff radius is chosen as high as 4.6 Å in the computations. The convergence tolerance of energy is 1.0 × 10−5 Ha, maximum force is 0.002 Ha Å−1, and maximum displacement is 0.005 Å in the geometry optimization. The k-point is set as 1 × 1 × 1. The transition state (TS) for CO2 adsorption is obtained by LST/QST tools in DMol3 code. In order to describe the van der Waals (vdW) interaction for CO2 adsorption, the DFT + D method within Grimme scheme is adopted.38 In calculations, the supercell models include three unit cells for (n, 0) zigzag SiC nanotubes, four unit cells for (n, n) armchair SiC nanotubes, and 5 × 5 supercell for single SiC layer. The minimal distance between the SiC nanotubes/sheet and their mirror images is set as 15 Å, which is sufficiently large to avoid the interaction between them. The Si–C bond length is 1.79 Å.The adsorption energies (Ead) of adsorbates on SiC are calculated through Ead = Eads + ESiC − Eads/SiC, where Eads, ESiC, and Eads/SiC are the total energies of an isolated adsorbate molecule, the SiC catalysts, and the adsorption systems, respectively. By these definitions, positive Ead values correspond to stably exothermic adsorption processes. In order to simulate the electrochemistry environment of CO2 electro reduction, a conductor-like screening model (COSMO) is introduced to simulate a H2O solvent environment throughout the whole process.39–41 Free energies of the intermediates involved in CO2 hydrogenation are calculated based on a computational hydrogen electrode (CHE) model suggested by Nørskov et al.21,42,43 The CHE model defines that the chemical potential of a proton/electron in solution is equal to a half of the chemical potential of a gas-phase H2. Free energy change (ΔG) of every elemental step is determined by ΔG = ΔE + ΔZPE − TΔS, where ΔE denotes the electronic energy change directly obtained from DFT calculations, ΔZPE is the change of zero point energies, T is the temperature (equals 298.15 K), and ΔS is the change in entropy. Zero point energy and entropy of the every intermediate are calculated based on the vibrational frequencies, where all the atoms are included.
Results and discussion
The CO2 adsorption properties of single SiC layer sheet without curvature and several SiC nanotubes with different surface curvatures are summarized in Fig. 1. Two main changes take place during the adsorption of CO2 on SiC, i.e., the bending of the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO skeleton and the binding to the surface. CO2 prefers to be adsorbed on SiC with C–O bond attacking the Si–C bond, forming a four-membered ring. CO2 molecule prefers to be adsorbed perpendicular to the tube axis for armchair SiC nanotubes, while it prefers to be parallel to the tube axis for zigzag SiC nanotubes (Fig. S1 of ESI†). It is found that the Ead value of CO2 on SiC nanotubes is a function of curvature. As shown in Fig. 1, the Ead of CO2 decreases gradually with decreased surface curvature of nanotube. When the diameter of the SiC nanotubes is larger than 13 Å, CO2 molecules are physically adsorbed. This is consistent with the variation of bond lengths for Si–O and C–C bonds (Table S1 of ESI†). The bonding distances for both Si–O and C–C bonds become longer as the diameters of SiC nanotubes increases, indicating the decrease of interactions between CO2 and SiC nanotubes. Note that the effect of the chirality on the CO2 adsorption is negligible as displayed in Fig. 1. For comparison, CO2 adsorption under the consideration of vdW interaction is also calculated, as shown in Fig. 1c. Similar with previously theoretical investigation,44 CO2 adsorption is strengthened after considering the effect of vdW bonding. Note that the Ead value of CO2 on single layer SiC sheet is very small even after considering the vdW interaction, indicating that CO2 cannot be adsorbed on single layer SiC sheet stably.
CO skeleton and the binding to the surface. CO2 prefers to be adsorbed on SiC with C–O bond attacking the Si–C bond, forming a four-membered ring. CO2 molecule prefers to be adsorbed perpendicular to the tube axis for armchair SiC nanotubes, while it prefers to be parallel to the tube axis for zigzag SiC nanotubes (Fig. S1 of ESI†). It is found that the Ead value of CO2 on SiC nanotubes is a function of curvature. As shown in Fig. 1, the Ead of CO2 decreases gradually with decreased surface curvature of nanotube. When the diameter of the SiC nanotubes is larger than 13 Å, CO2 molecules are physically adsorbed. This is consistent with the variation of bond lengths for Si–O and C–C bonds (Table S1 of ESI†). The bonding distances for both Si–O and C–C bonds become longer as the diameters of SiC nanotubes increases, indicating the decrease of interactions between CO2 and SiC nanotubes. Note that the effect of the chirality on the CO2 adsorption is negligible as displayed in Fig. 1. For comparison, CO2 adsorption under the consideration of vdW interaction is also calculated, as shown in Fig. 1c. Similar with previously theoretical investigation,44 CO2 adsorption is strengthened after considering the effect of vdW bonding. Note that the Ead value of CO2 on single layer SiC sheet is very small even after considering the vdW interaction, indicating that CO2 cannot be adsorbed on single layer SiC sheet stably.
 | ||
| Fig. 1 (a) Schematic profile of CO2 adsorbed on single SiC layer and SiC nanotubes. Gray, gold and red colors denote C, Si and O atoms, respectively. Adsorption energies of CO2 on single layer SiC sheet and nanotubes without (b) and with (c) the consideration of van der Waals interaction. | ||
CO2 adsorption and desorption under the experimental condition (at 298.15 K and 1 atm) are considered, as shown in Fig. S2 of ESI.† The activation barrier energy (Ea) of CO2 from gas phase to adsorbed is 0.92 eV on single layer SiC sheet, which is much larger than that on (4,4) SiC nanotube with Ea of 0.48 eV. Furthermore, the CO2 desorption on single layer SiC sheet is much easier than that on SiC nanotubes, with Ea of 0.51 eV compared with 0.95 eV. These suggest that single layer SiC sheet cannot capture CO2, while SiC nanotubes with suitable diameter can adsorb CO2 stably. This is because the curvature of SiC nanotubes walls causes the electron of the SiC layers to shift from the concave inner surface to the convex outer surface resulting in CO2 adsorption. Adsorption strength is directly proportional to curvature size. The capture capacity of CO2 on SiC nanotubes is also examined, as shown in Table S2 and Fig. S3 of ESI.† There are 32 CO2 molecules adsorbed on (4,4) SiC nanotube, corresponding to a coverage of 1 monolayer (ML) (1 ML is defined as one CO2 molecule per Si–C dimer). As the diameter increases from 6.833 Å for (4,4) SiC nanotube to 10.297 Å for (6,6) SiC nanotube, the adsorption strength decreases from strong physisorption to weak physisorption and the adsorption coverage decreases to 0.5 ML on (6,6) SiC nanotube, further testifying that the CO2–SiC nanotube interaction becomes weaker with the increase of the tube diameter.
CO2 hydrogenation under electrochemistry environment on single SiC layer and SiC nanotubes are studied. Producing formic acid by direct hydrogenation of CO2 is mainly performed through a three-step process. Firstly, CO2 is hydrogenated to a formate or a carboxyl. And then, the formate or the carboxyl is further hydrogenated to form formic acid. At last, the formic acid gets released from SiC nanotubes and sheet. As shown in Fig. 2, there exist four different routes for CO2 hydrogenation with H+: CO2 can be hydrogenated either at its oxygen atom for carboxyl (COOH) formation (Paths 1 and 2), or at its carbon atom for formate (HCOO) formation with the bidentate (Path 3) or the trans (Path 4) structure adsorbed on SiC nanotubes. Taking the (4,4) SiC nanotube as an example, the calculated free energy diagrams for the reduction of CO2 to HCOOH at 0 V vs. RHE are shown in Fig. 2. Our calculations suggest that carboxyl pathways (Paths 1 and 2) are disadvantageous compared to formate pathways (Paths 3 and 4). The ΔG values of CO2 hydrogenation to carboxyl via Paths 1 and 2 are 1.33 and 1.57 eV, respectively, which are too large to overcome. However, the ΔG values of CO2 hydrogenation to formate through Paths 3 and 4 are −0.19 and 0.26 eV, respectively. This is consistent with the adsorption of formate and carboxyl on (4,4) SiC nanotube, where the adsorption of formate is stronger than carboxyl by nearly 2 eV. Moreover, it is also found that CO2 hydrogenation to formate intermediate is more favorable than carboxyl intermediate on Ni(111).45 Due to the stronger adsorption of formate in bidentate structure with two O atoms binding to two Si atoms at atop sites compared with that in trans configuration with H pointing toward to the surface, the ΔG value for hydrogenation of formate with bidentate structure into formic acid (Path 3) is larger than that in trans configuration (Path 4) by 0.29 eV. In addition, desorption of formic acid in Path 4 on (4,4) SiC nanotube is easier than that in Path 3. Therefore, formate pathway with trans formate as intermediate (Path 4) is the most energy favorable one and will be further investigated for the purposes of this study.
 | ||
| Fig. 2 (a–d) Free energy diagrams of four pathways for CO2 hydrogenation on (4,4) SiC nanotube. Gray, gold and red colors denote C, Si and O atoms, respectively. | ||
The calculated free energy diagrams of the lowest-energy pathways for CO2 capture and reduction on single layer SiC sheet and nanotubes are summarized in Fig. 3, shown at 0 V vs. RHE. The overall process of CO2 capture and reduction is almost thermodynamically neutral. As the curvature of SiC nanotubes decreases, the ΔG values for CO2 reduction decrease. The energy needed for the whole reduction process of CO2 on (2,2) SiC nanotube are 7 times higher compared with (8,8) SiC nanotube. CO2 capture and reduction on single layer SiC sheet and nanotubes follow the Sabatier principle.46,47 The interactions between reactants and catalysts cannot be too strong or too weak.48–50 For SiC nanotubes (diameters smaller than 6 Å) that bind CO2 too strongly, the rate of CO2 reduction is limited by the removal of adsorbed reduction intermediates. For SiC nanotubes (diameters larger than 13 Å) and single layer SiC sheet that bind CO2 too weakly, the rate is limited by the activation of CO2, or more likely, the transfer of electrons and protons to adsorbed CO2. As seen in Fig. 3, SiC nanotubes with diameters of 7–12 Å bind CO2 intermediately as compared to other SiC nanotubes and single layer sheets. At (2,2) armchair SiC nanotube, the ΔG for CO2 adsorption is as large as −1.83 eV and the ΔG values for the three steps of the CO2 reduction are 0.60, 0.60 and 0.61 eV, respectively. At (6,6) armchair SiC nanotube, the ΔG for CO2 adsorption increases to −0.47 eV, and the Er values for the three steps of CO2 reduction decrease to 0.15, 0.10 and 0.26 eV, respectively, which are much smaller than that on CODH and similar to that on tin oxide nanoparticles,25,51 suggesting high activities of CO2 reduction. Upon further increasing the diameter of SiC nanotubes to 13 Å, although the ΔG value for CO2 reduction is very small, SiC nanotubes cannot capture CO2 efficiently due to the weak adsorption.
 | ||
| Fig. 3 Free energy diagrams of CO2 hydrogenation on single SiC layer and armchair (a) and zigzag (b) SiC nanotubes. | ||
Similar with CO2 adsorption, the effect of chirality on CO2 reduction is also very small, as shown in Fig. 3. Such a remarkable agreement can be attributed to the localized characteristics of the CO2–SiC interaction. This is consistent with the interaction between CO2 and boron-rich BN nanotubes, which also shows localized characteristics.17
In order to gain further insight into the origin of the interaction between CO2 and SiC, the electronic structures of the above materials are studied. Fig. 4 illustrates the spin-polarized partial density of states (PDOS) projected onto the CO2 and SiC. In free CO2 molecule, the highest occupied molecular orbital (HOMO) is the 1πg orbital, while the lowest unoccupied molecular orbital (LUMO) is the 2πu orbital. When the OCO skeleton was bent, the LUMO of 2πu split into 6a1 and 2b1.52–54 As a consequence, the 6a1 orbital becomes the LUMO. The resulting LUMO orbital shows σ-orbital and π-orbital characteristics, and can interact effectively with the p-orbitals for charge transfer from SiC to CO2. Since the C–O orbital in 6a1 has an anti-bonding characteristic, the stronger the electron transfer, the stronger the anti-bonding and therefore the longer the C–O bond is. The renowned electron donation-back-donation mechanism contributes to the interaction between the CO2 and SiC.17 During the adsorption process of CO2 on the surface, electrons are transferred from Si-2p states to CO2-6a1 orbital and are back donated from CO2-1πg orbital to C-2p states, which result in the shift of CO2-6a1 to lower energies below the Fermi level Ef and the degeneration of CO2-1πg, as shown in Fig. 4. The electron transfer to the anti-bonding LUMO orbital weakens the CO bonds in the adsorbed molecule. To mix with surface Si-2p and C-2p orbitals, the 2p states of the adsorbed CO2 broaden, compared to those of a free CO2. This signature is common for molecules interacting with solid surfaces as predicted by the Newns–Anderson model.55 From electron density difference (Fig. S4 in ESI†), one can therefore conclude that there must be a fairly substantial covalent contribution to the C–C bond and ionic contribution to the Si–O bond between CO2 and SiC. As shown in Tables S3 and S4 of ESI,† when the curvature of the SiC nanotubes decreases, the positive charge of the Si atoms increases while the charge transfer from Si atom to O–CO2 atom and from C–CO2 to C atom of SiC decrease, resulting in weaker interactions between SiC and CO2.
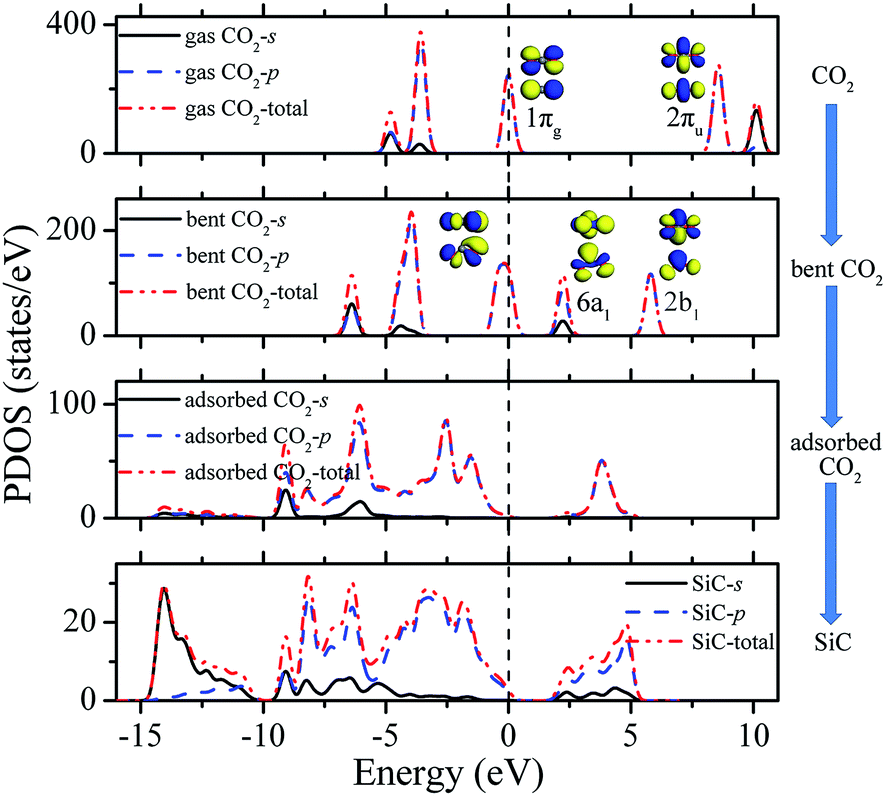 | ||
| Fig. 4 Partial density of states (PDOS) for CO2 adsorbed on (4,4) armchair SiC nanotubes. | ||
Conclusions
In conclusion, theoretical DFT calculations were performed on single layer SiC sheet and nanotubes to ascertain the catalytic activity toward CO2 adsorption and reduction. It is found that CO2 adsorption and reduction significantly depend on the surface curvature of SiC nanomaterials. CO2 cannot be adsorbed on single layer SiC sheet, while SiC nanotubes with diameter smaller than 13 Å can catch it stably. As the curvature of SiC nanotubes decreases, both the adsorption energy and reduction free energy values of CO2 decrease. These findings give insight into the unique structure–property relationship following Sabatier principle, which are particularly valuable for the design and development of new materials that would generate fuels with a carbon-neutral footprint using aqueous solutions of electrocatalysts for CO2 reduction at low over-potentials.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (no. 21403092), the Natural Science Foundation of Jiangsu (no. BK20130519), the China Postdoctoral Science Foundation (no. 2013M541611 and 2014M550270), the Senior Intellectuals Fund of Jiangsu University (no. 12JDG094 and 13JDG032) and the Danish National Research Foundation and the Danish Ministry of Science, Technology, and Innovation through Center for DNA Nanotechnology (CDNA), interdisciplinary Nanoscience Center (iNANO) and the Danish Research Councils.References
- T. R. Karl and K. E. Trenberth, Science, 2003, 302, 1719–1723 CrossRef CAS PubMed.
- D. W. Keith, Science, 2009, 325, 1654–1655 CrossRef CAS PubMed.
- R. S. Haszeldine, Science, 2009, 325, 1647–1652 CrossRef CAS PubMed.
- Z. L. Wang, J. M. Yan, H. L. Wang, Y. Ping and Q. Jiang, Sci. Rep., 2012, 2, 598 Search PubMed.
- Z. L. Wang, J. M. Yan, Y. Ping, H. L. Wang, W. T. Zheng and Q. Jiang, Angew. Chem., Int. Ed., 2013, 52, 4406–4409 CrossRef CAS PubMed.
- B. Wang, A. P. Cote, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nature, 2008, 453, 207–211 CrossRef CAS PubMed.
- O. K. Farha, A. Özgür Yazaydın, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 CrossRef CAS PubMed.
- Y. Xie, T. T. Wang, X. H. Liu, K. Zou and W. Q. Deng, Nat. Commun., 2013, 4, 1960 Search PubMed.
- H. A. Patel, S. H. Je, J. Park, D. P. Chen, Y. Jung, C. T. Yavuz and A. Coskun, Nat. Commun., 2013, 4, 1357 CrossRef PubMed.
- A. B. Vidal, L. Feria, J. Evans, Y. Takahashi, P. Liu, K. Nakamura, F. Illas and J. A. Rodriguez, J. Phys. Chem. Lett., 2012, 3, 2275–2280 CrossRef CAS.
- J. Wei, D. Zhou, Z. Sun, Y. Deng, Y. Xia and D. A. Zhao, Adv. Funct. Mater., 2013, 23, 2322–2328 CrossRef CAS.
- J. Park, H. Kim, S. S. Han and Y. Jung, J. Phys. Chem. Lett., 2012, 3, 826–829 CrossRef CAS.
- D. Feng, W. C. Chung, Z. Wei, Z. Y. Gu, H. L. Jiang, Y. P. Chen, D. J. Darensbourg and H. C. Zhou, J. Am. Chem. Soc., 2013, 135, 17105–17110 CrossRef CAS PubMed.
- M. M. Deshmukh, M. Ohba, S. Kitagawa and S. Sakaki, J. Am. Chem. Soc., 2013, 135, 4840–4849 CrossRef CAS PubMed.
- D. Liu, J. Gu, Q. Liu, Y. Tan, Z. Li, W. Zhang, Y. Su, W. Li, A. Cui, C. Gu and D. Zhang, Adv. Mater., 2014, 26, 1229–1234 CrossRef CAS PubMed.
- D. S. Zhang, Z. Chang, Y. F. Li, Z. Y. Jiang, Z. H. Xuan, Y. H. Zhang, J. R. Li, Q. Chen, T. L. Hu and X. H. Bu, Sci. Rep., 2013, 3, 3312 Search PubMed.
- H. Choi, Y. C. Park, Y. H. Kim and Y. S. Lee, J. Am. Chem. Soc., 2011, 133, 2084–2087 CrossRef CAS PubMed.
- Q. Sun, Z. Li, D. J. Searles, Y. Chen, G. M. Lu and A. Du, J. Am. Chem. Soc., 2013, 135, 8246–8253 CrossRef CAS PubMed.
- Y. Yang, M. G. White and P. Liu, J. Phys. Chem. C, 2012, 116, 248–256 CAS.
- K. Sekizawa, K. Maeda, K. Domen, K. Koike and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 4596–4599 CrossRef CAS PubMed.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 CAS.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52, 2459–2462 CrossRef CAS PubMed.
- J. H. Kwak, L. Kovarik and J. Szanyi, ACS Catal., 2013, 3, 2449–2455 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 CAS.
- J. B. Varley, H. A. Hansen, N. L. Ammitzbøll, L. C. Grabow, A. A. Peterson, J. Rossmeisl and J. K. Nørskov, ACS Catal., 2013, 3, 2640–2643 CrossRef CAS.
- D. J. Boston, C. Xu, D. W. Armstrong and F. M. Macdonnell, J. Am. Chem. Soc., 2013, 135, 16252–16255 CrossRef CAS PubMed.
- C. A. Huff and M. S. Sanford, ACS Catal., 2013, 3, 2412–2416 CrossRef CAS.
- A. Karelovic and P. Ruiz, ACS Catal., 2013, 3, 2799–2812 CrossRef CAS.
- A. S. Varela, C. Schlaup, Z. P. Jovanov, P. Malacrida, S. Horch, I. E. L. Stephens and I. Chorkendorff, J. Phys. Chem. C, 2013, 117, 20500–20508 CAS.
- J. Ye, C. Liu, D. Mei and Q. Ge, ACS Catal., 2013, 3, 1296–1306 CrossRef CAS.
- H. A. Hansen, J. B. Varley, A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2013, 4, 388–392 CrossRef CAS.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- M. Z. Ertem, S. J. Konezny, C. M. Araujo and V. S. Batista, J. Phys. Chem. Lett., 2013, 4, 745–748 CrossRef CAS.
- E. B. Cole, P. S. Lakkaraju, D. M. Rampulla, A. J. Morris, E. Abelev and A. B. Bocarsly, J. Am. Chem. Soc., 2010, 132, 11539–11551 CrossRef PubMed.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS PubMed.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- A. Klamt and G. Schuurmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- B. Delley, Mol. Simul., 2006, 32, 117–123 CrossRef CAS.
- J. Andzelm, C. Kölmel and A. Klamt, J. Chem. Phys., 1995, 103, 9312–9320 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- L. Yu, X. Pan, X. Cao, P. Hu and X. Bao, J. Catal., 2011, 282, 183–190 CrossRef CAS PubMed.
- P. Zhang, B. B. Xiao, X. L. Hou, Y. F. Zhu and Q. Jiang, Sci. Rep., 2014, 4, 3821 CAS.
- G. Peng, S. J. Sibener, G. C. Schatz, S. T. Ceyer and M. Mavrikakis, J. Phys. Chem. C, 2012, 116, 3001–3006 CAS.
- Q. T. Trinh, J. Yang, J. Y. Lee and M. Saeys, J. Catal., 2012, 291, 26–35 CrossRef CAS PubMed.
- J. L. Lin and I. Wheeldon, ACS Catal., 2013, 3, 560–564 CrossRef CAS.
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Angew. Chem., Int. Ed., 2006, 45, 2897–2901 CrossRef CAS PubMed.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- S. Zhang, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 1734–1737 CrossRef CAS PubMed.
- Q. L. Tang and Q. H. Luo, J. Phys. Chem. C, 2013, 117, 22954–22966 CAS.
- S. G. Wang, X. Y. Liao, D. B. Cao, C. F. Huo, Y. W. Li, J. Wang and H. Jiao, J. Phys. Chem. C, 2007, 111, 16934–16940 CAS.
- C. Cazorla, S. A. Shevlin and Z. X. Guo, J. Phys. Chem. C, 2011, 115, 10990–10995 CAS.
- D. M. Newns, Phys. Rev., 1969, 178, 1123–1135 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra07542k |
| This journal is © The Royal Society of Chemistry 2014 |