DOI:
10.1039/C4RA04976D
(Paper)
RSC Adv., 2014,
4, 44244-44252
Role of carbon coating in improving electrochemical performance of Li-rich Li(Li0.2Mn0.54Ni0.13Co0.13)O2 cathode†
Received
26th May 2014
, Accepted 1st September 2014
First published on 1st September 2014
Abstract
Li-rich Li(Li0.2Mn0.54Ni0.13Co0.13)O2 cathode coated with carbon layer has been prepared by a hydrothermal approach followed by a post annealing process. The cathode after surface modification exhibits both enhanced cyclability and improved rate capability compared with the pristine one without coating. The carbon coating process causes a phase transformation from Li2MnO3-like domain to cubic-spinel domain with Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m symmetry at surface regions of particles. As a consequence, the valence state of Mn on the surface accordingly varies. Such transformed surface spinels, as well as the wrapped carbon layers as a result of this coating strategy are believed to be responsible for the enhanced electrochemical performance.
m symmetry at surface regions of particles. As a consequence, the valence state of Mn on the surface accordingly varies. Such transformed surface spinels, as well as the wrapped carbon layers as a result of this coating strategy are believed to be responsible for the enhanced electrochemical performance.
Introduction
Lithium-rich manganese-based layer-structured cathodes, which could be recognized as either Li(Li1−x−yMnxMy)O2 or xLi2MnO3(1 − x)LiMO2 (M refers to commonly-used transition metals such as Ni and Co), have attracted extensive interests in recent years.1–5 The unique feature of this type of material is its high reversible capacities close to 270 mA h g−1,4 whereas the traditional layered Li1−xCoO2 is capable of maintaining structural stability only when a half of Li is deintercalated (x < 0.5). The integrated Li2MnO3 phase can be electrochemically activated by Li extraction accompanied with oxygen release when the potential difference between cathode and anode is higher than 4.5 V vs. Li/Li+ in a first charge process.6–8 In the following discharge process, the resultant defective MnO2 structure, which has both Li and O vacancies after first charge, can adopt the reinsertion of Li where Mn is reduced from tetravalent to trivalent.1,9 The ability of one or more Li per unit extraction/reinsertion without obvious collapse from structural instability compared to Li1−xCoO2 is the key for achieving high reversible capacity.
Although the high capacity is very attractive, this family of cathode materials still suffers from several drawbacks: poor cyclability and rate capability and a reducing output voltage during cycling. These drawbacks are believed to be partially associated with the sensitivity of particle surfaces according to recent discoveries on these materials. Gu et al.10 found that in the Li(Li0.2Ni0.2Mn0.6)O2 system, Ni prefers to segregate at the surface facets of synthesized particles, and the particle facets tend to terminate at transition metal (TM) layers, while the Li fast diffusion channels in these layers are almost perpendicular to the channels inside. In fact, both of the phenomena nearby surface regions could induce a high diffusion barrier against Li transportation, leading to poor rate capability. Concerning cyclability, the dissolution of TM ions in high potential ranges from layered structure is always recognized as a main reason for the capacity fade, as suggested by Amine11 and Song.12 Another surface initiated effect related to the capacity fade is a phase transformation caused by the migration of TM ions, which is driven by thermodynamic instability of Li and O vacancies.5,8,13 Because of the above reasons, such particle surfaces easily experience internal stress leading to local non-crystallization and further micro-cracks on surfaces during cycling.14
To address the issues associated to the sensitivity of the particles' surfaces, various methodologies have been used for surface modifications through various coatings of Li–Ni–PO4,15 AlPO4,16 Al17 and RuO218 as the conductive surface layers to enhance the rate capability. On the other hand, Al2O3,19 ZnO coating layers20 and surface nitridation21 were also applied to improve the cycle performance. Besides these traditional protection or conductivity effects that resulted from coating layers, AlF322 and MnO223 were also found to contribute to surface spinel formation after corresponding surface modifications. Such surface spinels initiating from original Li2MnO3-like domains were reasonably believed to be able to improve both cyclability and rate performance. As a matter of fact, similar improvements because of surface spinels were also observed in (NH4)2SO4,24 graphene25 and carbon black26 treated Li-rich layered systems. Nevertheless, due to the reduction concerns of carbon coating, only a few works are reported on the effects of carbon coating on such Li-rich layered systems,27 although carbon coating is always regarded as the most effective method for the enhancement in olivine systems. Therefore, in this work we intend to develop a facile way to achieve carbon coating layers on Li(Li0.2Mn0.54Ni0.13Co0.13)O2 particles to improve electrochemical performance without largely changing the chemical composition of the layered compounds.
Experimental section
Preparation of carbon coated Li(Li0.2Mn0.54Ni0.13Co0.13)O2
The pristine Li-rich compound Li(Li0.2Mn0.54Ni0.13Co0.13)O2 (LLNCM) was synthesized by a sol–gel method assisted with a spray-drying machine (YC-015, Shanghai Politech Instrument & Equipment Co. Ltd). The detailed procedures can be found elsewhere,25 whereas the carbon precursor was prepared based on Fang's work.28,29 In a typical synthesis, 0.6 g phenol, 15 ml NaOH aqueous solution (0.1 M) and 2.8 ml formalin solution were mixed and stirred at 70 °C for 30 min. Then, 15 ml deionized water containing triblock Pluronic F127 (1.28 g) was added. 2 h later, 50 ml deionized water was added, followed by reaction for 20 h. The mixture was diluted 4 times before obtaining the pink solution. 300 mg LLNCM and 25 ml pink solution were mixed together by stirring for 1 h. To obtain the carbon coated LLNCM, the mixture was transferred into an autoclave, and then heated at 130 °C for 3 h or 15 h, which are assigned to C-3 or C-15. After that, the obtained powders were post annealed at 350 °C for 2 h in air with both heating and cooling rates at 5 °C min−1. The corresponding products after post annealing were respectively named as C-3-H and C-15-H.
Structure and morphology characterizations
Crystal structures of all the powders were characterized by a powder X-ray diffraction machine (Shimadzu XRD-6000 Cu-Kα radiation, λ = 1.5418 Å). Diffraction data were collected at a scan rate of 0.5° min−1. Powder morphologies were investigated using a scanning electron microscope (Fe-SEM) (S-4300 Shimadzu, 15 kV). A high-resolution transmission electron microscope (HRTEM) (JEOL 2200FS, operating at 200 kV) was employed to reveal the detailed lattice information before and after the annealing process. Another scanning transmission electron microscope (JEOL JEM-2010F, operating at 200 kV) coupled with electron dispersive X-ray spectroscopy (EDX) mapping was applied for elemental identification after the coating process. The variations in the valence states of transition metals before and after surface treatment were investigated by X-ray photoelectron spectroscopy (XPS) (Kratos AXIS Ultra DLD, Kratos Analytical Ltd) with a monochromatized Al Kα X-ray source (1486.6 eV photons) at a constant dwelling time of 100 ms. The binding energy was ranged from −5 to 1100 eV. Thermogravimetric analysis (TGA) was applied on C-3 and C-15 samples to reveal the carbon content in air at a heating rate of 5 °C min−1 from room temperature to 800 °C using a DTG-60H Shimadzu.
Electrochemical characterizations
Electrochemical behaviors of the pristine and modified samples were tested using CR-2016 type coin cells. To prepare the coin cells, the electrode slurries were prepared by mixing active cathode materials, carbon black (Super P) and polyvinylidene fluoride (PVDF) in the weight ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 in a n-methyl-2-pyrrolidone (NMP) solution and stirred overnight. The slurries were coated on an aluminum foil followed by drying overnight at 120 °C. The dried electrodes were punched into pieces with 10 mm diameters with a typical loading of 2–3 mg. Half cells were assembled in an argon-filled glove box. Li foil was used as the count and reference electrode. Two pieces of membrane (Celgard 2500) were used as the adapted separator, and the electrolyte in the form of 1 M LiPF6 in EC:DEC = 1:1 organic solutions was used as the electrolyte. All cycling tests were performed in the potential window of 2.0–4.8 V vs. Li/Li+ as 1 C refers to 250 mA g−1. Plots of dQ/dV vs. voltage were obtained based on the testing data of charge/discharge cycling.
:10:10 in a n-methyl-2-pyrrolidone (NMP) solution and stirred overnight. The slurries were coated on an aluminum foil followed by drying overnight at 120 °C. The dried electrodes were punched into pieces with 10 mm diameters with a typical loading of 2–3 mg. Half cells were assembled in an argon-filled glove box. Li foil was used as the count and reference electrode. Two pieces of membrane (Celgard 2500) were used as the adapted separator, and the electrolyte in the form of 1 M LiPF6 in EC:DEC = 1:1 organic solutions was used as the electrolyte. All cycling tests were performed in the potential window of 2.0–4.8 V vs. Li/Li+ as 1 C refers to 250 mA g−1. Plots of dQ/dV vs. voltage were obtained based on the testing data of charge/discharge cycling.
Results and discussion
Fig. 1 compares X-ray diffraction spectra of the pristine and the surface modified Li(Li0.2Mn0.54Ni0.13Co0.13)O2 powders. All the strong peaks in the patterns could be easily indexed to a rhombohedral layered phase (subscript of R) with an Rm space group. In addition to these signal reflection peaks, several weak peaks in the range of 20–23° are consistent with a super-ordering structure among Li and transition metals (Mn, Ni and Co) in the transition metal layers of Li2MnO3-like monoclinic phase (subscript of M).4 Apparently, no impurity phase could be observed either in carbon coated samples (C-3 and C-15) or post annealed ones (C-3-H and C-15-H). However, the intensity of the super-ordering peaks (020)M and (110)M obviously decreases after carbon coating and post annealing, suggesting that the modification process may affect the local ordering of Li2MnO3-like phases in crystal lattice, in addition to the influence of carbon coating itself. Furthermore, the (003)R/(001)M and (101)R/(−201)M peaks become broader and shift to higher degrees in the modified samples compared with the peaks in the pristine sample. A fitting of C-3-H and C-15-H spectra using Rietveld refinement was successful based on an assumption of the Li2MnO3 and LiNi1/3Co1/3Mn1/3O2 composite structure, implying only the lattice shrinks in c – direction after modification rather than new phase formation, which is consistent with the trend observed from acid-treated Li-rich layered cathodes.30 The hump related to the spinel-like ordering phase located at lower than 36.9°, labeled by a diamond, could be identified in selective XRD patterns in Fig. 1.22 Based on the above observations, it is likely that the surface coating of carbon leads to a phase transformation from the layer-ordering phase to the spinel-like phase.
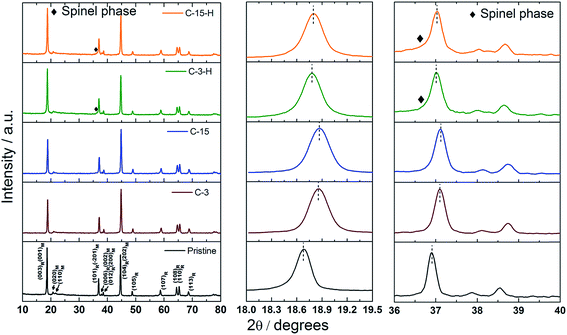 |
| | Fig. 1 Powder XRD patterns of the pristine carbon coated (C-3, C-15) and post annealed (C-3-H, C-15-H) LLNCM samples. | |
Fig. 2 compares the morphologies of the pristine carbon coated (C-3, C-15) and post annealed (C-3-H, C-15-H) particle. The distribution of particle size of about 100 to 200 nm with facets could be observed in the pristine sample, while the particles tend to aggregate and grow in size without a clear facet after coating process, as seen in both C-3 and C-15 samples. The increase in particle size is caused by a hydrothermal reaction and might also be associated with carbon coatings which interconnect small crystals. A thin carbon layer about 3 nm was evenly coated on the particles from the TEM observation (Fig. 3), whereas parts of carbon were accumulated to form spheres. Elemental mappings of the C-3 particle reveal uniform distribution of C, Mn, Ni and Co (Fig. 4) after carbon coating. Similar but not equal, with the increase in duration time of the reaction from 3 to 15 h, more carbon layers in the C-15 sample tend to grow into carbon spheres, (Fig. 2) which was confirmed in the bright field TEM image (Fig. 3). The more transparent spheres around a LLNCM particle represent the carbon spheres. According to TGA results (Fig. S1†), the carbon contents in C-3 and C-15 are approximately 6.3 wt% and 6.6 wt%, respectively. A sudden loss in weight from 150 to 300 °C was caused by loss of functional groups on these synthesized carbons. After a post annealing process, the particle morphologies of C-3-H and C-15-H samples barely change, but the average particle sizes are apparently reduced compared to those before annealing, as observed in Fig. 2. Moreover, the corresponding TEM images of both C-3-H and C-15-H samples (Fig. S2†) exhibit non-uniform transparency in the form of nano-domains compared to dissimilar characters in transparency of the pristine sample (Fig. S2†). It may indicate a local disorder or rearrangement of the original layered structure as a result of surface treatment, which is highly related to local spinel-like transformation.
 |
| | Fig. 2 SEM images of the pristine, carbon coated (C-3, C-15) and post annealed (C-3-H, C-15-H) LLNCM particles. Arrows inside indicate the carbon spheres in the C-15 sample. | |
 |
| | Fig. 3 Bright field TEM images of C-3 and C-15 samples. Inset of C-3 reveals nano carbon layers of 3 nm thickness on LLNCM particles. C-15 exhibits carbon spheres around a LLNCM particle instead of coating layers. | |
 |
| | Fig. 4 Carbon, nickel, cobalt, and manganese elemental mapping using a STEM on the C-3 sample confirms a uniform distribution of all the types of elements after the coating process. Insets show carbon coating layer and EDX spectrum. | |
HRTEM characterization of the C-15-H sample is shown in Fig. 5. Fig. 5(a) reveals the general appearance of the particles with electron diffraction pattern (EDP) and diffraction index in Fig. 5(b) and (c). Two sets of diffraction can be indexed, layered triclinic structure and a spinel cubic structure as confirmed. Fig. 5(d) shows HRTEM on the surface of a particle. Further FFT images for sub areas as shown in Fig. 5(e) and (f) reveal the spatial relationship between these two phases. Green dash lines in Fig. 5(d) highlighting the layered domains tend to be enclosed by spinel domains, which are terminated on the surface of the particle. The zone axis with respect to two phases are [1−11]layered and [110]spinel.
 |
| | Fig. 5 TEM identification of the C-15-H sample: (a) low magnification TEM bright field image of the modified LLNCM particles, (b) electron diffraction pattern (EDP) corresponding to (a) and (c) index of the electron diffraction rings of (b), revealing two types of phases, i.e., a layered triclinic structure and spinel cubic structure, (d) HRTEM image showing surface regions of the particle composed of mixed spinel and layered structures inside, (e) and (f) fast Fourier transforms (FFT) to sub areas in (d) with indexes shown in (h) and (i), (g) FFT to (d) with two-phase index shown in (j). The zone axis with respect to the two phases are [1−11]layered and [110]spinel. Green dash lines in (d) highlight the layered domain, which is enclosed by the spinel domain. | |
The transformation on the surface regions is believed to be induced during the hydrothermal processing where the presence of carbon coating increases the acidity of the hydrothermal solution resulting in ions exchanged between H+ and Li+ at high pressure. The final annealing may assist the phase transformation and aggregation of carbon. On the other hand, the further reduced carbon layers as confirmed by both TGA results (Fig. S1†) and the TEM image (Fig. S3†) could be partially sustained on particle surfaces after heat treatment. Based on the above observations and the synthesis procedure, the mechanisms of carbon coating and transformation on the particles' surfaces in C-3 (C-15) and C-3-H (C-15-H) are schematically illustrated in Fig. 6.
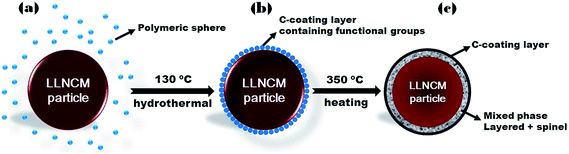 |
| | Fig. 6 Schematic illustrations of the formation process: (a) mixture of pristine particles and the polymeric solution before the hydrothermal process, (b) carbon coating with functional groups during hydrothermal processing, and (c) formation of carbon coating and surface transformation. | |
XPS was applied to investigate the solid-state chemistry of particle surfaces of all the pristine and modified C-3, C-3-H samples. Spectra of C 1s, O 1s, Li 1s, Mn 2p, Ni, 2p and Co 2p orbital are shown in Fig. 7. Curve fittings were performed based on the binding energy of C 1s (C–C sp2 bond) at 284.5 eV. As shown in C 1s spectra, several peaks corresponding to 284.5, 285.5 and 287.6 eV could be recognized as C–C, C–OH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively.31,32 The higher intensities of both C–OH and CO peaks in the C-3 sample indicate the presence of functional oxygen groups on the surface carbon layers rather than highly-carbonized layers after the coating process. However, these functional groups partially decompose after the post annealing process judged from the drastic decrease in the intensities of the C-3-H sample. The similar trend is also observed in the O 1s spectra. A larger amount of functional oxygen groups on the particle surface for the C-3 sample compared to the amount of the pristine sample was able to function as the protection layer on particles to mitigate the corrosion effects of the electrolyte, and thus enhances the cyclability. Moreover, these functional oxygen groups could also decrease the surface electrical conductivity of particles. Consequently, these characteristics of the functional groups significantly affect the electrochemical performance and will be shown later. On the other hand, the surface modification has a fewer effects on the variations of the valence states of both Ni and Co, according to the comparable peaks fitted in Ni 2p and Co 2p spectra. Ni2+ (854.6 eV) and Co3+ (780.1 eV) dominate in all the samples with trace amounts of Ni3+ (855.9 eV) and Co4+ (781.1 eV) at the synthesized state. It is interesting to note that the valence state of Mn substantially varies as a result of carbon coating and post annealing, as seen from both the Mn 2p and Mn 3p (close to Li 1s) spectra. Mn4+ with respect to 642.9 eV peak is the majority constituent in the pristine sample, while the majority constituent switches to Mn3+ in both the C-3 and C-3-H samples. The decrease in average valence state of Mn may imply formation of spinel, or at least a change in electronic configuration related to the Mn at surface regions. As also suggested in HRTEM results in Fig. 5, it is likely to deduce that the transformed spinel is the LiMn2O4-like cubic spinel, despite that the accurate chemical composition in the local structure is hardly detected. However, the statistical feature of the chemical compositions on surface regions of the particle could be confirmed by XPS.
O, respectively.31,32 The higher intensities of both C–OH and CO peaks in the C-3 sample indicate the presence of functional oxygen groups on the surface carbon layers rather than highly-carbonized layers after the coating process. However, these functional groups partially decompose after the post annealing process judged from the drastic decrease in the intensities of the C-3-H sample. The similar trend is also observed in the O 1s spectra. A larger amount of functional oxygen groups on the particle surface for the C-3 sample compared to the amount of the pristine sample was able to function as the protection layer on particles to mitigate the corrosion effects of the electrolyte, and thus enhances the cyclability. Moreover, these functional oxygen groups could also decrease the surface electrical conductivity of particles. Consequently, these characteristics of the functional groups significantly affect the electrochemical performance and will be shown later. On the other hand, the surface modification has a fewer effects on the variations of the valence states of both Ni and Co, according to the comparable peaks fitted in Ni 2p and Co 2p spectra. Ni2+ (854.6 eV) and Co3+ (780.1 eV) dominate in all the samples with trace amounts of Ni3+ (855.9 eV) and Co4+ (781.1 eV) at the synthesized state. It is interesting to note that the valence state of Mn substantially varies as a result of carbon coating and post annealing, as seen from both the Mn 2p and Mn 3p (close to Li 1s) spectra. Mn4+ with respect to 642.9 eV peak is the majority constituent in the pristine sample, while the majority constituent switches to Mn3+ in both the C-3 and C-3-H samples. The decrease in average valence state of Mn may imply formation of spinel, or at least a change in electronic configuration related to the Mn at surface regions. As also suggested in HRTEM results in Fig. 5, it is likely to deduce that the transformed spinel is the LiMn2O4-like cubic spinel, despite that the accurate chemical composition in the local structure is hardly detected. However, the statistical feature of the chemical compositions on surface regions of the particle could be confirmed by XPS.
 |
| | Fig. 7 XPS spectra for pristine, C-3 and C-3-H samples as indicated by A, B and C, respectively. Black solid, blue solid and red dash lines represent original graphs, fitted peaks and fitted graphs, respectively. | |
As shown in Table 1, two important observations are summarized: (1) compositional ratio of Li relative to a sum of TM ions reduces from 1.04 of the pristine sample to 0.21 of C-3 and to 0.73 of C-3-H and (2) the relative contents of Ni, Co and Mn also varies. It is important to note that although the above data indicate the spinel formation in the C-3 sample, a subsequent heat treatment is still necessary to complete such transformations according to distinct electrochemical behaviors in the following section.
Table 1 Surface chemical compositions of the pristine, C-3 and C-3-H samples based on the XPS results
| |
Li |
Transition metals |
O |
| Mn |
Ni |
Co |
Total |
| Pristine |
1.02 |
0.54 |
0.22 |
0.22 |
0.98 |
2.8 |
| C-3 |
0.35 |
0.91 |
0.45 |
0.30 |
1.65 |
5.9 |
| C-3-H |
0.84 |
0.84 |
0.18 |
0.13 |
1.15 |
2.6 |
Fig. 8 compares the first cycle charge/discharge curves at a current density of 50 mA g−1 and the respective dQ/dV plots of all the samples. The charge/discharge capacities, as well as the coulombic efficiencies are tabulated in Table 2. The pristine LLNCM delivers a charge capacity of 300 mA h g−1 and discharge capacity of 253 mA h g−1 with 84% efficiency, while these values drastically reduce for C-3 and C-15 samples. It is noted that the coated layers of carbon or spheres are of poor electronic conductivity because of their poor carbonization feature, as supported by the presence of functional oxygen groups in XPS C 1s and O 1s results. However, the charge capacities decrease, whereas both discharge capacities and coulombic efficiencies increase for C-3-H and C-15-H samples, i.e. 263 mA h g−1 with 96.7% and 264 mA h g−1 with 96.4%, respectively. These improvements in discharge capacity and initial coulombic efficiency after post annealing could be ascribed to two reasons: the formation of spinel by consuming the amount of the Li2MnO3-like component, which is confirmed by the reduced 4.5 V charge plateau, but elongated the 2.7 V discharge plateau and the better carbonized layers or spheres as a result of post annealing as also supported by XPS results in Fig. 7. Although the pristine LLNCM exhibits a typical deintercalation/intercalation profile of Li rich layered structures,2 the typical electrochemical profile apparently changed after modification (samples C-3, C-15, C-3-H and C-15-H). The dQ/dV plot of the pristine sample first shows an oxidation of Ni2+ and Co3+ to higher states related to the 4.1 V peak (O1), followed by a redox-like peak at about 4.6 V, which is commonly recognized because of the oxygen release. Both of the two peaks clearly shift to higher potentials after coating (C-3, C-15) because of the polarization effects from coating layers, but they shift back to the comparable positions of the pristine sample after the further heating process (C-3-H, C-15-H). In a typical discharge process of the pristine sample, three reduction peaks noted as R1, R2 and R3 were observed. The origin of R1 is still unclear at the present,33 while R2 and R3 are believed to be associated with Ni4+/Ni2+ in addition to Co4+/Co3+ redox couples in a layered structure and Mn4+/Mn3+ redox in a layered structure, respectively. The only peaks appearing in C-3-H and C-15-H samples are R4 and R5 and are attributed to the Mn4+/Mn3+ redox in a layered-spinel-mixed complex structure and in a surface-formed spinel structure.34 It is worth pointing out that (1) R3 contributions in C-3 and C-15 samples significantly reduce compared to the pristine sample, suggesting a less activation of the Li2MnO3-like component, which is responsible for reduced discharge capacities, and (2) the existence of only R4 and R5 peaks confirms phase transformation after the heating process, which not only took place at surface regions, but also possibly initiated in bulk regions because of the significant contributions to the capacity. Although XRD and XPS results imply the possible spinel formation in the coated samples C-3 and C-15, the disappearance of R4 and R5 peaks still indicates that the leaching of Li incorporated with the rearrangement of electronic configuration related to Mn occurred, but the local structure was sustained in the layered form before the subsequent heating process.
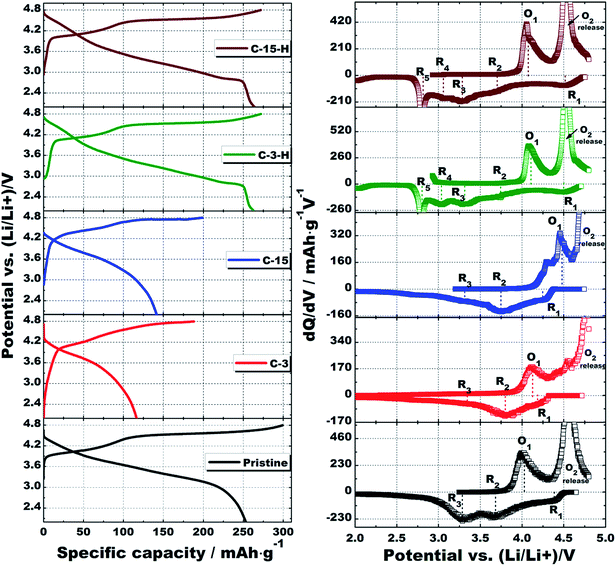 |
| | Fig. 8 First charge/discharge curves with corresponding dQ/dV plots of the pristine, C-3, C-15, C-3-H and C-15-H cathodes tested at a current density of 50 mA g−1, 2.0–4.8 V at room temperature. | |
Table 2 First charge/discharge capacities and corresponding coulombic efficiency of pristine, C-3, C-15, C-3-H and C-15-H cathodes cycled in a voltage window of 2.0–4.8 V at 50 mA g−1
| |
Charge capacity (mA h g−1) |
Discharge capacity (mA h g−1) |
First coulombic efficiency (%) |
| Pristine |
300 |
253 |
84.3 |
| C-3 |
188 |
116 |
61.7 |
| C-15 |
199 |
142 |
71.4 |
| C-3-H |
272 |
263 |
96.7 |
| C-15-H |
273 |
264 |
96.4 |
Fig. 9 shows the cycle performance and rate capability of all the samples. At 0.2 C (Fig. 9(a)), the pristine sample is able to provide 112 mA h g−1 discharge capacity after 100 cycles with 44% capacity retention, whereas a continuous increase in the capacities for both C-3 and C-15 samples were observed in the first 8 charge/discharge cycles, and 92% for C-3 and 91% for C-15 can be obtained. The increase in capacity during charge/discharge upon the first 8 cycles is because of the continuous activation of the Li2MnO3 component. As discussed in the previous XPS section, the apparent functional oxygen groups in surface carbon layers may slow down the kinetics of the related activation of Li2MnO3. However, it could be further electrochemically reduced to more conductive carbon layers by the gradual formation of Li2O during Li intercalation and deintercalation.35,36 The further reduced carbon layers not only facilitate the activation of Li2MnO3, but also act as protection layers to prevent thick formation of SEI layers and dissolution of transition metals because of HF attacking.37–39 In fact, after the initial activation, C-3 and C-15 materials are capable of delivering capacities as high as 42 and 54 mA h g−1, respectively, at a high rate of 20 C compared with only 13 mA h g−1 for the pristine sample as shown in Fig. 9(b). On the other hand, the post annealed samples C-3-H and C-15-H possess not only similar initial capacities compared to the pristine sample, but also the enhanced ability of capacity retention, i.e. 70% and 68%, respectively. It suggests that the sustained carbon layers at particle surfaces after post annealing are still able to function as the protection layers. Furthermore, more than 100 mA h g−1 capacity could be achieved at 10 C from both the samples, which might be attributed to increased extrinsic conductivity of carbon coating, as well as the transformed surface spinels providing fast diffusion channels of Li.40,41 Moreover, it is very important to note that the surface spinels may not be associated with the improved cycleability. On the contrary, they may even negatively contribute to the cyclability since spinel is an unstable phase when exposed to electrolyte (Mn2+ dissolution). To better understand the structural evolution process upon cycling, Fig. S4† compares the charge/discharge and dQ/dV curves at various cycling stages of all the samples. As widely accepted, the family of Li-rich layered cathodes is inevitably involved with a phase transformation to the spinel ordered structure during deintercaltion/intercalation of Li.12,13,42 The pristine material underwent phase transformation in the first 100 cycles. All the other cathodes after surface modification exhibit similar behaviors, indicating that neither surface coating of carbon nor subsequent heat treatment could contribute to the suppression of this structural evolution process, as also supported by arrows in dQ/dV plots.
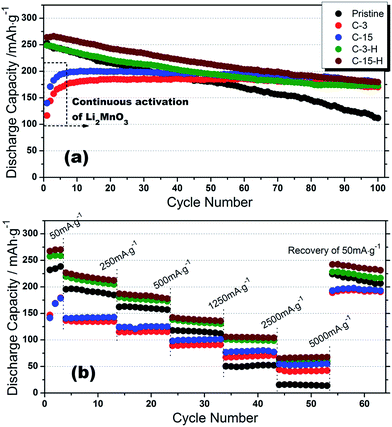 |
| | Fig. 9 Cycle performance and rate capability of pristine, C-3, C-15, C-3-H and C-15-H cathodes at different testing conditions: (a) 0.2 C, (b) at incremental C rates of 0.2, 1, 2, 5, 10 and 20 C (same current densities for charge/discharge), where 1 C stands for 250 mA h g−1. | |
Conclusions
A carbon coating on Li(Li0.2Mn0.54Ni0.13Co0.13)O2 has been realized by a hydrothermal approach. The carbon coated Li(Li0.2Mn0.54Ni0.13Co0.13)O2 cathode material exhibits enhanced cyclability with discharge capacity of 182 mA h g−1 corresponding to 92% capacity retention after 100 cycles at 0.2 C. After a post annealing of the carbon coated material, an increased conductivity of the carbon layers leads to the enhanced rate capability, as well as an acceptable cycle performance, i.e. 100 mA h g−1 at 10 C rate and 70% capacity retention, compared to 50 mA h g−1 and 44% for the pristine material. XRD, HRTEM, XPS and electrochemical testing confirm the formation of spinel domains initiated from the Li2MnO3-like component, accompanied by the variation of the valence state of Mn after this surface modification. The spinel benefits from the fast diffusion of Li+ at surface regions, and thus the rate capability. However, it may negatively affect the cyclability. Most importantly, this work provides convincing evidence to support the prospective that the surface characters of the Li-rich layered cathodes are sensitive to the impact of the electrochemical performance not only for cyclability, but also rate capability.
Acknowledgements
This work was supported by National Research Foundation, Singapore. The authors are grateful to TIMCAL Ltd. for providing Super PTM conductive carbon black. The authors also acknowledge grant R265-000-426-731/112, Dr Hongwei Liu for assistance of HRTEM characterizations.
References
- Z. H. Lu, D. D. MacNeil and J. R. Dahn, Electrochem. Solid-State Lett., 2001, 4, A191–A194 CrossRef CAS PubMed.
- B. H. Song, M. O. Lai and L. Lu, Electrochim. Acta, 2012, 80, 187–195 CrossRef CAS PubMed.
- M. M. Thackeray, C. S. Johnson, J. T. Vaughey, N. Li and S. A. Hackney, J. Mater. Chem., 2005, 15, 2257–2267 RSC.
- M. M. Thackeray, S. H. Kang, C. S. Johnson, J. T. Vaughey, R. Benedek and S. A. Hackney, J. Mater. Chem., 2007, 17, 3112–3125 RSC.
- B. Xu, C. R. Fell, M. F. Chi and Y. S. Meng, Energy Environ. Sci., 2011, 4, 2223–2233 CAS.
- A. R. Armstrong, M. Holzapfel, P. Novak, C. S. Johnson, S. H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694–8698 CrossRef CAS PubMed.
- J. Hong, H. D. Lim, M. Lee, S. W. Kim, H. Kim, S. T. Oh, G. C. Chung and K. Kang, Chem. Mater., 2012, 24, 2692–2697 CrossRef CAS.
- N. Yabuuchi, K. Yoshii, S. T. Myung, I. Nakai and S. Komaba, J. Am. Chem. Soc., 2011, 133, 4404–4419 CrossRef CAS PubMed.
- A. Ito, Y. Sato, T. Sanada, M. Hatano, H. Horie and Y. Ohsawa, J. Power Sources, 2011, 196, 6828–6834 CrossRef CAS PubMed.
- M. Gu, I. Belharouak, A. Genc, Z. G. Wang, D. P. Wang, K. Amine, F. Gao, G. W. Zhou, S. Thevuthasan, D. R. Baer, J. G. Zhang, N. D. Browning, J. Liu and C. M. Wang, Nano Lett., 2012, 12, 5186–5191 CrossRef CAS PubMed.
- K. Amine, Z. H. Chen, Z. Zhang, J. Liu, W. Q. Lu, Y. Qin, J. Lu, L. Curtis and Y. K. Sun, J. Mater. Chem., 2011, 21, 17754–17759 RSC.
- B. H. Song, Z. W. Liu, M. O. Lai and L. Lu, Phys. Chem. Chem. Phys., 2012, 14, 12875–12883 RSC.
- M. Gu, I. Belharouak, J. M. Zheng, H. M. Wu, J. Xiao, A. Genc, K. Amine, S. Thevuthasan, D. R. Baer, J. G. Zhang, N. D. Browning, J. Liu and C. M. Wang, ACS Nano, 2013, 7, 760–767 CrossRef CAS PubMed.
- A. Ito, D. C. Li, Y. Sato, M. Arao, M. Watanabe, M. Hatano, H. Horie and Y. Ohsawa, J. Power Sources, 2010, 195, 567–573 CrossRef CAS PubMed.
- S. H. Kang and M. M. Thackeray, Electrochem. Commun., 2009, 11, 748–751 CrossRef CAS PubMed.
- Y. Wu, A. V. Murugan and A. Manthiram, J. Electrochem. Soc., 2008, 155, A635–A641 CrossRef CAS PubMed.
- J. Liu, B. Reeja-Jayan and A. Manthiram, J. Phys. Chem. C, 2010, 114, 9528–9533 CAS.
- J. Liu and A. Manthiram, J. Mater. Chem., 2010, 20, 3961–3967 RSC.
- Y. S. Jung, A. S. Cavanagh, Y. F. Yan, S. M. George and A. Manthiram, J. Electrochem. Soc., 2011, 158, A1298–A1302 CrossRef CAS PubMed.
- G. Singh, R. Thomas, A. Kumar, R. S. Katiyar and A. Manivannan, J. Electrochem. Soc., 2012, 159, A470–A478 CrossRef CAS PubMed.
- H. Z. Zhang, Q. Q. Qiao, G. R. Li, S. H. Ye and X. P. Gao, J. Mater. Chem., 2012, 22, 13104–13109 RSC.
- Y. K. Sun, M. J. Lee, C. S. Yoon, J. Hassoun, K. Amine and B. Scrosati, Adv. Mater., 2012, 24, 1192–1196 CrossRef CAS PubMed.
- F. Wu, N. Li, Y. F. Su, H. Q. Lu, L. J. Zhang, R. An, Z. Wang, L. Y. Bao and S. Chen, J. Mater. Chem., 2012, 22, 1489–1497 RSC.
- D. Y. W. Yu, K. Yanagida and H. Nakamura, J. Electrochem. Soc., 2010, 157, A1177–A1182 CrossRef CAS PubMed.
- B. H. Song, M. O. Lai, Z. W. Liu, H. W. Liu and L. Lu, J. Mater. Chem. A, 2013, 1, 9954–9965 CAS.
- B. H. Song, H. W. Liu, Z. W. Liu, P. F. Xiao, M. O. Lai and L. Lu, Sci. Rep., 2013, 3, 3094, DOI:10.1038/srep03094.
- J. Liu, Q. Y. Wang, B. Reeja-Jayan and A. Manthiram, Electrochem. Commun., 2010, 12, 750–753 CrossRef CAS PubMed.
- Y. Fang, D. Gu, Y. Zou, Z. X. Wu, F. Y. Li, R. C. Che, Y. H. Deng, B. Tu and D. Y. Zhao, Angew. Chem., Int. Ed., 2010, 49, 7987–7991 CrossRef CAS PubMed.
- Y. Chen, B. H. Song, M. Li, L. Lu and J. M. Xue, Adv. Funct. Mater., 2014, 24, 319–326 CrossRef CAS.
- S. H. Kang, C. S. Johnson, J. T. Vaughey, K. Amine and M. M. Thackeray, J. Electrochem. Soc., 2006, 153, A1186–A1192 CrossRef CAS PubMed.
- K. Parvez, S. B. Yang, Y. Hernandez, A. Winter, A. Turchanin, X. L. Feng and K. Mullen, ACS Nano, 2012, 6, 9541–9550 CrossRef CAS PubMed.
- L. Roldan, I. Santos, S. Armenise, J. M. Fraile and E. Garcia-Bordeje, Carbon, 2012, 50, 1363–1372 CAS.
- H. J. Yu, H. J. Kim, Y. R. Wang, P. He, D. Asakura, Y. Nakamura and H. S. Zhou, Phys. Chem. Chem. Phys., 2012, 14, 6584–6595 RSC.
- J. R. Croy, D. Kim, M. Balasubramanian, K. Gallagher, S. H. Kang and M. M. Thackeray, J. Electrochem. Soc., 2012, 159, A781–A790 CrossRef CAS PubMed.
- Y. Chen, B. H. Song, X. S. Tang, L. Lu and J. M. Xue, J. Mater. Chem., 2012, 22, 17656–17662 RSC.
- D. Y. Pan, S. Wang, B. Zhao, M. H. Wu, H. J. Zhang, Y. Wang and Z. Jiao, Chem. Mater., 2009, 21, 3136–3142 CrossRef CAS.
- J. F. Ni, L. J. Gao and L. Lu, J. Power Sources, 2013, 221, 35–41 CrossRef CAS PubMed.
- K. Edstrom, T. Gustafsson and J. O. Thomas, Electrochim. Acta, 2004, 50, 397–403 CrossRef PubMed.
- S. J. Shi, J. P. Tu, Y. J. Mai, Y. Q. Zhang, C. D. Gu and X. L. Wang, Electrochim. Acta, 2012, 63, 112–117 CrossRef CAS PubMed.
- M. Park, X. C. Zhang, M. D. Chung, G. B. Less and A. M. Sastry, J. Power Sources, 2010, 195, 7904–7929 CrossRef CAS PubMed.
- J. Molenda and J. Marzec, Funct. Mater. Lett., 2009, 2, 1–7 CrossRef CAS.
- D. Mohanty, S. Kalnaus, R. A. Meisner, K. J. Rhodes, J. L. Li, E. A. Payzant, D. L. Wood and C. Daniel, J. Power Sources, 2013, 229, 239–248 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional information includes characterizations by TGA and TEM of carbon-coated samples. See DOI: 10.1039/c4ra04976d |
| ‡ Present address: Department of Engineering Science, University of Oxford, Parks Road, Oxford OX1 3 PJ, UK. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 








