DOI:
10.1039/C4RA08823A
(Paper)
RSC Adv., 2014,
4, 49374-49385
Towards high-performance poly(L-lactide)/elastomer blends with tunable interfacial adhesion and matrix crystallization via constructing stereocomplex crystallites at the interface
Received
22nd August 2014
, Accepted 29th September 2014
First published on 29th September 2014
Abstract
In this work, we report a facile strategy to prepare super-tough and heat-resistant poly(L-lactide) (PLLA) blends by constructing stereocomplex (sc) crystallites with dual interfacial adhesion enhancer/matrix crystallization accelerator functionality at the interface of the blends of PLLA/ethylene copolymer. To exploit the dual functionality, poly(D-lactide) grafted ethylene–acrylic ester copolymer (EMA-g-PDLA) capable of collaborating with the PLLA matrix to form the sc crystallites was first prepared via melt coupling reaction between end groups (carboxyl and hydroxyl) of PDLA and excess epoxy group of EMA–glycidyl methacrylate copolymer (EMA–GMA). During subsequent melt-blending of PLLA with the prepared EMA-g-PDLA, sc crystallites are formed at the interface. The results show that, compared with PLLA/EMA–GMA and PLLA/EMA-g-PLLA blends, injection molded PLLA/EMA-g-PDLA blends have much higher impact toughness and heat resistance because the interface-localized sc crystallites can induce substantial enhancement in both interfacial adhesion and matrix crystallinity. More interestingly, by modulating the amount of sc crystallites at the interface of the blends, optimum impact toughness can be achieved due to the optimization of interfacial strength and matrix crystallinity. This work provides a new concept for the fabrication of high-performance PLLA blends by tailoring matrix and interface properties with the aid of sc crystallites.
1. Introduction
With growing awareness of environmental concerns and sustainability, developing biodegradable polymers completely derived from renewable resources to replace conventional non-biodegradable polymers derived from petroleum has attracted considerable attention from both academia and the industrial community in the past two decades.1,2 One such polymer is poly(L-lactide) (PLLA), which has been widely recognized as one of the most promising environmentally friendly candidates due to its renewability, biodegradability, good biocompatibility, excellent transparency, favorable mechanical strength and stiffness, and easy processability.3–5 Nowadays, it has been applied to various biomedical and industrial applications, such as drug delivery devices, sutures, agricultural films, food packages, automotive interiors, and electronics.1,4 Unfortunately, its brittleness and poor heat resistance (low crystallization rate makes it difficult to crystallize during melt processing6) greatly limit the use of PLLA in large-scale commercial applications. Thus, there is a great deal of current interest in simultaneously improving these properties to broaden its utility.7–9
Among various strategies used for toughening modification of PLLA, melt blending with flexible polymers, such as natural rubber,10 ethylene-co-vinyl acetate (EVA),11 poly(butylene succinate) (PBS),12 poly(ether)urethane (PU) elastomer,13–16 and poly(ε-caprolactone) (PCL),8,9,17 is the most elegant and powerful approach. Nevertheless, even though most of these blends exhibit super tensile toughness (or ductility), only a slight enhancement in notched impact toughness can be obtained.18 The low resistance of the toughened blends against impact loading arises from the poor compatibility between PLLA matrix and these toughening modifiers as well as the resulting weak interfacial adhesion due to the insufficient chain entanglement density across the interface.19–21 Therefore, several compatibilization protocols including adding pre-synthesized block or graft copolymers and reactive compatibilization (producing compatibilizers in situ) have been developed to enhance the interfacial strength, and thus to improve toughening effect of dispersed modifier particles on PLLA matrix.18,20–27 Because copolymers with special structure are often difficult to synthesize and effective compatibility can be achieved only if the copolymer used is adequately diffused to the interface and then tightly entangled with the chains of the blend components, in situ reactive compatibilization has been frequently used as a much more simple and effective way to improve the compatibility and reinforce the interface of immiscible polymer blends.20,21,27 Reactive blending strategies have been widely employed for modifying PLLA by blending it with elastomers containing functional groups capable of reacting with its end groups (carboxyl and hydroxyl), such as maleic anhydride20 and glycidyl methacrylate,21,27 or by introducing catalysts/coupling agents into the elastomer toughened PLLA blends.17,24–26 For example, Liu et al.21,25 reported a super toughened PLLA blend system consisting of PLLA, ethylene–butyl acrylate–glycidyl methacrylate terpolymer (EBA–GMA), and zinc ionomer of ethylene–methyacrylic acid copolymer (EMAA–Zn). They found that zinc ions can catalyze the reactive compatibilization occurred at the interface between PLLA matrix and EBA–GMA dispersed phase, and blending temperature plays a key role in the toughening. Blending the ternary system at 240 °C gives rise to a much more effective interfacial reactive compatibilization than at 185 °C, so notched impact strength increases dramatically from 94.5 J m−1 to 777.2 J m−1.
Undoubtedly, direct reactive blending is a simple way towards super toughened PLLA/elastomer blends. However, this method may be less attractive for the fabrication of blends with both super toughness and excellent heat resistance because all the blending inevitably leads to an undesirable deterioration in the originally poor heat resistance of PLLA. On the other hand, adding small amounts (0.3–0.5 wt%) of organic nucleating agent (NA) can be used as a facile strategy to induce notable enhancements in PLLA matrix crystallinity and subsequent heat deflection temperature of melt-processed PLLA/elastomer blends without reactive functional groups while maintaining or even further improving their impact toughness,8,9 but it seems not suitable for enhancing the heat resistance of PLLA reactive blends because chemical groups (e.g., amides) of NA molecules could react with plentiful functional groups (e.g., maleic anhydride and epoxides) existed in the blends during melt blending process and eventually lose their ability to nucleate matrix crystallization.
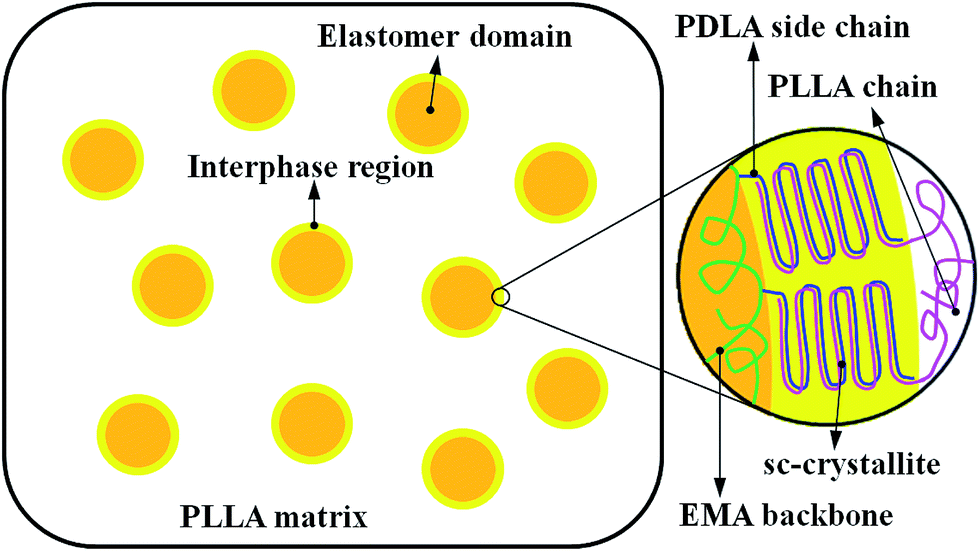
Therefore, we will report our effort to search for an alternative strategy to simultaneously enhance the interfacial adhesion and matrix crystallization of immiscible PLLA/elastomer blends in the present work. In recent years, PLLA/poly(D-lactide) (PDLA) stereocomplex (sc) crystallites, formed by incorporating PDLA into PLLA, have attracted much academic and industrial interest.28–37 Due to their high melting temperature (about 50 °C higher than that of homochiral PLLA or PDLA crystallites28) and multifunctionality, numerous applications have been developed for sc crystallites, such as rheological modifiers,29,30 reinforcer,31,32 and highly active nucleating agent for PLLA crystallization.29,33 Herein, we employed PDLA grafted ethylene–acrylic ester copolymer (EMA-g-PDLA) as a multifunctional modifier to improve the performance of PLLA. The melt bending temperature used for the preparation of PLLA/EMA-g-PDLA blends is selected as 190 °C, which is one of the optimum melt processing temperatures for the exclusive formation of sc crystallites.35,36 It is expected that PDLA side chains and PLLA matrix chains could arrange side by side at the interface of the blends under the drive of strong shear stress during the melt blending process, forming sc crystallites with dual functionality (Scheme 1) as both highly active nucleating agent and effective interfacial adhesion enhancer to simultaneously enhance PLLA matrix crystallization and interfacial strength. Then high-performance PLLA blends with both super impact toughness and high heat resistance could be achieved after subsequent injection molding. In particular, roles of the interface-localized sc crystallites in controlling the matrix crystallization and impact fracture behaviors of PLLA/EMA-g-PDLA blends were highlighted by comparing with those of PLLA/EMA–GMA and PLLA/EMA-g-PLLA blends. To the best of our knowledge, there have been no reports in the literature addressing the sc crystallites tailored matrix crystallization and impact behaviors of immiscible PLLA blends.
 |
| | Scheme 1 Schematic representation showing the interfacial structure of PLLA/EMA-g-PDLA blends with a sea-island phase morphology after melt-blending at a temperature between the melting temperature (Tm) of PLLA crystallites and that of sc crystallites. | |
2. Experimental section
2.1 Materials
Poly(L-lactide) (PLLA, trade name 4032D) was purchased from NatureWorks LLC, U.S.A. It has a weight-averaged molecular weight (Mw) of 1.7 × 105 g mol−1 and an optical purity of 98.6%. Poly(D-lactide) (PDLA) with a Mw of 1.2 × 105 g mol−1 and an optical purity of 99.5% was kindly supplied by Zhejiang Hisun Biomaterial Co. Ltd., China. Ethylene–acrylic ester–glycidyl methacrylate random copolymer (EMA–GMA, E/MA/GMA = 68/24/8 (wt%), trade name LOTADER AX 8900) used as the impact modifier for PLLA was provided by Arkema Inc., France. N,N-Dimethylstearylamine (DMSA) was obtained from Aldrich and used as received. Isotactic polypropylene (PP, T30S) with a Mw of 4.0 × 105 g mol−1 was supplied by Dushanzi Petrochemical Co., Ltd., China. Prior to use, all the plastic pellets were dried overnight in a vacuum oven at 60 °C.
2.2 Sample preparation
Grafting of PDLA onto EMA–GMA was realized through coupling reaction between epoxy group of EMA–GMA copolymer and end groups (carboxyl and hydroxyl) of PDLA. The reaction was carried out by melt-blending of EMA–GMA and PDLA (2.5–12.5 wt%) in the presence of small amount (0.3 wt%) of catalyst DMSA using a Haake Rheomix 600 internal mixer (Germany) at a temperature of 190 °C and a rotor speed of 80 rpm for 8 min. To ensure good dispersion and high catalytic efficiency, DMSA was firstly dissolved in absolute alcohol under nitrogen atmosphere and then mixed with PDLA pellets prior to the melt-blending. Because the EMA–GMA was used in excess in the reactive blending (grafting) procedure, the resultants should be composed of EMA-g-PDLA graft copolymer and unreacted EMA–GMA copolymer. For convenience, the prepared reactive blends are labeled as EMA-g-xPDLA in the present work, where x indicates the PDLA content. EMA–GMA/PLLA reactive blends were also prepared using the same procedure as the EMA–GMA/PDLA blends and the resultant copolymers are labeled as EMA-g-xPLLA.
Blends of PLLA with various amounts (5–30 wt%) of EMA-g-xPDLA were prepared using the Rheomix 600 internal mixer at a temperature of 190 °C and a rotor speed of 80 rpm for 5 min. For comparison purpose, PLLA/EMA–GMA and PLLA/EMA-g-xPLLA blends were also prepared. Standard specimens of the blends for mechanical testing were fabricated with a mini-injection molder (HAAKE MiniJet II, Germany) at a barrel temperature of 200 °C and a mold temperature of 130 °C. To tailor the crystallinity of PLLA matrix, several isothermal annealing time (0.5–25 min, depending on the weight percentage of PDLA in the EMA-g-PDLA copolymers and the content of EMA-g-PDLA copolymers in the PLLA/EMA-g-PDLA blend) of the blend melts in the hot mold were applied according to the DSC results obtained from isothermal crystallization.
2.3 Fourier transform infrared spectroscopy (FT-IR)
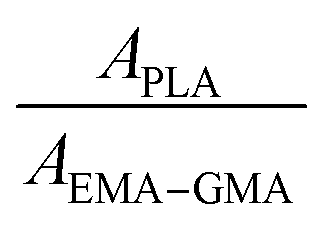
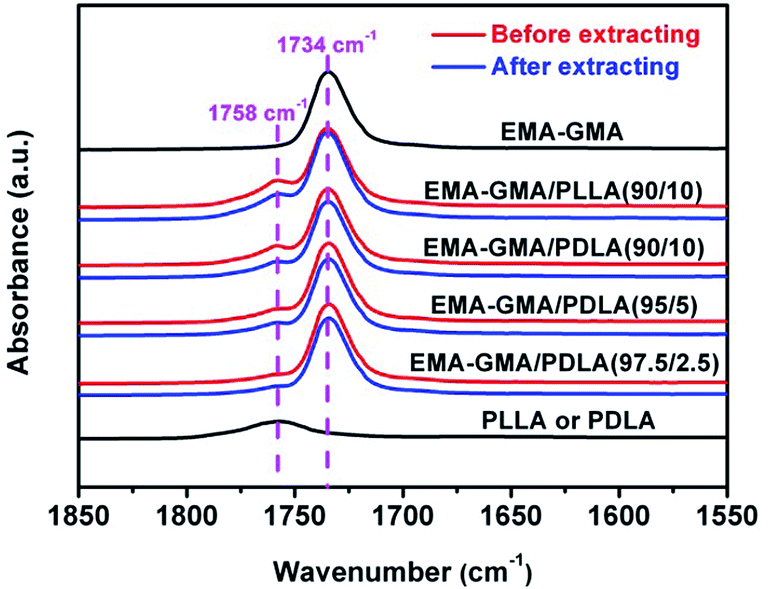
In order to confirm the formation of EMA-g-PDLA (or EMA-g-PLLA) graft copolymers during reactive blending and determine the graft efficiency of PDLA (or PLLA) onto EMA–GMA, Fourier transform infrared spectroscopy (FT-IR) spectra were recorded using a Thermo Nicolet 3700 spectrometer (U.S.A.) with a resolution of 4 cm−1 and an accumulation of 32 scans. Thin films (∼20 μm) of PLLA and EMA–GAM used for the FT-IR measurement were prepared by spin-coating from their chloroform solutions. For EMA–GMA/PDLA and EMA–GMA/PLLA blends, slices with a thickness of ∼100 μm were firstly extracted with dioxane at room temperature for a week to selectively remove unreacted PDLA or PLLA from the blends thoroughly, followed by spin-coating from their chloroform solutions into thin films (∼20 μm). Prior to the FT-IR measurements, all the films were dried in a vacuum oven to completely remove the residual solvent. After baseline correction, deconvolution of the bands at ∼1758 and ∼1734 cm−1 (attributed to the stretching vibration of carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in PLA and EMA–GMA, respectively25) was made using a Gaussian–Lorentzian mixed function.
O) in PLA and EMA–GMA, respectively25) was made using a Gaussian–Lorentzian mixed function.
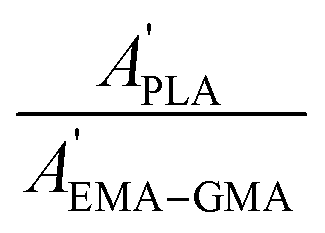
Considering that the amount of EMA–GMA component remains constant during the extracting process, the graft efficiency (GE) of PLA can be estimated by the amount of grafted PLA per unit of PLA used using the following equation:
| |
 | (1) |
where
Ai and
A′i are the absorption peak areas of component
i before and after extracting with dioxane, respectively.
2.4 Scanning electron microscopy (SEM)
The phase morphology of prepared blends was studied using an FEI Inspect F scanning electron microscope (SEM, U.S.A.) at an accelerating voltage of 5 kV. Specimens used for the SEM observation were prepared by cryogenically fracture of the blended samples. In order to quantify the dispersed particle size and particle size distribution, at least 350 particles from several individual SEM images were measured using an Image-Pro Plus software for each sample, neglecting those particles having the diameters smaller than 50 nm. Weight-average particle diameter (dw) and particle diameter distribution parameter (σ) were calculated using the following relationships:| |
 | (2) |
| |
 | (3) |
where ni is the number of particles with the diameter of di. In the case of polydispersity, σ is greater than 1, whereas for monodispersity, σ is equal to 1.
The fracture mechanisms of the impact-fractured samples were also investigated with the SEM. The impact-fractured surfaces obtained from the Notched Izod impact testing were observed, focusing especially on the regions of crack initiation. To get more in-depth information on the fracture mechanisms, the injection molded bars were cryo-fractured in liquid nitrogen along a plane perpendicular to the thickness direction and the deformation zones underneath the impact-fractured surfaces were examined. Prior to the SEM observations, all the fractured surfaces were sputtered with a thin layer of gold.
2.5 Crystalline structure and morphology
Crystalline structure was analyzed using a Philips X'Pert pro MPD X-ray diffractometer (Holland) with a CuKα radiation generated at 40 kV and 40 mA. The wide-angle X-ray diffraction (WAXD) patterns were recorded in the reflection mode at a scanning rate of 5° min−1 from 5° to 40°.
Crystalline morphology was observed using a Leica DMLP polarized optical microscope (POM, Germany) equipped with a Linkam THMS 600 hot stage (Germany). Specimens used for the observation were prepared according to the following procedure. Firstly, about 0.5 mg of a sample was placed between two microscope cover slips and then pressed at 200 °C to obtain a slice (∼20 μm in thickness). Subsequently, the as-prepared slice was quickly cooled down to a pre-determined temperature of 136 °C for isothermal crystallization after achieving thermal equilibrium at 200 °C. The POM micrographs upon crystallization processes were recorded in real time with a Cannon digital camera.
2.6 Thermal analysis
Thermal analysis was conducted on a Perkin-Elmer pyris-1 differential scanning calorimeter (DSC, U.S.A.) under a dry nitrogen atmosphere. For the analysis of isothermal crystallization behavior, about 5 mg of sample sealed in a aluminum pan was firstly melted at 200 °C for 3 min to erase any thermal history and then rapidly cooled down to various temperatures (ranging from 124 to 138 °C) at a cooling rate of 100 °C min−1 and maintained at this temperature for certain time to allow complete crystallization. The crystallinity of PLLA matrix (Xc,PLLA) in the injection molded blends was evaluated by first DSC heating runs at a heating rate of 10 °C min−1 from 30 °C to 200 °C according to the following equation:| |
 | (4) |
where ΔHm and ΔHc are the melting enthalpy and the cold crystallization enthalpy during the heating run, respectively; ΔH°m is the melting enthalpy of 100% crystalline PLLA (93.6 J g−1 (ref. 38)), and wf is the weight percent of PLLA matrix in the sample.
2.7 Mechanical testing
Notched Izod impact strength was measured using a pendulum impact tester (XJU-5.5, China) in accordance with the ISO 180/179 standard and the tensile properties were evaluated using a tensile testing machine (SANS, China) at a cross-head speed of 5.0 mm min−1 according to the ISO 527-3 standard. The mechanical testing was performed at room temperature (23 °C) and the average value reported was obtained from five independent specimens for each sample.
2.8 Dynamic mechanical analysis (DMA)
Dynamic mechanical properties were measured using a TA Q800 instrument (USA) in the single-cantilever mode with a sinusoidal oscillating strain of 10 μm and a frequency of 1 Hz. The measurement was performed from 0 to 150 °C at a heating rate of 3 °C min−1. For each sample, at least two independent specimens were tested in order to ensure the reproducibility of results obtained.
3. Results and discussion
3.1 Synthesis of EMA-g-PLA graft copolymers
EMA-g-PLA graft copolymers containing either PDLA or PLLA branches were synthesized by melt coupling reaction between epoxy group of EMA–GMA copolymer and end groups (carboxyl and hydroxyl) of PLA using DMSA as catalyst, as illustrated in Scheme 2. The reaction was performed by melt-blending PDLA or PLLA with excess EMA–GMA at 180 °C. Graft efficiency (GE) of PDLA and PLLA onto EMA–GMA, defined as the amount of grafted PLA per unit of PLA used, was estimated using FT-IR and the calculated results are given in Table 1. Fig. 1 shows the FT-IR absorption spectra of EMA–GMA, PLLA (or PDLA) and their binary blends before and after extracting with dioxane in the range of 1550–1850 cm−1. The absorption peaks at ∼1758 and ∼1734 cm−1 are attributed to the CO stretching vibration in the PLA and EMA–GMA, respectively.25 Clearly, all EMA–GMA/PLA blends still exhibit strong characteristic absorption at ∼1758 cm−1 after complete removal of free PLA component by dioxane extraction, confirming the success in grafting PLA molecules onto EMA–GMA to form EMA-g-PLA graft copolymers. More importantly, although GE decreases evidently with increasing PDLA content from 2.5 to 10 wt% (Table 1), the GE in EMA–GMA/PLA(90/10) blend is as high as 85.5–86.7%, indicating that more than 85% PDLA or PLLA molecules used are successfully grafted onto EMA–GMA chains during reactive blending process.
 |
| | Scheme 2 Schematic illustration of the approach used for melt-grafting of PDLA onto EMA–GMA copolymer. | |
Table 1 FT-IR peak-resolving results of EMA–GMA/PLA blends before and after extracting with dioxane
| Samples |
Before |
After |
GE (%) |
| APLA |
AEMA–GMA |

|
A′PLA |
A′EMA–GMA |

|
| EMA–GMA/PDLA(97.5/2.5) |
0.69 |
21.05 |
0.033 |
0.66 |
20.48 |
0.032 |
96.9 |
| EMA–GMA/PDLA(95/5) |
1.61 |
21.47 |
0.075 |
1.41 |
20.37 |
0.068 |
90.7 |
| EMA–GMA/PDLA(90/10) |
3.22 |
20.25 |
0.159 |
2.72 |
20.02 |
0.136 |
85.5 |
| EMA–GMA/PLLA(90/10) |
3.92 |
21.72 |
0.180 |
3.15 |
20.41 |
0.156 |
86.7 |
 |
| | Fig. 1 FT-IR absorption spectra of EMA–GMA, PLLA (or PDLA) and their binary blends before and after extracting with dioxane. | |
3.2 Microstructure and matrix crystallization behavior of the PLLA blends
In order to reveal the role of interfacial microstructure in determining the performance of PLLA/elastomer blends, PLLA/EMA-g-PDLA blends as well as their counterparts (PLLA/EMA–GMA and PLLA/EMA-g-PLLA blends) were prepared for comparison. To prevent the occurrence of potential reactive compatibilization, no catalyst was incorporation into these blends. WAXD analysis provides a direct evidence for the formation of sc crystallites in the PLLA/EMA-g-PDLA blends. As presented in Fig. 2, the WAXD pattern of melt-quenched PLLA/EMA-g-5PDLA (85/15) blend exhibits two weak characteristic diffraction peaks of sc crystallites at around 12.0° and 20.9°, corresponding to the (110) and (300)/(030) planes.28 With increasing weight percentage of PDLA in the EMA-g-PDLA graft copolymers up to 10 wt%, the pattern shows not only the enhanced intensity of the two characteristic diffraction peaks but also the presence of a new characteristic peak of sc crystallites at around 24.0°, indicating greatly increased amount of sc crystallites. As for PLLA/EMA–GMA (85/15) and PLLA/EMA-g-10PLLA (85/15) blends, no diffraction peaks of sc crystallites are observed due to the absence of PDLA chains in these blends.
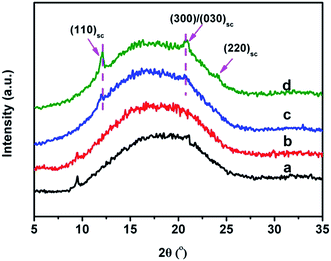 |
| | Fig. 2 WAXD patterns of melt-quenched blends: (a) PLLA/EMA–GMA (85/15), (b) PLLA/EMA-g-10PLLA (85/15), (c) PLLA/EMA-g-5PDLA (85/15), and (d) PLLA/EMA-g-10PDLA (85/15). | |
Phase morphology of the as-prepared blends was observed with SEM and some representative SEM images are presented in Fig. 3. Noticeably, all blends exhibit an identical “sea-island” morphology (with spherical elastomer domains dispersed in continuous PLLA matrix) and a similar particle size of dispersed elastomer domains (statistical results of weight-average particle diameter and its distribution are summarized in Table 2), differing only in interfacial adhesion. For PLLA/EMA–GMA (85/15) blend, many evident holes and particles resulting from the debonding of dispersed EMA–GMA particles from PLLA matrix are visible on the whole cryo-fractured surface (Fig. 3a), indicating poor interfacial adhesion between matrix and dispersed phase due to the absence of effective compatibilization. PLLA/EMA-g-10PLLA (85/15) blend also shows extensive debonding at interfaces (Fig. 3b). Only a slightly improved wetting of the dispersed EMA-g-PLLA particles by the PLLA matrix is detected when compared to the PLLA/EMA–GMA (85/15) blend. Very interestingly, PLLA/EMA-g-5PDLA (85/15) blend displays a much better interface wetting (Fig. 3c) than the PLLA/EMA-g-10PLLA (85/15) blend, demonstrating a good interfacial adhesion. The markedly enhanced interfacial adhesion might be attributed to the formation of sc crystallites between PDLA side chains of EMA-g-PDLA graft copolymers and PLLA matrix chains at the interfaces. The interface-localized sc crystallites have a much stronger interaction with both PLLA matrix and dispersed elastomer phase as compared with the low-density chain entanglement across the interface of PLLA/EMA–GMA and PLLA/EMA-g-PLLA blends, which has been demonstrated by He and coworkers in their attempt to improve the toughening efficiency of PLLA/rubber blends by enhancing interfacial adhesion.39,40 Increasing weight percentage of PDLA in the EMA-g-PDLA graft copolymers from 5 wt% to 10 wt% gives rise to a further enhancement in interfacial adhesion (Fig. 3d) because of the increased amount of interface-localized sc crystallites. In general, the introduced compatibilizer can lead to simultaneous occurrence of strengthening interface adhesion and decreasing droplet size of the dispersed phase.20,26 Unexpectedly, PLLA/EMA-g-PDLA blends exhibit much stronger interfacial adhesion than the PLLA/EMA–GMA and PLLA/EMA-g-PLLA blends, but no evident difference in the dispersed elastomer particle size is observed (Table 2). This finding suggests that, besides reducing the interfacial tension and suppressing droplet coalescence as an effective compatibilizer, sc crystallites formed at the interface of PLLA/EMA-g-PDLA blends may also play a role in promoting droplet coalescence of the dispersed EMA-g-PDLA phase by changing rheology behavior of the blend melts or/and inducing aggregation of EMA-g-PDLA droplets as some inorganic nanoparticles (e.g., nano-silica) with a strong self-networking capability.13,41 An in-depth investigation is underway to get a clear understanding of the underlying mechanisms and the results will be reported in our future work.
 |
| | Fig. 3 SEM images showing the phase morphologies of melt-blended blends: (a) PLLA/EMA–GMA (85/15), (b) PLLA/EMA-g-10PLLA (85/15), (c) PLLA/EMA-g-5PDLA (85/15), and (d) PLLA/EMA-g-10PDLA (85/15). | |
Table 2 Weight-average diameter of dispersed phase (dw) and diameter distribution parameter (σ) in the injection molded blends
| Samples |
dw (μm) |
σ |
| PLLA/EMA–GMA (85/15) |
0.45 |
1.47 |
| PLLA/EMA-g-10PLLA (85/15) |
0.46 |
1.57 |
| PLLA/EMA-g-5PDLA (85/15) |
0.42 |
1.51 |
| PLLA/EMA-g-10PDLA (85/15) |
0.40 |
1.53 |
To demonstrate the effectiveness of the sc crystallites formed at the interface for nucleating PLLA matrix crystallization, isothermal crystallization behaviors of PLLA/EMA–GMA, PLLA/EMA-g-PLLA, and PLLA/EMA-g-PDLA blends were comparatively investigated using DSC and POM. Please be noted that all the samples were melted at 200 °C (above the melting temperature of homochiral PLLA and PDLA crystallites but below the melting temperature of sc crystallites) prior to PLLA matrix crystallization. In this case, homochiral crystallites were completely melted but sc crystallites were reserved in the melt of the PLLA blends. Fig. 4 shows the DSC thermograms of four blends upon isothermal crystallization at various temperatures. Obviously, all blends exhibit symmetric exothermic peaks and the crystallization time (t) increases evidently with increasing crystallization temperature because of the increased nucleation barrier. The values obtained for the isothermal crystallization peak time (tp), defined as the time at which exotherm reaches a maximum, are given in the profiles. Apparently, the crystallization rate of PLLA/EMA–GMA (85/15) blend is very slow (Fig. 4a). For PLLA/EMA-g-10PLLA (85/15) blend, no apparent difference in the values of tp can be observed at the same crystallization temperatures as compared with PLLA/EMA–GMA (85/15) blend (Fig. 4b), implying a similar low crystallization rate. However, once some sc crystallites are formed at the interface, the crystallization rate of PLLA/EMA-g-5PDLA (85/15) blend is enhance dramatically (Fig. 4c). For example, at the crystallization temperature of 130 °C, the value of tp decreases significantly from 17.1 min for PLLA/EMA–GMA (85/15) blend to 2.4 min for PLLA/EMA-g-5PDLA (85/15) blend, indicating strong heterogeneous nucleating effect of sc crystallites on PLLA crystallization. Such heterogeneous nucleating effect becomes more evident in the PLLA/EMA-g-10PDLA (85/15) blend due to the increased amount of sc crystallites at the interface, and the value of tp at 130 °C is further decreased to 2.0 min (Fig. 4d). More importantly, the tp values of PLLA/EMA-g-10PDLA (85/15) blend are comparable to those of PLLA containing various commercial nucleating agents, such as talc,6 1,3,5-benzene-tricarboxylamide derivatives,42 and homogeneously dispersed sc crystallites,29 demonstrating that the interface-localized sc crystallites can serve as highly active nucleating agent for PLLA matrix crystallization.
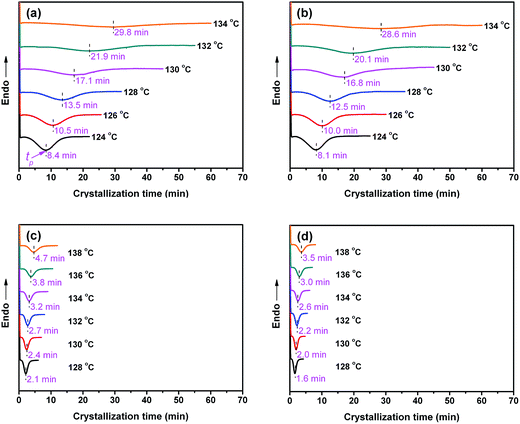 |
| | Fig. 4 DSC thermograms recorded during isothermal crystallization of (a) PLLA/EMA–GMA (85/15), (b) PLLA/EMA-g-10PLLA (85/15), (c) PLLA/EMA-g-5PDLA (85/15), and (d) PLLA/EMA-g-10PDLA (85/15) blends at different temperatures. | |
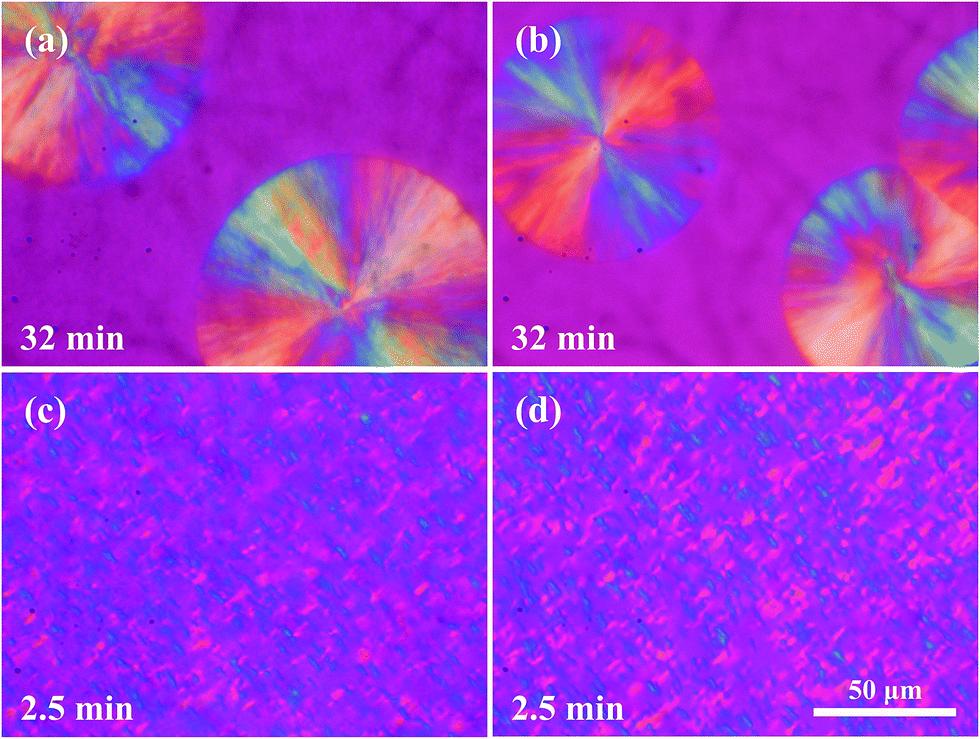
The effectiveness of the formed sc crystallites as highly active nucleating agent for PLLA matrix crystallization can be further proved by POM observations on the crystalline morphology. For PLLA/EMA–GMA (85/15) and PLLA/EMA-g-10PLLA (85/15) blends, large PLLA spherulites with average diameter of 100–150 μm are observed (Fig. 5a and b). However, with the formation of sc crystallites at the interface, crystalline morphology of PLLA/EMA-g-5PDLA (85/15) and PLLA/EMA-g-10PDLA (85/15) blends are markedly changed as expected. The size of PLLA spherulites decreases dramatically (Fig. 5c and d) and it is hardly to differentiate them one by one, confirming the high nucleating efficiency of sc crystallites on PLLA matrix crystallization. This is in good agreement with the DSC results obtained from isothermal crystallization process.
 |
| | Fig. 5 POM images showing the crystalline morphologies of (a) PLLA/EMA–GMA (85/15), (b) PLLA/EMA-g-10PLLA (85/15), (c) PLLA/EMA-g-5PDLA (85/15), and (d) PLLA/EMA-g-10PDLA (85/15) blends after isothermal crystallization at 136 °C for a certain time. | |
From the above results, it is evident that sc crystallites formed at the interface of PLLA/EMA-g-PDLA blends can serve as both highly active nucleating agent and effective interfacial adhesion enhancer to simultaneously tailor the PLLA matrix crystallization behavior and the interfacial adhesion.
3.3 Impact toughness and toughening mechanisms
To explore the effect of sc crystallites tailored interfacial adhesion and matrix crystallization on the impact toughness, a series of PLLA/ethylene copolymer blends with different interfacial strengths and matrix crystallinities were prepared by injection molding. Fig. 6 shows the DSC melting curves of representative injection molded PLLA/EMA–GMA, PLLA/EMA-g-10PLLA and PLLA/EMA-g-10PDLA blends fabricated at different conditions. As presented in Fig. 6a and b, both PLLA/EMA–GMA and PLLA/EMA-g-10PLLA blends exhibit multiple transitions upon heating: a glass transition bump, a cold crystallization peak (Pcc), a dominant melting peak (Pm), and a small exothermic peak (Pex) before the Pm arising from the phase transition from disordered α′-form crystallites to ordered α-form crystallites during DSC heating scan.43 Moreover, Xc,PLLA in both the blends is at the same low level (<6%), indicating an almost amorphous PLLA matrix in these blends. PLLA/EMA-g-10PDLA blends with a comparable amorphous matrix also show the similar multiple transitions in the DSC melting curves (Fig. 6c). However, the melting behaviors of PLLA/EMA-g-10PDLA blends with a highly crystalline PLLA matrix (Xc,PLLA is as high as 40%) are considerably different from those with an almost amorphous matrix. All the melting curves display only Pm while Pcc and Pex disappear (Fig. 6d). Additionally, all the injection molded blends have the similar domain size of dispersed elastomer phase (weight-average particle diameter is about 0.45–0.49 μm, SEM images are not shown here for brevity). The above results demonstrate that we have successfully prepared a series of PLLA/ethylene copolymer blends differing only in interfacial strength and matrix crystallinity, i.e., PLLA/EMA–GMA blends with a poor interfacial adhesion and an amorphous matrix, PLLA/EMA-g-10PLLA blends with a weak interfacial adhesion and an amorphous matrix, PLLA/EMA-g-10PDLA blend with a strong interfacial adhesion and an amorphous matrix, and PLLA/EMA-g-10PDLA blend with a strong interfacial adhesion and a highly crystalline matrix.
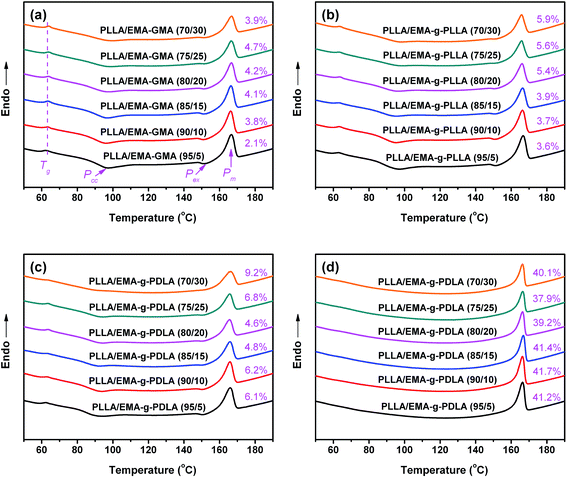 |
| | Fig. 6 DSC melting curves of PLLA/ethylene copolymer blends prepared by injection molding and subsequent annealing in a preheated mold of 130 °C for various amounts of time: (a) 0.5 min for PLLA/EMA–GMA, (b) 0.5 min for PLLA/EMA-g-10PLLA, (c) 0.5 min for PLLA/EMA-g-10PDLA, and (d) 5 min for PLLA/EMA-g-10PDLA. The values of PLLA matrix crystallinity (Xc,PLLA) are presented in the profiles. | |
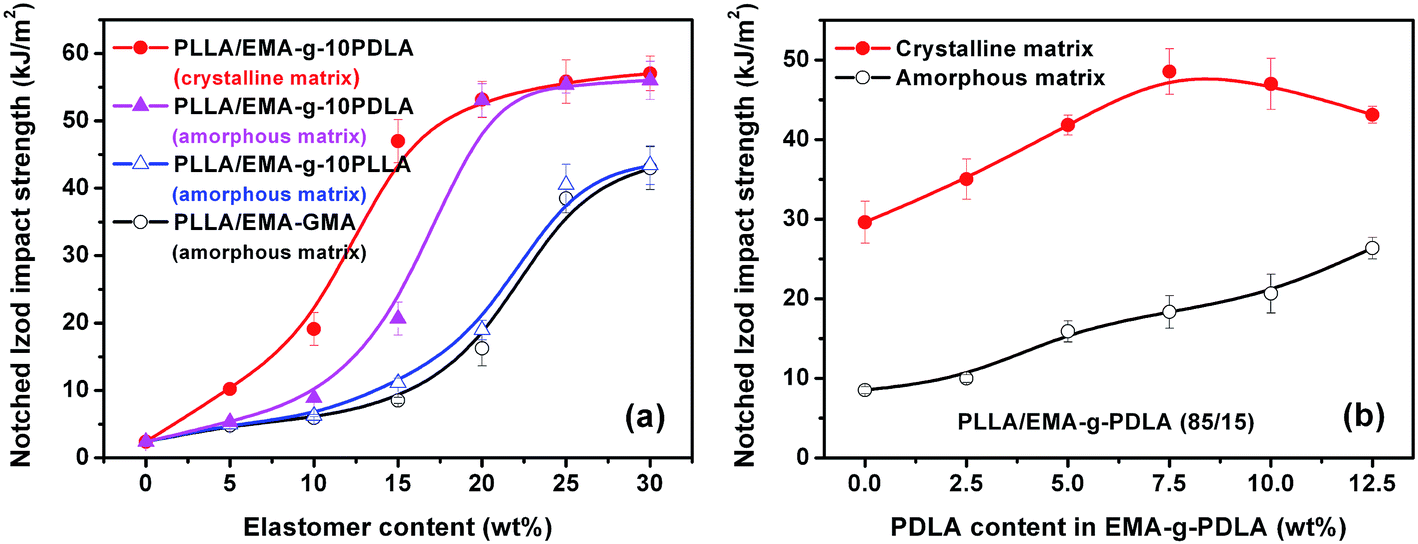
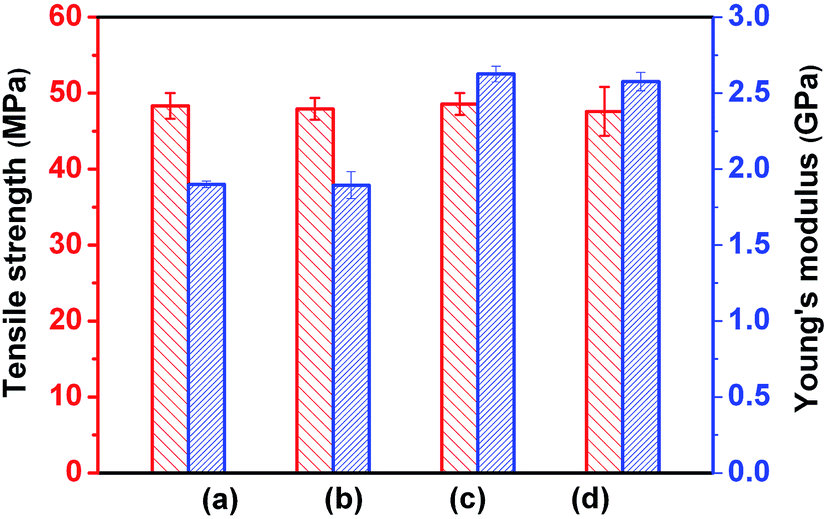
Fig. 7a shows the effects of interfacial strength and matrix crystallinity on impact toughness of the as-prepared blends. For PLLA/EMA–GMA blends with a poor interfacial adhesion and an amorphous matrix, impact toughness undergoes an obvious brittle–ductile (B–D) transition when EMA–GMA content increases from 20 to 25 wt%, but no noticeable toughening effect can be obtained with further increasing EMA–GMA content up to 30 wt%. The notched Izod impact toughness of PLLA/EMA-g-10PLLA blends only exhibits slight enhancement with respect to that of PLLA/EMA–GMA blends at the same elastomer contents because of the limited improvement in the interfacial adhesion. Very interestingly, with the substantial improvement in the interfacial adhesion, PLLA/EMA-g-10PDLA blends display not only a tremendous enhancement in toughening efficiency but also an apparent shift of B–D transition towards lower elastomer content (15–20 wt%). The PLLA/EMA-g-10PDLA (80/20) blend shows an impact strength of 53.0 kJ m−2, more than 3-fold over that of PLLA/EMA-g-10PLLA (80/20) blend. Furthermore, with the change of matrix crystalline state from almost amorphous to highly crystalline, toughening becomes much easier to achieve, evidenced by the notably decreased elastomer content (10–15 wt%) required for the B–D transition. The detailed relationship between matrix crystallinity and impact toughness of elastomer toughened PLLA blends has been discussed in our previous work.8 Herein, to highlight the role of interfacial strength in the toughening of PLLA blends with the two different matrix crystalline states, PLLA/EMA-g-PDLA (85/15) blends were chosen as model blends and the interfacial strength was tailored by varying the weight percentage of PDLA in the EMA-g-PDLA graft copolymers, which determines the amount of sc crystallites formed at the interfaces of the blends. The variations of impact strength as a function of PDLA content in EMA-g-PDLA is shown Fig. 7b. It can be clearly seen that the impact toughness of PLLA/EMA-g-PDLA (85/15) blends with an amorphous matrix increases linearly with increasing PDLA content up to 12.5 wt%, implying a linear relationship between interfacial strength and toughening efficiency. However, for the blends with a highly crystalline matrix, toughening efficiency starts to decline when PDLA content is higher than 7.5 wt%. It seems that an optimum interfacial adhesion is required for toughening highly crystalline PLLA. In particular, the impact strength of PLLA/EMA-g-10PDLA (85/15) blend with a strong interfacial adhesion and a highly crystalline matrix is as high as 47.0 kJ m−2, in sharp contrast with 29.6 kJ m−2 for PLLA/EMA–GMA (85/15) blends with a poor interfacial adhesion and a highly crystalline matrix as well as 20.6 kJ m−2 for PLLA/EMA-g-10PDLA (85/15) blend with a strong interfacial adhesion and an amorphous matrix. The contribution of matrix crystallinity or/and interfacial strength in the toughening can be evaluated by the difference between the impact strength of these blends and that of PLLA/EMA-g-GMA (85/15) blend with a poor interfacial adhesion and an amorphous matrix (8.5 kJ m−2). It is very interesting to find that the combined contribution (47.0–8.5) is higher than the sum of the individual contribution of matrix crystallinity (29.6–8.5) and that of interfacial strength (20.6–8.5), indicating that there is a pronounced synergistic effect between interfacial strength and matrix crystallinity in achieving super-tough PLLA/elastomer blends. However, although Young’ modulus of the toughened blends enhances greatly with the increase in the matrix crystallinity, both interfacial crystallization and matrix crystallinity have no apparent effect on tensile strength (Fig. 8).
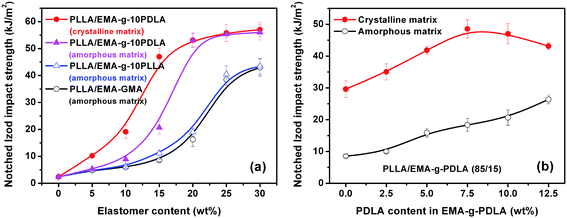 |
| | Fig. 7 Notched Izod impact strength of injection molded PLLA/EMA–GMA, PLLA/EMA-g-PLLA and PLLA/EMA-g-PDLA blends. | |
 |
| | Fig. 8 Tensile properties of injection molded blends: (a) PLLA/EMA–GMA (85/15) with a poor interfacial adhesion and an amorphous PLLA matrix, (b) PLLA/EMA-g-10PDLA (85/15) with a strong interfacial adhesion and an amorphous PLLA matrix, (c) PLLA/EMA–GMA (85/15) with a poor interfacial adhesion and a highly crystalline PLLA matrix, and (d) PLLA/EMA-g-10PDLA (85/15) with a strong interfacial adhesion and a highly crystalline PLLA matrix. | |
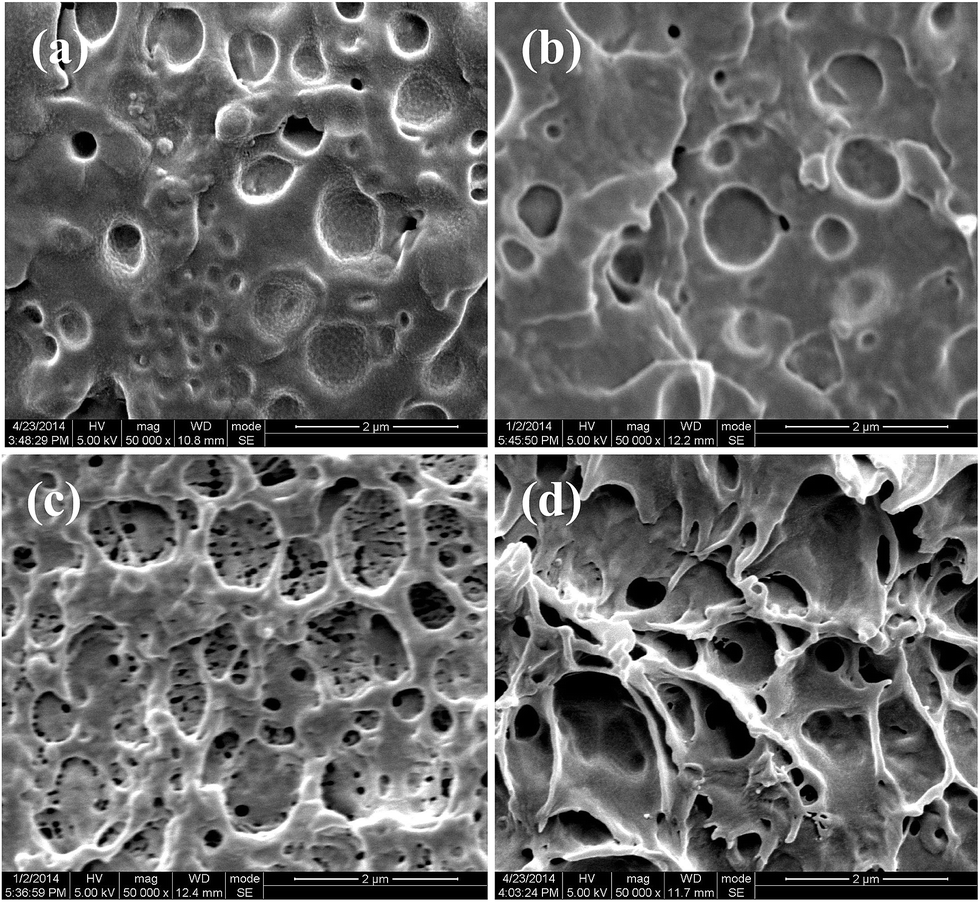
To understand the synergistic role of interfacial adhesion and matrix crystallinity in the toughening of PLLA/elastomer blends, the impact fractured surfaces of the blends with different interfacial strengths and matrix crystallinities were examined using SEM and the results are shown in Fig. 9. For PLLA/EMA–GMA (85/15) blend with an amorphous matrix (Fig. 9a), the fractured surface exhibits many cavities resulting from debonding of dispersed EMA–GMA particles from PLLA matrix due to the poor interfacial adhesion, but no obvious plastic deformation is observed in the PLLA matrix. With a slight improvement in the interfacial adhesion, the number of cavities decreases apparently and a certain degree of plastic deformation appears in the matrix of PLLA/EMA-g-10PLLA (85/15) blend (Fig. 9b). It seems that enhancing interfacial adhesion facilitates the initiation of matrix plastic deformation during impact fracture. As expected, extensive matrix plastic deformation can be clearly observed on the whole fractured surface of PLLA/EMA-g-10PDLA (85/15) blend with a strong interfacial adhesion (Fig. 9c). PLLA/EMA–GMA (85/15) blend with a poor interfacial adhesion and a highly crystalline matrix also displays a similar impact fracture characteristic (not shown here). More interestingly, the extent of plastic deformation is found to be further increased with the simultaneous enhancement in both the interfacial strength and matrix crystallinity (Fig. 9d), suggesting that the initiation of massive plastic deformation in the matrix becomes much easier to achieve in this case. In order to get more in-depth understanding of the synergistic toughening mechanism, SEM was further used to observe the microstructural differences underneath the impact fractured surfaces of the above blends with a special attention on the cavitation of elastomer particles. Depending on the interfacial strength, two types of cavitations could be induced by impact loading: cavitations inside the elastomer particles for the blends with a strong interface and cavitations resulting from interfacial adhesion for those with a poor interface.44,45 As shown in Fig. 10a and b, both PLLA/EMA–GMA (85/15) and PLLA/EMA-g-10PLLA (85/15) blends exhibit pervasive interfacial cavitations around the dispersed elastomer particles due to the insufficient interfacial adhesion, but no internal cavitations. However, a remarkably different situation can be observed in the PLLA/EMA-g-10PDLA (85/15) blend with a strong interfacial adhesion, where only large amounts of internal cavitations are visible (Fig. 10c). Despite cavitation itself does make a small contribution to the fracture energy, cavitated particles can release the triaxial stress state and then initiate massive plastic deformation of the matrix around them.46–48 Compared with the interfacial debonding, internal cavitations seem to be much more effective in initiating matrix plastic deformation and subsequently considerable energy dissipation because interfacial debonding may readily develop into premature cracks. Therefore, internal cavitation of elastomer particles followed by the serious plastic deformation of PLLA matrix around them is most likely the predominant synergistic toughening mechanism. As for PLLA/EMA-g-10PDLA (85/15) blend with a highly crystalline matrix, besides internal cavitations, another important characteristic is the presence of massive matrix plastic deformation since the plastic deformation of glassy amorphous matrix is much difficult to achieve in comparison with that of the crystalline one.8
 |
| | Fig. 9 SEM images of impact fractured surfaces of injection molded blends: (a) PLLA/EMA–GMA (85/15) with an amorphous PLLA matrix, (b) PLLA/EMA-g-10PLLA (85/15) with an amorphous PLLA matrix, (c) PLLA/EMA-g-10PDLA (85/15) with an amorphous PLLA matrix, and (d) PLLA/EMA-g-10PDLA (85/15) with a highly crystalline PLLA matrix. | |
 |
| | Fig. 10 SEM images of cross-sections underneath the impact fractured surfaces of injection molded blends: (a) PLLA/EMA–GMA (85/15) with an amorphous PLLA matrix, (b) PLLA/EMA-g-10PLLA (85/15) with an amorphous PLLA matrix, (c) PLLA/EMA-g-10PDLA (85/15) with an amorphous PLLA matrix, and (d) PLLA/EMA-g-10PDLA (85/15) with a highly crystalline PLLA matrix. | |
3.4 Heat resistance
PLLA/EMA-g-10PDLA (85/15) blend with an amorphous matrix and that with a highly crystalline one were prepared by injection molding and subsequent annealing in a hot mold of 130 °C for 0.5 min and 5 min, respectively. Amorphous and crystalline PLLA samples were also prepared with the same procedures. Note that, for the preparation of highly crystalline PLLA, 0.3 wt% highly active nucleating agent N,N′,N′′-tricyclohexyl-1,3,5-benzene-tricarboxylamide was introduced into PLLA before the injection molding. The heat resistance of the toughened PLLA blends was analyzed with DMA. Because interfacial strength has no influence on the heat resistance, only the curves of storage modulus vs. temperature obtained from the PLLA/EMA-g-10PDLA (85/15) blend with an amorphous matrix and that with a highly crystalline matrix are presented in Fig. 11. For comparison, the results of PLLA and PP were also plotted in the figure. Clearly, both amorphous PLLA and the blend with an amorphous matrix exhibit poor heat resistance, evidenced by the sharply decreased storage modulus above the glass transition temperature (Tg) of PLLA. The increase in the storage modulus at higher temperatures (90–100 °C) is caused by the cold crystallization of PLLA. Compared with the amorphous PLLA, the apparently deteriorated storage modulus at low temperatures (below Tg) and the slightly enhanced storage modulus in the temperature range of 65–110 °C observed in the toughened blend can be attributed to the locally plasticizing effect of flexible EMA-g-PDLA molecules on solid-like PLLA matrix at the interface and the restricting effect of rigid sc crystallites on the chain mobility of liquid-like PLLA matrix, respectively. However, for the highly crystalline PLLA and the blend with a highly crystalline PLLA matrix, the modulus decrease above Tg becomes much lesser. More importantly, the modulus is comparable to that of PP at temperatures higher than Tg, indicating a good heat resistance. The enhancement in heat resistance for PLLA is highly desirable in various industrial applications.
 |
| | Fig. 11 Temperature dependence of storage modulus of injection molded PLLA and PLLA/ethylene copolymer blends. PP specimens were prepared with the same method as PLLA/EMA-g-10PDLA (85/15) blend with a highly crystalline matrix. | |
4. Conclusions
In conclusion, we have demonstrated a feasible route to fabricate PLLA/elastomer blends with both super toughness and high heat resistance via constructing multifunctional sc crystallites at the interface. A series of EMA-g-PLLA and EMA-g-PDLA graft copolymers with various compositions were prepared by reactive blending PLLA or PDLA with excess EMA–GMA copolymer, and sc crystallites involving PDLA segments and PLLA chains are in situ formed at the interface during subsequent melt-blending of PLLA and the EMA-g-PDLA. The amount of sc crystallites can be adjusted by tuning the weight percentage of PDLA in the EMA-g-PDLA copolymer. The results show that the interface-localized sc crystallites serve as both effective interfacial adhesion enhancer and highly active nucleating agent to simultaneously enhance interfacial strength and matrix crystallinity. Compared with the PLLA/EMA–GMA and PLLA/EMA-g-PLLA blends, sc crystallites tailored interfacial strength and matrix crystallinity can impart the blends not only a significantly enhanced toughening efficiency but also a good heat resistance. We believe that this work provides a promising approach toward industrial-scale fabrication of high-performance PLA products.
Acknowledgements
This work was supported by National Natural Science Foundation of China (51121001, 21404075, 21034005) and China Postdoctoral Science Foundation (2013M530399, 2014T70869).
References
- M. M. Reddy, S. Vivekanandhan, M. Misra, S. K. Bhatia and A. K. Mohanty, Prog. Polym. Sci., 2013, 38, 1653–1689 CrossRef CAS PubMed.
- M. J. L. Tschan, E. Brule, P. Haquette and C. M. Thomas, Polym. Chem., 2012, 3, 836–851 RSC.
- L. T. Lim, R. Auras and M. Rubino, Prog. Polym. Sci., 2008, 33, 820–852 CrossRef CAS PubMed.
- K. M. Nampoothiri, N. R. Nair and R. P. John, Bioresour. Technol., 2010, 101, 8493–8501 CrossRef PubMed.
- R. M. Rasal, A. V. Janorkar and D. E. Hirt, Prog. Polym. Sci., 2010, 35, 338–356 CrossRef CAS PubMed.
- H. B. Li and M. A. Huneault, Polymer, 2007, 48, 6855–6866 CrossRef CAS PubMed.
- K. Hashima, S. Nishitsuji and T. Inoue, Polymer, 2010, 51, 3934–3939 CrossRef CAS PubMed.
- H. W. Bai, H. Xiu, J. Gao, H. Deng, Q. Zhang, M. B. Yang and Q. Fu, ACS Appl. Mater. Interfaces, 2012, 4, 897–905 CAS.
- H. W. Bai, C. M. Huang, H. Xiu, Y. Gao, Q. Zhang and Q. Fu, Polymer, 2013, 54, 5257–5266 CrossRef CAS PubMed.
- Y. K. Chen, D. S. Yuan and C. H. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 3811–3816 CAS.
- P. Ma, D. G. Hristova-Bogaerds, J. G. P. Goossens, A. B. Spoelstra, Y. Zhang and P. J. Lemstra, Eur. Polym. J., 2012, 48, 146–154 CrossRef CAS PubMed.
- K. Y. Zhang, A. K. Mohanty and M. Misra, ACS Appl. Mater. Interfaces, 2012, 4, 3091–3101 CAS.
- H. Xiu, C. M. Huang, H. W. Bai, J. Jiang, F. Chen, H. Deng, K. Wang, Q. Zhang and Q. Fu, Polymer, 2014, 55, 1593–1600 CrossRef CAS PubMed.
- Y. J. Li and H. Shimizu, Macromol. Biosci., 2007, 7, 921–928 CrossRef CAS PubMed.
- Y. S. He, J. B. Zeng, G. C. Liu, Q. T. Li and Y. Z. Wang, RSC Adv., 2014, 4, 12857–12866 RSC.
- G. C. Liu, Y. S. He, J. B. Zeng, Y. Xu and Y. Z. Wang, Polym. Chem., 2014, 5, 2530–2539 RSC.
- M. Harada, K. Iida, K. Okamoto, H. Hayashi and K. Hirano, Polym. Eng. Sci., 2008, 48, 1359–1368 CAS.
- H. Z. Liu and J. W. Zhang, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1051–1083 CrossRef CAS.
- T. Ge, G. S. Grest and M. O. Robbins, ACS Macro Lett., 2013, 2, 882–886 CrossRef CAS.
- W. M. Gramlich, M. L. Robertson and M. A. Hillmyer, Macromolecules, 2010, 43, 2313–2321 CrossRef CAS.
- H. Liu, W. Song, F. Chen, L. Guo and J. Zhang, Macromolecules, 2011, 44, 1513–1522 CrossRef CAS.
- K. S. Anderson and M. A. Hillmyer, Polymer, 2004, 45, 8809–8823 CrossRef CAS PubMed.
- K. H. Chang, M. L. Robertson and M. A. Hillmyer, ACS Appl. Mater. Interfaces, 2009, 1, 2390–2399 CAS.
- Y. Feng, G. Zhao, J. Yin and W. Jiang, Polym. Int., 2014, 63, 1263–1269 CrossRef CAS.
- H. Liu, F. Chen, B. Liu, G. Estep and J. Zhang, Macromolecules, 2010, 43, 6058–6066 CrossRef CAS.
- V. Ojijo, S. S. Ray and R. Sadiku, ACS Appl. Mater. Interfaces, 2013, 5, 4266–4276 CAS.
- W. Y. Dong, F. H. Jiang, L. P. Zhao, J. C. You, X. J. Cao and Y. J. Li, ACS Appl. Mater. Interfaces, 2012, 4, 3667–3675 CAS.
- H. Tsuji, Macromol. Biosci., 2005, 5, 569–597 CrossRef CAS PubMed.
- X. F. Wei, R. Y. Bao, Z. Q. Cao, W. Yang, B. H. Xie and M. B. Yang, Macromolecules, 2014, 47, 1439–1448 CrossRef CAS.
- H. Yamane, K. Sasai and M. Takano, J. Rheol., 2004, 48, 599–609 CrossRef CAS.
- X. S. Fan, M. A. Wang, D. Yuan and C. B. He, Langmuir, 2013, 29, 14307–14313 CrossRef CAS PubMed.
- H. Zhao, Y. Bian, Y. Li, Q. Dong, C. Han and L. Dong, J. Mater. Chem. A, 2014, 2, 8881–8892 CAS.
- N. Rahman, T. Kawai, G. Matsuba, K. Nishida, T. Kanaya, H. Watanabe, H. Okamoto, M. Kato, A. Usuki, M. Matsuda, K. Nakajima and N. Honma, Macromolecules, 2009, 42, 4739–4745 CrossRef CAS.
- H. W. Bai, H. L. Liu, D. Y. Bai, Q. Zhang, K. Wang, H. Deng, F. Chen and Q. Fu, Polym. Chem., 2014, 5, 5985–5993 RSC.
- R. Y. Bao, W. Yang, W. R. Jiang, Z. Y. Liu, B. H. Xie, M. B. Yang and Q. Fu, Polymer, 2012, 53, 5449–5454 CrossRef CAS PubMed.
- Y. L. Liu, J. R. Sun, X. C. Bian, L. D. Feng, S. Xiang, B. Sun, Z. M. Chen, G. Li and X. S. Chen, Polym. Degrad. Stab., 2013, 98, 844–852 CrossRef CAS PubMed.
- H. Tsuji and T. Tajima, Macromol. Mater. Eng., 2014, 299, 1089–1105 CAS.
- D. Garlotta, J. Polym. Environ., 2001, 9, 63–84 CrossRef CAS.
- Y. Sun and C. B. He, Macromolecules, 2013, 46, 9625–9633 CrossRef CAS.
- S. M. Ye, T. T. Lin, W. W. Tjiu, P. K. Wong and C. B. He, J. Appl. Polym. Sci., 2013, 128, 2541–2547 CrossRef CAS.
- G. Z. Wu, B. P. Li and J. D. Jiang, Polymer, 2010, 51, 2077–2083 CrossRef CAS PubMed.
- H. W. Bai, W. Y. Zhang, H. Deng, Q. Zhang and Q. Fu, Macromolecules, 2011, 44, 1233–1237 CrossRef CAS.
- J. Zhang, K. Tashiro, H. Tsuji and A. J. Domb, Macromolecules, 2008, 41, 1352–1357 CrossRef CAS.
- D. Dompas, G. Groeninckx, M. Isogawa, T. Hasegawa and M. Kadokura, Polymer, 1995, 36, 437–441 CrossRef CAS.
- G. M. Kim and G. H. Michler, Polymer, 1998, 39, 5689–5697 CrossRef CAS.
- A. Galeski and Z. Bartczak, Macromol. Symp., 2003, 194, 47–62 CrossRef CAS.
- Y. Kayano, H. Keskkula and D. R. Paul, Polymer, 1998, 39, 2835–2845 CrossRef CAS.
- G. M. Kim and G. H. Michler, Polymer, 1998, 39, 5699–5703 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.