Chemoselective flow hydrogenation approaches to isoindole-7-carboxylic acids and 7-oxa-bicyclio[2.2.1]heptanes†
L. Hizartzidis,
M. Tarleton,
C. P. Gordon and
A. McCluskey*
Chemistry, Centre for Chemical Biology, The University of Newcastle, University Drive, Callaghan NSW 2308, Australia. E-mail: Adam.McCluskey@newcastle.edu.au; Fax: +61 (0)249 215472; Tel: +61 (0)249 216486
First published on 6th January 2014
Abstract
Two libraries of highly decorated norcantharidin analogues were accessed via a series of sequential chemoselective flow hydrogenations and solvent-free transformations. Utilising a 10% Pd/C catalyst, modifications to reaction parameters (H2 pressure, temperature and flow rate conditions) allowed facile access to effect selective direct reductive aminations and olefin reductions in the presence of furan, benzyl and nitrile moieties were established. The use of 20% Pd(OH)2/C; Pd tetrakis; 5% Pt/C (sulfided) gave mixtures of furan and olefin (both reduced) and olefin reduced products. RuO2; 0.5% Re/C and Re2O7 resulted in no reduction. Concurrent olefin and nitrile reduction was achieved in the presence of furan moieties by employing a RANEY® nickel catalyst. In total, 31 reaction conditions were examined using less than 200 mg of reagents allowing optimised conditions to be efficiently determined. These optimised hydrogenation conditions afforded desired analogues in near quantitative yields thus removing the requirements of reaction workup and chromatography.
Introduction
Adult members of the Meloidae family of Coleoptera (beetles) deter attacks from many predators by discharging droplets of cantharidin-laden blood reflexively from their hind leg joints.1 These cantharidin (1) rich secretions are also used as a copulatory gift to protect the fertilised eggs from predation.2,3Cantharidin, in the form of the dried body of the Mylabris beetle, has a long history of use in Chinese traditional medicine for treatment of dermal conditions and tumours with the first chemotherapeutic application reported in 1264.3 Cantharidin and norcantharidin are potent inhibitors of the serine/threonine protein phosphatases, especially protein phosphatases 1 and 2A.4,5 The interplay between protein kinases, which add phosphate to mainly serine and threonine residues, and phosphatases, which remove phosphate moieties, modulates the vast majority of cellular signal transduction events including neurotransmission, muscle contraction, glycogen synthesis, T-cell activation, and cell proliferation.4,6 Cantharidin is nephrotoxic, and this has prevented its widespread use in Western medicine where current use is limited to topical applications.7,8 However, the demethylated norcantharidin (2) has no such nephrotoxicity issues and has thus been the subject of a considerable number of anti-tumour studies (Fig. 1).9–17
 | ||
| Fig. 1 Chemical structures of cantharidin (1) and the demethylated analogue norcantharidin (2). | ||
We and others have exploited the 7-oxa-bicyclo[2.2.11]heptane scaffold in drug development programs geared towards developing tumor suppressing agents.9–17 We have also demonstrated that the scaffold exhibits promising levels of activity against Plasmodium falciparum,18 and Haemonchus contortus.19 Fig. 2 shows representative examples of analogues investigated in these studies, and include the benzoyloxymethyl-substituted norcantharidins (3 & 4),20 the acid amides (5),21–23 anhydride modified ethers (6),24,25 norcantharimides (7 & 8),36,37 the bis-norcantharimides (9 & 10),23,26 the tetracyclic norcantharimides (11 & 12),12 along with the tetracyclic-bisnorcantharimides (13 & 14).26
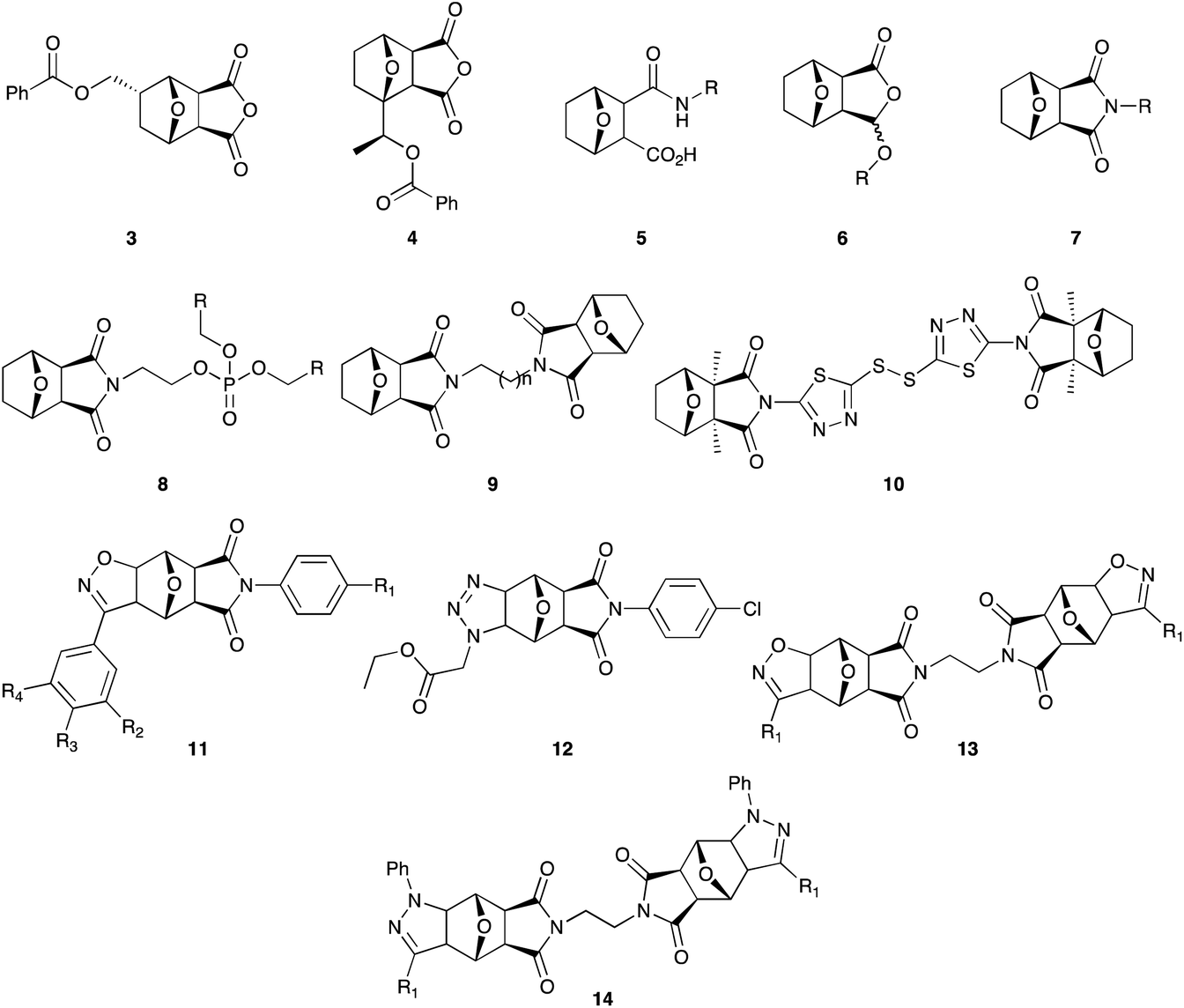 | ||
| Fig. 2 Illustrative examples of previously reported 7-oxa-bicyclo[2.2.1]heptane analogues.12,14,21,37 | ||
In addition to our medicinal chemistry interest we have a keen interest in the application of green chemistry approaches to the development of focused compound libraries. This, coupled with our prior report of the pivotal nature of the 7-oxa-bicyclo[2.2.1]heptane moiety,27,28 prompted us to examine potential expedient and green approaches to analogues such as 20 and 24 (Fig. 3). We envisaged that analogue series' based on 20 (isoindole-7-carboxylic acids) and 24 (7-oxabicyclo[2.2.1]heptane carboxylic acids) could be expediently accessed through the development of sequential flow pathways i.e. Path A and Path B, respectively. Path A, which is adapted from a number of previously reported protocols, would allow interrogation of the scaffold adjacent the 7-oxo-bridge head,29–32 a region which is poorly described in the literature,33 whereas Path B would provide a means of incorporating analogues of cyanoamide 22, which displays cytotoxicity against a range of carcinoma cells,34–36 into the norcantharidin scaffold.
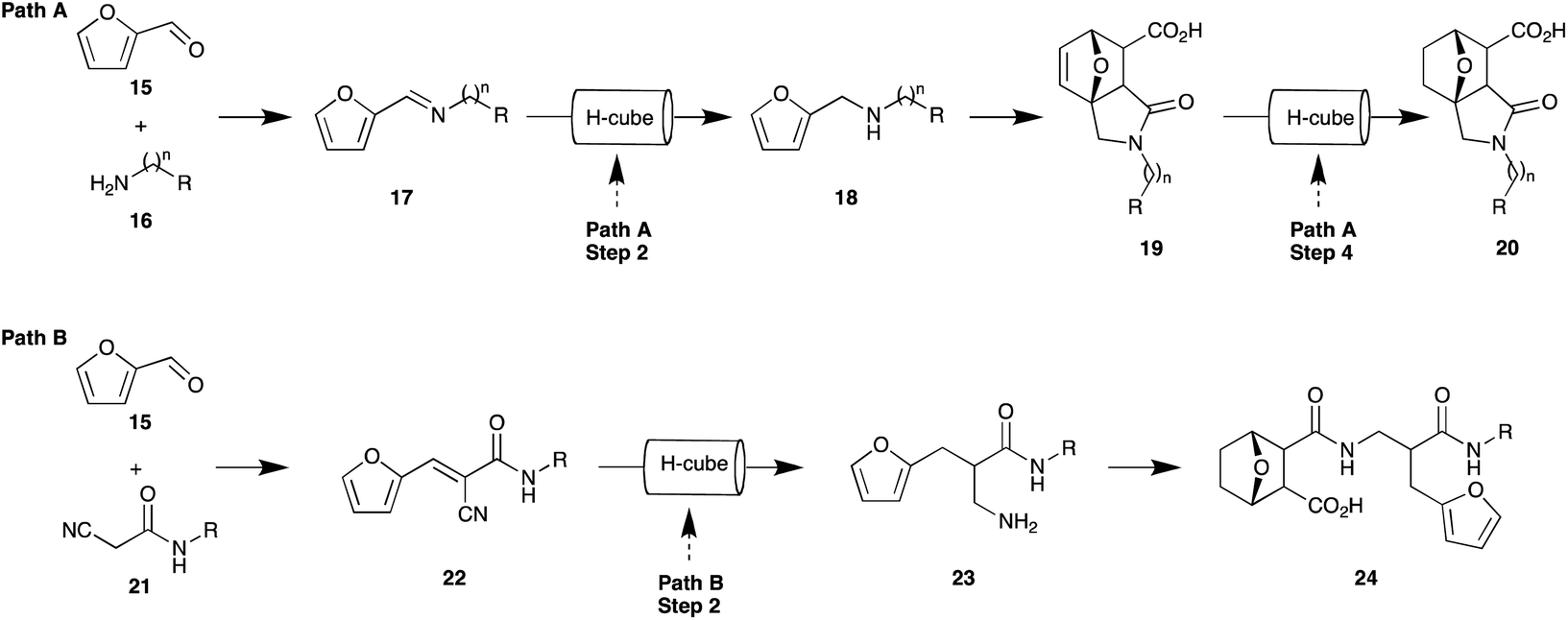 | ||
| Fig. 3 Proposed sequential flow pathways Path A and Path B to provide access to analogues of compound 20 and 24, respectively. (R-groups defined for Path A and Path B in Table 1 and Fig. 4, respectively). | ||
Common to both pathways is a series of reductive approaches including reductive amination (Path A, Step 2), olefin (Path A, Step 4 & Path B, Step 2), and nitrile reductions (Path B, Step 2). We envisaged that each of these reductions could be effected and rapidly optimised using minimum reagents quantities by careful manipulation of the flow hydrogenation conditions, i.e. judicious choice of catalysts, H2 pressure, temperature, and flow rate. However at the outset, we were also cognisant that furan reduction and de-benzylation were potential undesirable outcomes. Protocols to reduce furan analogues using platinum group metals have been well documented,38,39 and recently a number of highly diasteroselective protocols to access optically active tetrahydrofurans from furans have been reported40–43 in addition to asymmetric hydrogenation of thiophenes and benzothiophenes.41,44,45 These protocols use a range of platinum based catalysts, high pressures, typically in the range of 30–90 bar H2 pressure, temperatures up to 80 °C and long reaction times. We anticipated that, by screening milder reduction conditions and exploiting the exquisite control of temperature, pressure, and catalyst exposure provided by the ThalesNano H-cube™ (H-Cube), reaction conditions could be tuned to afford the desired selective reductions. Additionally the reduced contact (residence) times afforded by flow chemistry potentially allowed for the isolation of partially reduced intermediates unlike the corresponding batch chemistry approaches. While flow chemistry can be considered inherently green as a consequence of low material usage during process optimisation, we also sought to use easy to recycle solvents46,47 and processes that facilitated ease of product access and purification.48,49
Results and discussion
Our investigation commenced with a model coupling of furan-2-carbaldehyde (15) with 4-methoxybenzylamine (16a) Step 1 of Path A (Fig. 3 and Scheme 1), using our previously reported direct reductive amination protocol.50 Briefly, 0.05 M methanolic solution of furan-2-carbaldehyde (15) and 4-methoxybenzylamine (16a) were passed through the H-Cube equipped with a 30 mm 10% Pd/C catcart™, under 50 bar H2 at 50 °C, with a flow rate of 1 mL min−1. Each optimisation step was conducted for 10 min allowing collection of a 10 mL sample for 1H NMR and GC-MS analysis. Analysis indicated quantitative conversion to the desired adduct 18a (imine 17a was not isolated) with no evidence of furan reduction or de-benzylation evident. | ||
| Scheme 1 Reagents and conditions: (i) H-Cube hydrogenation, 0.05 M 15 and 0.05 M 16a–h (for details of R, see Table 1) in MeOH, 10% Pd/C, 50 °C, 50 bar H2 pressure, 1 mL min−1. | ||
With 18a in hand, our attention turned to the sequential Diels–Alder and lactam formation (Step 2, Path A, Scheme 2). This two-step transformation proceeded smoothly and in excellent overall yield (69%) with the product collected by filtration after trituration with diethyl ether. The diethyl ether was recovered and reused in the trituration of the hydrogenated product (below). Access to compounds such as 19a–h allow, on treatment with protic ionic liquids, entry to N-substituted 5-hydroxy-4-methyl-3-oxoisoindoline-1-carboxamides and N-substituted 3-oxisoindoline-4-carboxylic acids. This current approach allows greener access to these 7-oxoisoindole analogues.29
 | ||
| Scheme 2 Reagents and conditions: (i) diethyl ether, rt, 24 h; (ii) H-Cube, 0.05 M 20a in MeOH, 10% Pd/C, 50 °C, 50 bar H2 pressure, 1 mL min−1. | ||
The final step of reaction Path A, olefin reduction, was again carried out using the H-Cube charged with a 30 mm 10% Pd/C catcart™ (50 bar H2, 50 °C, and a flow rate of 1 mL min−1). Trituration of the eluent with diethyl ether afforded the desired analogue 20a in >95% purity (Scheme 2).
With our model system, the sequential flow reaction protocol of Path A afforded the desired bicyclo[2.2.1]heptane analogue 20a in excellent overall yield (51%, 3 steps) with no chromatography requirement. More broadly the two 10 min flow chemistry steps and one batch reaction allowed rapid access to a diverse library of analogues with a range for functional groups (Table 1, 20a–g) and anilines tolerated (Table 1, 20h) and afford 25–50 mg (sufficient for all preliminary biological screening) of the desired products in >95% purity by trituration. This protocol offers dramatic reductions in reaction times and eliminating the requirement for catalyst preparation, reaction work-up, and chromatography typically used to access compounds of this nature.22,29 In keeping with previous reports on the use of Pd-based catcarts™ we detected no Pd-leakage or residue in the products isolated.51
|
|||||
|---|---|---|---|---|---|
| Compound | R | Step 1 (%) | Step 2 (%) | Step 3 (%) | Overall (%) |
| 20a |  |
89 | 69 | 100 | 51 |
| 20b |  |
85 | 66 | 88 | 47 |
| 20c |  |
88 | 62 | 100 | 53 |
| 20d |  |
84 | 44 | 100 | 37 |
| 20e |  |
74 | 33 | 85 | 24 |
| 20f | 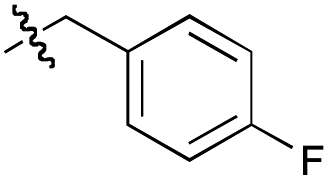 |
82 | 46 | 100 | 38 |
| 20g |  |
63 | 57 | 100 | 36 |
| 20h | 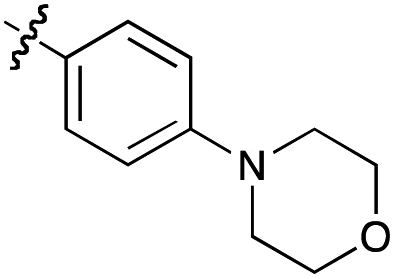 |
76 | 40 | 100 | 30 |

Having successfully developed a small library of analogues using Path A, our attention turned to the second proposed sequential flow pathway, Path B (Fig. 3). Access to the desired furan acrylamides (22a–22c) was via a solvent free condensation of methyl cyanoacetate and small library of benzylamines. The initial synthesis examined the use of 4-methoxybenzylamine (16a) to afford cyanoamide 21a which was used directly in the Knoevenagel condensation with furan-2-carbaldehyde (15) at room temperature giving 22a in a 71% yield (2 steps) with the product collected by filtration in >95% purity. Cyanoamides 22b and 22c were accessed in a similar manner from 4-chlorobenzylamine and 4-methylbenzylamine (Scheme 3).
 | ||
| Scheme 3 Reagents and conditions: (i) EtOH, piperidine, 0.5 h, 25 °C. | ||
With 22a we next investigated the potential selective reduction of the olefin and the nitrile moieties as outlined in Step 2, Path B (Fig. 3). Excluding possible debenzylation products, hydrogenation of 22a had the potential to give seven different reduction products (23a, 25a–30a) arising from various combinations of furan, olefin and nitrile moiety reductions (Scheme 5). Hence access to the analogues desired for subsequent focused library development was potentially a significant challenge.
A 0.05 M solution of 22a was subjected to H-Cube conditions of 50 bar H2 pressure, 50 °C, and a flow rate of 1 mL min−1 for 10 minutes using a 30 mm 10% Pd/C catcart™ (Scheme 5). Analysis showed quantitative reduction of the furan and olefin double bond moieties giving 28a (Scheme 4 and Table 2). No nitrile reduction was observed.
 | ||
| Scheme 4 Reagents and conditions: (i) H-Cube, 0.05 M 22a (R = p-OCH3PhCH2–) in MeOH, 10% Pd/C, at 1 mL min−1 and the reduction products (23a, 25a–30a) potentially obtainable by choice of hydrogenation conditions (see Table 2 for detail). | ||
| Entry | T (°C) | H2 P (bar) | 25a (%) | 28a (%) |
|---|---|---|---|---|
| a 1 mL min−1 is equivalent to a 4 min residence time (http://www.thalesnano.com). | ||||
| 1 | 100 | 0 | 0 | 100 |
| 2 | 80 | 0 | 0 | 100 |
| 3 | 60 | 60 | 0 | 100 |
| 4 | 60 | 0 | 0 | 100 |
| 5 | 50 | 50 | 0 | 100 |
| 6 | 50 | 0 | 0 | 100 |
| 7 | 40 | 40 | 12 | 88 |
| 8 | 40 | 0 | 4 | 96 |
| 9 | 30 | 30 | 15 | 85 |
| 10 | 25 | 40 | 18 | 82 |
| 11 | 25 | 30 | 15 | 85 |
| 12 | 25 | 20 | 11 | 89 |
| 13 | 25 | 10 | 5 | 95 |
| 14 | 25 | 0 | 1 | 99 |
| 15 | 20 | 0 | 11 | 89 |
| 16 | 15 | 0 | 11 | 89 |
| 17 | 10 | 0 | 21 | 79 |
Efforts to effect nitrile reduction using standard conditions (70 °C and 50 bar H2 pressure‡) resulted only in clean conversion to 28a. A similar outcome was noted with all optimisations with T ≥ 50 °C regardless of the H2 pressure (Table 2, entries 1–6). Easing of the reduction conditions (10 °C, 0 bar H2 pressure) did show the first evidence of olefin vs. furan reduction selectivity with both the olefin reduction product (25a, 21%) and 28a (79%) evident (Table 2, entry 17).
The production of 25a suggested that 28a resulted from over reduction of this compound. We believed that this is most likely related to catalyst residence time. Reducing residence time from 4 min (1 mL min−1) to 3 min resulted in an improvement in the 25a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :28a ratio to 64:36 (Table 3, entry 1). Further residence time reduction to 0.5 min saw clean generation of 25a (Table 3, entry 4).
:28a ratio to 64:36 (Table 3, entry 1). Further residence time reduction to 0.5 min saw clean generation of 25a (Table 3, entry 4).
| Entry | Catalyst | Residence time (min) | 25a (%) | 28a (%) |
|---|---|---|---|---|
| 1 | 10% Pd/C | 3.0 | 64 | 36 |
| 2 | 10% Pd/C | 1.5 | 80 | 20 |
| 3 | 10% Pd/C | 0.8 | 89 | 11 |
| 4 | 10% Pd/C | 0.5 | 100 | 0 |
To examine whether this selective reduction could be performed on related heterocycles we investigated the pyrrole (31) and thiophene (32) analogues of 22a (Scheme 5). Analogues 31 and 32 were synthesised as per 22a from pyrrole-2-carbaldehyde and thiophene-2-carbaldehyde respectively. In contrast to 22a, both 31 and 32 showed exclusive reduction of the olefin moiety affording 33 and 34, respectively. No change in this reduction was evident with 31 and 32 with reaction conditions of 100 °C and 100 bar H2 pressure and once again no evidence of nitrile reduction was evident.
 | ||
| Scheme 5 Reagents and conditions: (i) 0.05 M 31 or 32 (MeOH), 10% Pd/C, 100 °C, 100 bar H2, 1 mL min−1, H-Cube™. | ||
As no evidence of nitrile reduction was obtained using Pd/C, for any of the analogues examined thus far, we next conducted a rapid catalyst scan using 22a as the model compound (Table 4). At 60 °C and 60 bar H2 pressure the Pd-based catalysts gave varying ratios of 25a and 28a, with no evidence of the desired 23a (Table 4, entries 1–4). Of the Pd-based catalysts both the 10% Pd–C and 20% Pd(OH)2 showed preference for reduction of the furan and olefin moiety (Table 4, entries 1 and 2). The Pd tetrakis and 5% Pt/C (sulfided) showed ca. 2:1 preference for the formation of the olefin reduced 25a over the furan and olefin reduced 28a (Table 4, entries 3 and 4). While no evidence of the desired nitrile reduction was evident, these data suggest that further optimisation may afford exclusive access to both 25a and 28a. We observed no reduction products at 1.0 mL min−1, 60 °C and 60 bar H2 pressure with the RuO2, 0.5% Ir/C, 5% Re/C or Re2O7 (Table 4, entries 5–8). However using the same conditions with RaNi we observed global reduction to 30a (see Scheme 5). However repeating the RaNi reaction at 10 bar H2 pressure and 50 °C reduced the olefin and nitrile moieties giving the desired 23a (91%, Table 4 entry 10).
| Entry | Catalyst | 23a (%) | 25a (%) | 28a (%) | 30a (%) |
|---|---|---|---|---|---|
| a Reaction conducted at 10 bar H2 pressure, 50 °C and 1.0 mL min−1. | |||||
| 1 | 10% Pd/C | 0 | 10 | 90 | 0 |
| 2 | 20% Pd(OH)2/C | 0 | 4 | 96 | 0 |
| 3 | Pd tetrakis | 0 | 71 | 29 | 0 |
| 4 | 5% Pt/C (sulfided) | 0 | 65 | 35 | 0 |
| 5 | RuO2 | 0 | 0 | 0 | 0 |
| 6 | 0.5% Ir/C | 0 | 0 | 0 | 0 |
| 7 | 5% Re/C | 0 | 0 | 0 | 0 |
| 8 | Re2O7 | 0 | 0 | 0 | 0 |
| 9 | RaNi | 0 | 0 | 0 | 100 |
| 10 | RaNi | 91a | 9 | 0 | 0 |
The RaNi reduction also proved compatible with a range of substituted pyrrole analogues allowing access to a series of novel amines for subsequent addition to norcantharidin (2) in the final step of Path B. Stirring in acetone at room temperature for 4 hours afforded the desired product 7-oxabiclcyo[2.2.1]heptane 24a in a 78% yield upon filtration of the crude reaction mixture (Scheme 6).52 Pleasingly, as with reaction Path A, reaction Path B proved amenable to various functional group alterations and was utilised to access a small library of furan (Fig. 4, 24a–24c) and pyrrole (24d–24g), and thiophene (24h and 24i) based norcantharidin analogues.
 | ||
| Scheme 6 Reagents and conditions: (i) acetone, rt, 4 h. | ||
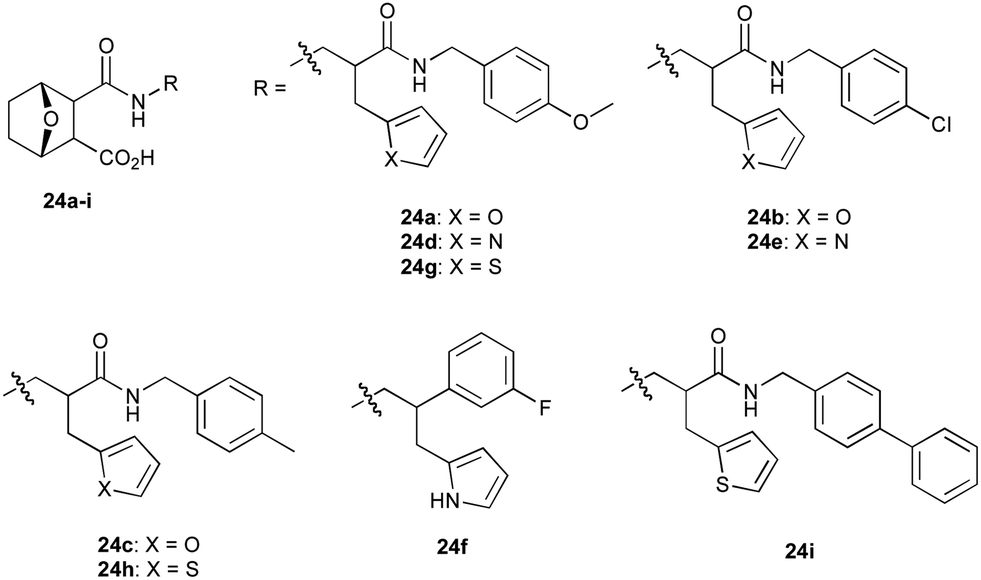 | ||
| Fig. 4 Small library of analogues access by sequential flow reaction Path B including a number of furan (24a–24c), pyrrole (24d–24g), and thiophene (24h and 24i) based norcantharidin analogues. | ||
Conclusion
From the outset of this study we were aware that step 1 of Path A, reduction of the imine 17, couple potentially could yield the desired reductive amination product (18, Fig. 3) or the equivalent tetrahydrofuran; and that Path B was likely to be more complex with the reduction of 22 potentially yielding seven reduction products (Scheme 5, plus additional debenzylation products). However by judicious choice of flow reduction reductive amination of 15 with a range of amines was effected at 50 °C, 50 bar H2 pressure and 1.0 mL min−1 to give exclusively the reductive amination product 18. Interestingly the same conditions with furan 22, which could give rise to seven reduction products (excluding debenzylation), resulted in reduction only of the furan and olefin moieties with clean generation of 28, suggesting that the amino/imine moieties of 18 drove affected the flow reduction outcomes. Reducing catalyst residence times from 4 min to 0.5 min allowed access to exclusively the furan olefin reduced product (25). Catalyst switching from Pd/C to RaNi and manipulation of the flow conditions to 50 °C, 10 bar and 1 mL min−1 allowed access to the desired furanyl amides (reduction of the olefin and nitrile moieties) (23) for subsequent synthesis of the desired 7-oxabicyclo[2.2.1]heptane carboxylic acid analogues (24).With the desired amines in hand the Diels–Alder addition of 18 with maleic anhydride followed by an intramolecular lactamisation followed by a second flow reduction (of the resultant C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) gave a focused library of isoindole-7-carboxylic acids (20a–h) in modest to good yields for the three steps. Importantly these products were access in 50–100 mg quantities through two flow chemistry (10 minute) and one batch reaction in >95% purity requiring no purification. Similarly, with 23 in hand, addition of norcantharidin (2) saw smooth conversion to the desired focused library of 7-oxabicyclo[2.2.1]heptane carboxylic acids (24a–i). In this instance analogues 24a–i were rapidly accessed via a solvent free reaction, one 10 minute flow hydrogenation reaction and nucleophilic addition to the anhydride moiety of 2. These analogues were isolated by filtration and/or trituration in 50–100 mg quantities in >95% purity.
C) gave a focused library of isoindole-7-carboxylic acids (20a–h) in modest to good yields for the three steps. Importantly these products were access in 50–100 mg quantities through two flow chemistry (10 minute) and one batch reaction in >95% purity requiring no purification. Similarly, with 23 in hand, addition of norcantharidin (2) saw smooth conversion to the desired focused library of 7-oxabicyclo[2.2.1]heptane carboxylic acids (24a–i). In this instance analogues 24a–i were rapidly accessed via a solvent free reaction, one 10 minute flow hydrogenation reaction and nucleophilic addition to the anhydride moiety of 2. These analogues were isolated by filtration and/or trituration in 50–100 mg quantities in >95% purity.
Herein we have applied the principles of green chemistry where possible. The use of flow chemistry approaches has allowed reagent minimisation (10 mL of a 0.05 M solution) and rapid reaction optimisation. Careful control of residence times, temperature and H2 pressure permitted exclusive reduction of 22 to 23a, 26a or 28a, a level of control difficult to envisage under traditional batch hydrogenation approaches (Scheme 7). Batch reduction approaches typically only afford the global reduction products.43–45 The high conversion rates minimised purification requirements typically to filtration and/or trituration. In instances were diethyl ether was the solvent of choice, the recovered solvent was directly for product trituration in subsequent steps. The combined use of flow an batch chemistry provided facile access to the two series of bicycle[2.2.1]heptane analogues, the isoindole-7-carboxylic acids (20) and the 7-oxabicyclo[2.2.1]0heptane carboxylic acids (24). We are currently developing an extended series of these analogues which will be assessed against a panel of human cancer cell lines and the results of these studies will be reported in due course.
 | ||
| Scheme 7 Reagents and conditions: (i) H-cube RANEY® Ni, 50 °C, 10 bar H2, 1 mL min−1; (ii) H-Cube Pd/C, 25 °C, 0 bar, 10% H2, 3.0 mL min−1 (iii) H-Cube Pd/C, 50 °C, 50 bar H2, 1 mL min−1; (iv) H-cube RANEY® Ni, 60 °C, 60 bar H2, 1.0 mL min−1. | ||
We believe that with rapid reaction optimisation, reduced work-up and chromatography requirements, sequential flow methodologies integrated with batch chemistry has the potential to aid sustainable practices in medicinal chemistry.
Experimental
General experimental
All reagents were purchased from Sigma Aldrich and were used without purification, with the exception of furfural, which was distilled from glass prior to use. Solvents were bulk, and distilled from glass prior to use.1H and 13C NMR spectra were recorded on a Brüker Advance™ AMX 400 MHz spectrometer at 400.1 and 100.1 MHz, respectively. Chemical shifts (δ) are reported in parts per million (ppm) measured to relative the internal standards. Coupling constants (J) are expressed in Hertz (Hz). Mass spectra were recorded on a Shimadzu LCMS 2010 EV using a mobile phase of 1:1 acetonitrile:H2O with 0.1% formic acid. Gas chromatography-mass spectrometry (GC-MS) was performed on a Shimadzu GC-MS QF2010 EI/NCI System equipped with a ZB-5MS capillary column of 5% phenylarylene stationary phase.
Melting points were recorded on a BUCHI Melting Point M-565. IR spectra were recorded on a PerkinElmer Spectrum Two™ FTIR Spectrometer. Thin layer chromatography (TLC) was performed on Merck 60 F254 pre-coated aluminium plates with a thickness of 0.2 mm. Column chromatography was performed under ‘flash’ conditions on Merck silica gel 60 (230–400 mesh). Microwave irradiations were conducted using a CEM Discover® Benchmate microwave, and hydrogenations were performed either using a ThalesNano H-Cube™ or a ThalesNano H-CubePro™ (H-Cube™) continuous-flow hydrogenation reactor. All reactions were passed through the H-Cube™ reactor once, unless otherwise specified.
General procedure 1 – direct reductive amination
GC-MS 4.06 r.t.; LRMS (ESI+) m/z 218 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 7.45 (d, J = 1.1 Hz, 1H), 7.35 (d, J = 8.6 Hz, 2H), 6.92 (d, J = 8.6 Hz, 2H), 6.38 (dd, J = 2.0, 1.2 Hz, 1H), 6.27 (d, J = 3.2 Hz, 1H), 3.84 (s, 3H), 3.70 (s, 2H), 3.61 (m, 2H). 13C NMR (CDCl3, 101 MHz): δ 158.7, 152.6, 142.0, 130.9, 130.2, 113.7, 110.1, 108.8, 56.5, 55.3, 49.2. IR (cm−1) 3330 (NH), 2960, 2763 (CH), 1615, 1513 (CC), 1463, 1432 (CH2), 1247 (CH3), 1218 (C–O), 830 (p-C Ph).
LRMS (ESI+) m/z 188 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 7.30 (dd, J = 4.2, 3.3 Hz 1H), 7.23 (d, J = 4.1, Hz, 2H), 7.19–7.11 (m, 1H), 6.24–6.22 (m, 1H), 6.14 (d, J = 3.1 Hz, 1H), 3.70 (s, 1H), 3.58 (s, 2H), 3.53 (s, 1H). 13C NMR (CDCl3, 101 MHz): δ 152.5, 142.1, 129.0, 128.5, 128.3, 127.0, 110.1, 108.8, 57.1, 49.4. IR (cm−1) 3330 (NH), 2976, 2929, 2838 (CH), 1602, 1507 (CC), 1453, 1360 (CH2).
LRMS (ESI+) m/z 204 (M + 1). 1H NMR (MeOD, 400 MHz): δ 7.49 (d, J = 1.0 Hz, 1H), 7.19 (d, J = 8.4 Hz, 1H), 6.76 (d, J = 8.5 Hz, 1H), 6.40–6.38 (m, 1H), 6.30 (d, J = 3.0 Hz, 1H), 3.62 (s, 2H), 3.51 (s, 1H), 3.33 (s, 1H). 13C NMR (MeOD, 101 MHz): δ 156.3, 152.0, 141.9, 130.2, 128.9, 114.6, 109.8, 108.8, 56.3, 48.4. IR (cm−1) 3290 (OH), 2963, 2923, 2858 (CH), 1613, 1514 (CC), 1448, 1360 (CH2), 1225 (C–O), 819 (p-C Ph).
LRMS (ESI+) m/z 244 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 7.36–7.33 (m, 3H), 7.25 (d, J = 8.3 Hz, 2H), 6.31 (dd, J = 3.1, 1.9 Hz, 1H), 6.18 (d, J = 3.1 Hz, 1H), 3.79 (s, 2H), 3.76 (s, 2H), 1.31 (s, 9H). 13C NMR (CDCl3, 101 MHz): δ 154.0, 149.9, 141.8, 137.0, 128.0, 125.3, 125.3, 110.1, 107.0, 52.5, 45.5, 34.5, 31.4. IR (cm−1) 3330 (NH), 2961, 2904, 2868 (CH), 1597, 1508 (CC), 1460, 1362 (CH2), 1269 (CH3), 804 (p-C Ph).
LRMS (ESI+) m/z 238 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 8.06 (d, J = 8.2 Hz, 1H), 7.88–7.84 (m, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.52–7.40 (m, 5H), 6.35 (dd, J = 3.1, 1.9 Hz, 1H), 6.24 (d, J = 3.2 Hz, 1H), 4.23 (s, 2H), 3.89 (s, 2H). 13C NMR (CDCl3, 101 MHz): δ 153.9, 141.9, 135.5, 133.9, 131.9, 128.7, 127.9, 126.3, 126.2, 125.6, 125.4, 123.6, 110.2, 107.2, 50.4, 45.9. IR (cm−1) 3321 (NH), 3047, 2925, 2832, (CH), 1597, 1508 (CC), 1450, 1396 (CH2), 790 (p-C Ph).
LRMS (ESI+) m/z 206 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 7.37 (d, J = 1.0 Hz, 1H), 7.30–7.27 (m, 2H), 6.99 (d, J = 8.7 Hz, 2H), 6.32 (dd, J = 3.1, 1.9 Hz, 1H), 6.18 (d, J = 3.1 Hz, 1H), 3.77 (s, 2H), 3.75 (s, 2H). 13C NMR (CDCl3, 101 MHz): δ 162.0 (d, J = 245.5 Hz), 152.3, 142.1, 130.5, 130.4, 115.2, 115.0, 110.1, 108.9, 56.3, 49.3. IR (cm−1) 3317 (NH), 2971, 2925, 2836, (CH), 1602, 1507 (CC), 1416, 1358 (CH2), 1009 (F–C Ph), 821 (p-C Ph).
LRMS (ESI+) m/z 222 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 7.41 (d, J = 1.1 Hz, 1H), 7.36–7.25 (m, 4H), 6.34 (dd, J = 3.1, 1.9 Hz, 1H), 6.23 (d, J = 3.1 Hz, 1H), 3.77 (d, J = 9.2 Hz, 1H), 3.66 (s, 2H), 3.58 (s, 1H). 13C NMR (CDCl3, 101 MHz): δ IR (cm−1) 3312 (NH), 2921, 2777, 22730 (CH), 1601, 1573 (CC), 1452, 1428 (CH2), 810 (p-C Ph), 599 (Cl–C Ph).
LRMS (ESI+) m/z 259 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 7.36 (d, J = 1.1 Hz, 1H), 6.84 (d, J = 8.9 Hz, 2H), 6.66 (d, J = 8.9 Hz, 2H), 6.31 (dd, J = 3.1, 1.9 Hz, 1H), 6.21 (d, J = 2.8 Hz, 1H), 4.28 (s, 2H), 3.85 (t, J = 4.5 Hz, 4H), 3.02 (t, J = 4.6 Hz, 4H). 13C NMR (CDCl3, 101 MHz): δ 153.0, 144.1, 142.1, 141.9, 118.2, 114.4, 110.3, 106.9, 67.1, 51.1, 42.2. IR (cm−1) 3395 (NH), 2964, 2851, 2824 (CH), 1615 (CC), 1512 (OC–N), 1458, 1408 (CH2), 1220 (C–O), 1117 (C–O), 918 (CC bend) 824 (p-C Ph).
General procedure 2 – reaction path A
1H NMR (CDCl3, 400 MHz): δ 7.72 (s, 1H), 7.20 (d, J = 8.6 Hz, 2H), 6.89 (d, J = 8.6 Hz, 2H), 4.87 (d, J = 5.6 Hz, 1H), 4.52 (d, J = 14.8 Hz, 1H), 4.42 (d, J = 14.8 Hz, 1H), 3.80 (s, 3H), 3.62 (d, J = 11.8 Hz, 1H), 3.53 (d, J = 11.8 Hz, 1H), 3.21–3.13 (m, 2H), 1.97 (dt, J = 13.3, 6.9 Hz, 1H), 1.79–1.69 (m, 2H), 1.64–1.55 (m, 1H). 13C NMR (CDCl3, 101 MHz): δ 172.9, 159.2, 129.4, 127.4, 114.3, 86.3, 79.9, 55.3, 54.4, 52.4, 48.7, 46.4, 29.5, 29.1. IR (cm−1) 3592 (N), 3528 (OH), 2983, 2955, 2892, 2834 (CH), 1730 (CO), 1644 (CO), 1612 (CC), 1513 (OC–N), 1473, 1422 (CH2), 1355 (CH3), 1173 (C–O), 842 (p-C Ph).
MS (ESI+) m/z 288 (M + 1). 1H NMR (MeOD, 400 MHz): δ 7.37–7.28 (m, 5H), 4.70 (d, J = 5.6 Hz, 1H), 4.58 (d, J = 15.2 Hz, 1H), 4.42 (d, J = 15.1 Hz, 1H), 3.63 (d, J = 11.7 Hz, 1H), 3.55 (d, J = 11.7 Hz, 1H), 3.19 (d, J = 9.6 Hz, 1H), 3.09 (d, J = 9.6 Hz, 1H), 2.00–1.90 (m, 1H), 1.84 (td, J = 11.3, 7.2 Hz, 1H), 1.76–1.66 (m, 2H). 13C NMR (MeOD, 101 MHz): δ 173.6, 173.0, 135.9, 128.3, 127.5, 127.2, 86.3, 79.8, 54.0, 51.6, 48.3, 45.9, 29.0, 28.6. IR (cm−1) 3378 (OH), 2972, 2935, 2878 (CH), 1713 (CO), 1685 (CO), 1652 (CC), 1474, 1428 (CH2), 1259 (C–O), 1163 (C–O).
LRMS (ESI+) m/z 304 (M + 1). 1H NMR (DMSO, 400 MHz): δ 11.71 (s, OH), 9.33 (s, NH), 7.05 (d, J = 8.4 Hz, 2H), 6.70 (d, J = 8.4 Hz, 2H), 4.56 (d, J = 4.8 Hz, 1H), 4.31 (d, J = 14.9 Hz, 1H), 4.21 (d, J = 14.9 Hz, 1H), 3.48 (d, J = 11.5 Hz, 1H), 3.38 (d, J = 11.5 Hz, 1H), 3.07 (d, J = 9.7 Hz, 1H), 2.86 (d, J = 9.7 Hz, 1H), 1.73 (dd, J = 11.6, 5.3 Hz, 2H), 1.56 (t, J = 10.4 Hz, 2H). 13C NMR (DMSO, 101 MHz): δ 172.7, 171.6, 157.0, 129.3, 127.1, 115.7, 86.1, 79.2, 53.9, 51.7, 48.2, 45.3, 29.6, 29.0. IR (cm−1) 3257 (OH), 2979, 2955, 2950 (CH), 1713 (CO), 1650 (CO), 1620 (CC), 1437, 1422 (CH2), 1261 (CO), 1162 (CO), 822 (p-C Ph).
LRMS (ESI+) m/z 338 (M + 1). 1H NMR (MeOD, 400 MHz): δ 7.40 (d, J = 8.3 Hz, 2H), 7.24 (d, J = 8.3 Hz, 2H), 4.69 (d, J = 5.6 Hz, 1H), 4.51 (d, J = 15.0 Hz, 1H), 4.41 (d, J = 15.0 Hz, 1H), 3.61 (d, J = 11.7 Hz, 1H), 3.55 (d, J = 11.7 Hz, 1H), 3.17 (d, J = 9.6 Hz, 1H), 3.08 (d, J = 9.6 Hz, 1H), 1.97–1.90 (m, 1H), 1.87–1.80 (m, 1H), 1.70 (ddd, J = 10.7, 9.1, 4.3 Hz, 2H), 1.32 (s, 9H). 13C NMR (MeOD, 101 MHz): δ 173.8, 172.9, 150.2, 132.9, 127.3, 125.2, 86.2, 79.8, 54.0, 51.7, 48.3, 45.6, 33.9, 30.4, 29.0, 28.6. IR (cm−1) 3250 (OH), 2967, 2870 (CH), 1748 (CO), 1674 (CO), 1656 (CC), 1475, 1409 (CH2), 1401, 1389, 1354 (CH3), 1269 (CO), 1148 (CO), 840 (p-C Ph).
LRMS (ESI+) m/z 338 (M + 1). 1H NMR (MeOD, 400 MHz): δ 8.09 (d, J = 8.0 Hz, 1H), 7.90 (d, J = 7.5 Hz, 1H), 7.85 (d, J = 8.1 Hz, 1H), 7.57–7.44 (m, 4H), 5.01 (d, J = 15.1 Hz, 1H), 4.86 (d, J = 15.1 Hz, 1H), 4.67 (d, J = 5.6 Hz, 1H), 3.53 (d, J = 11.8 Hz, 1H), 3.44 (d, J = 11.8 Hz, 1H), 3.16 (d, J = 9.6 Hz, 1H), 3.07 (d, J = 9.6 Hz, 1H), 1.93–1.83 (m, 1H), 1.79–1.71 (m, 1H), 1.68–1.58 (m, 2H). 13C NMR (MeOD, 101 MHz): δ 173.5172.5, 134.0, 131.4, 131.2, 128.3, 128.3, 126.5, 126.2, 125.6, 125.0, 123.2, 86.2, 79.8, 54.1, 51.6, 48.4, 44.2, 28.9, 28.5. IR (cm−1) 3480 (OH), 3060, 3044, 2999, 2983, 2939 (CH), 1732 (CO), 1649 (CO), 1612 (CC), 1485, 1425 (CH2), 1264 (C–O), 1232 (C–O), 794(m-C Ph), 782 (o-C Ph).
LRMS (ESI+) m/z 306 (M + 1). 1H NMR (MeOD, 400 MHz): δ 7.34 (dd, J = 8.7, 5.4 Hz, 2H), 7.07 (t, J = 8.8 Hz, 2H), 4.69 (d, J = 5.6 Hz, 1H), 4.61 (d, J = 15.2 Hz, 1H), 4.35 (d, J = 15.2 Hz, 1H), 3.64 (d, J = 11.7 Hz, 1H), 3.54 (d, J = 11.7 Hz, 1H), 3.18 (d, J = 9.6 Hz, 1H), 3.09 (d, J = 9.6 Hz, 1H), 1.95 (dd, J = 5.6, 3.6 Hz, 1H), 1.89–1.79 (m, 1H), 1.77–1.65 (m, 2H). 13C NMR (MeOD, 101 MHz): δ 173.6, 173.0, 162.3 (d, J = 245.1 Hz), 132.0, 129.4, 129.3, 115.1, 114.8, 86.3, 79.8, 53.9, 51.6, 48.1, 45.1, 29.0, 28.6. IR (cm−1) 3390 (OH), 3003, 2979, 2955, 2894 (CH), 1726 (CO), 1682 (CO), 1508 (CC), 1436, 1415 (CH2), 1262 (C–O), 1219 (C–O), 1003 (F–C Ph), 841 (p-C Ph).
LRMS (ESI+) m/z 322 (M + 1). 1H NMR (MeOD, 400 MHz): δ 7.24–7.15 (m, 4H), 4.57 (d, J = 5.6 Hz, 1H), 4.50 (d, J = 15.4 Hz, 1H), 4.20 (d, J = 15.4 Hz, 1H), 3.52 (d, J = 11.7 Hz, 1H), 3.42 (d, J = 11.7 Hz, 1H), 3.06 (d, J = 9.6 Hz, 1H), 2.96 (d, J = 9.6 Hz, 1H), 1.87–1.77 (m, 1H), 1.76–1.68 (m, 1H), 1.64–1.53 (m, 2H). 13C NMR (MeOD, 101 MHz): δ 173.5, 173.0, 134.8, 132.9, 129.0, 128.4, 86.3, 79.8, 53.9, 51.6, 48.3, 45.1, 29.0, 28.6. IR (cm−1) 3439 (NH), 3322 (OH), 2975, 2906 (CH), 1721 (CO), 1659 (CO), 1489, 1426 (CH2), 1275 (C–O), 1215 (C–O), 841 (p-C Ph), 567 (Cl–C Ph).
LRMS (ESI+) m/z 359 (M + 1). 1H NMR (DMSO-d,6 400 MHz): δ 12.06 (s, OH), 7.49 (d, J = 9.1 Hz, 2H), 6.93 (d, J = 9.2 Hz, 2H), 4.59 (d, J = 5.1 Hz, 1H), 4.15 (d, J = 11.5 Hz, 1H), 3.90 (d, J = 11.4 Hz, 1H), 3.76–3.70 (m, 4H), 3.26 (d, J = 9.7 Hz, 1H), 3.10–3.04 (m, 4H), 2.93 (d, J = 9.7 Hz, 1H), 1.88–1.73 (m, 2H), 1.74–1.55 (m, 2H). 13C NMR (MeOD, 101 MHz): δ 171.8, 169.9, 147.0, 131.2, 119.8, 114.5, 84.2, 78.4, 65.5, 54.0, 51.1, 49.0, 48.1, 28.5, 27.9. IR (cm−1) 3300 (OH), 2999, 2955, 2922, 2882 (CH), 1740 (CO), 1689 (CO), 1606 (CC), 1511 (OC–N), 1455, 1394 (CH2), 1210 (C–O), 1170 (C–O), 824 (p-C Ph).
General procedure 3 – Knoevenagel condensation
LRMS (ESI+) m/z 205 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 7.23 (d, J = 8.5 Hz, 2H), 6.90 (d, J = 8.6 Hz, 2H), 6.45 (s, 1H), 4.41 (d, J = 5.5 Hz, 2H), 3.82 (s, 3H), 3.39 (s, 2H). 13C NMR (CDCl3, 101 MHz): δ 160.7, 159.4, 129.4, 128.9, 114.7, 114.3, 55.3, 43.9, 25.9. IR (cm−1) 3284 (NH), 3073, 2939, 2841 (CH), 2258 (CN), 1640 (CO), 1614, 1515 (CC), 1515 (NH bend), 1462 (CH2), 1368 (CH3), 1240 (CO), 1031 (C–O–C), 815 (p-C Ph).
Furan-2-carboxaldehyde (15) (0.11 g, 1.2 mmol) and 2-cyano-N-(4-methoxybenzyl)acetamide (21a) (0.24 g, 1.2 mmol) were added together in EtOH (4 mL). Piperidine (cat.) was added and the reaction was left to stir at room temperature for 30 min and then placed in the freezer for 60 min. The solution was filtered and washed with cold EtOH and dried under suction to afford 22a as an orange solid, 71%; mp 119–121 °C. GC–MS (r.t.) 15.46 min.
LRMS (ESI+) m/z 283 (M + 1). 1H NMR (acetone, 400 MHz): δ 8.04 (s, 1H), 7.96 (d, J = 1.4 Hz, 1H), 7.92 (br s, NH), 7.38 (d, J = 3.6 Hz, 1H), 7.31 (d, J = 8.6 Hz, 2H), 6.89 (d, J = 8.6 Hz, 2H), 6.78 (dd, J = 3.5, 1.7 Hz, 1H), 4.50 (d, J = 6.0 Hz, 1H), 3.78 (s, 3H), 13C NMR (acetone, 101 MHz): δ 160.3, 159.1, 149.2, 148.0, 136.2, 131.0, 129.1, 120.7, 115.9, 113.7, 113.6, 101.1, 54.6, 43.1. IR (cm−1) 3323 (NH), 3128, 3038, 3006, 2833 (CH), 2226 (CN), 1657 (CO), 1605 (CC), 1532 (NH bend), 1436 (CH2), 1351 (CH3), 1298 (C–O), 1244 (C–O), 1029 (C–O–C), 826 (p-C Ph).
LRMS (ESI+) m/z 209 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 7.39–7.27 (m, 2H), 7.22 (d, J = 8.5 Hz, 2H), 6.47 (s, 1H), 4.44 (d, J = 5.8 Hz, 2H), 3.41 (s, 2H). 13C NMR (CDCl3, 101 MHz): δ 160.8, 135.3, 134.0, 129.3, 129.1, 114.6, 43.7, 25.9. IR (cm−1) 3281 (NH), 3083, 2965, 2919 (CH), 1648 (CO), 1565 (CC), 1456 (CH2), 1220 (C–O), 1204 (C–O), 804 (p-C Ph), 533 (Cl–C Ph).
Furan-2-carboxaldehyde (15) and 2-cyano-N-(4-chlorobenzyl)acetamide (21b) were reacted as described in general procedure 3 to afford 22b as an orange solid 67%; mp 149–151 °C.
LRMS (ESI+) m/z 287 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 8.10 (s, 1H), 7.73 (d, J = 1.6 Hz, 1H), 7.35–7.31 (m, 2H), 7.27–7.25 (m, 2H), 7.20 (d, J = 3.6 Hz, 1H), 6.64 (dd, J = 3.6, 1.7 Hz, 2H), 4.56 (d, J = 5.9 Hz, 2H). 13C NMR (CDCl3, 101 MHz): δ 160.4, 148.9, 147.9, 137.7, 135.8, 133.8, 129.2, 129.0, 121.4, 116.7, 113.6, 99.5, 43.8. IR (cm−1) 3375 (NH), 3115, 3036, (CH), 2211 (CN), 1668 (CO), 1610 (CC), 1540 (NH bend), 1463 (CH2), 1283 (C–O), 1253 (C–O), 1022 (C–O–C), 826 (p-C Ph), 591 (Cl–C Ph).
LRMS (ESI+) m/z 287 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 7.17 (s, 4H), 6.34 (s, 1H), 4.43 (d, J = 5.6 Hz, 2H), 3.38 (s, 2H), 2.35 (s, 3H). 13C NMR (CDCl3, 101 MHz): δ 160.6, 137.9, 133.7, 129.6, 128.0, 114.6, 44.2, 25.8, 21.1. IR (cm−1) 3289 (NH), 3058, 2928 (CH), 2261 (CN), 1644 (CO), 1545 (CC), 1517 (NH bend), 1463 (CH2), 1364 (CH3), 1229 (C–O), 1062 (C–O–C), 809 (p-C Ph).
Furan-2-carboxaldehyde (15) and 2-cyano-N-(4-methylbenzyl)acetamide (21c) were reacted as described in general procedure 3 to afford 22c as a white solid, 50%; mp 130–132 °C.
LRMS (ESI+) m/z 267 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 8.10 (s, 1H), 7.71 (d, J = 1.6 Hz, 1H), 7.19 (dt, J = 8.0, 3.7 Hz, 4H), 6.62 (dd, J = 3.6, 1.7 Hz, 1H), 6.57 (s, 1H), 4.55 (d, J = 5.7 Hz, 2H), 2.35 (s, 3H). 13C NMR (CDCl3, 101 MHz): δ 160.2, 149.0, 147.7, 137.7, 137.5, 134.1, 129.6, 127.9, 121.1, 116.7, 113.5, 99.8, 44.4, 21.1. IR (cm−1) 3327 (NH), 3117, 3037 (CH), 2225 (CN), 1655 (CO), 1604, 1531 (CC), 1512 (NH bend), 1425 (CH2), 1392 (CH3), 1261 (C–O), 1016 (C–O–C), 810 (p-C Ph).
GC-MS (r.t.) 19.08 min. LRMS (ESI+) m/z 282 (M + 1). 1H NMR (acetone, 400 MHz): δ 8.07 (s, 1H), 7.38 (d, J = 3.5 Hz, 1H), 7.27 (d, J = 8.5 Hz, 2H), 7.22 (s, 1H), 6.90 (d, J = 8.6 Hz, 2H), 6.49–6.38 (m, 1H), 4.45 (s, 2H), 3.79 (s, 3H) 13C NMR (Acetone, 101 MHz): δ 163.2, 159.0, 140.3, 130.6, 128.5, 126.7, 126.3, 117.2, 117.2, 113.5, 112.29, 93.9, 54.3, 42.8. IR (cm−1) 3360 (NH), 3089, 3008, 2936 (CH), 2202 (CN), 1647 (CO), 1611, 1550 (CC), 1510 (NH bend), 1426 (CH2), 1395 (CH3), 1254 (C–O), 1050 (C–O–C), 821 (p-C Ph).
LRMS (ESI+) m/z 286 (M + 1). 1H NMR (DMSO, 400 MHz): δ 11.90 (s, NH), 8.68 (t, J = 5.9 Hz, 1H), 8.07 (s, 1H), 7.45–7.33 (m, 2H), 7.33–7.29 (m, 3H), 6.42 (dd, J = 3.5, 2.6 Hz, 1H), 4.37 (d, J = 6.0 Hz, 2H). 13C NMR (DMSO, 101 MHz): δ 162.4, 140.8, 138.8, 131.8, 129.7, 128.7, 127.0, 126.8, 118.2, 116.1, 113.1, 94.9, 42.9. IR (cm−1) 3349 (NH), 3097, 3025, 2971 (CH), 2205 (CN), 1645 (CO), 1574, 1524 (CC), 1490 (NH bend), 1421 (CH2), 1231 (C–O), 1012 (C–O–C), 810 (p-C Ph), 557 (Cl–C Ph).
LRMS (ESI+) m/z 213 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 9.72 (br s, NH), 7.33–7.28 (m, 2H), 7.22–7.18 (m, 2H), 7.02 (d, J = 1.2 Hz, 1H), 6.99–6.90 (m, 1H), 6.65 (t, J = 3.7 Hz, 1H), 6.32–6.26 (m, 1H). 13C NMR (CDCl3, 101 MHz): δ 164.5, 136.3, 131.9, 127.5, 124.6, 120.8, 119.9, 115.1, 114.9, 111.9, 111.7, 111.1, 100.1. IR (cm−1) 3330 (NH), 3028, 2972 (CH), 2220 (CN), 1608, 1542 (CC), 1512 (NH bend), 1050 (F–C Ph), 759 (m-C Ph).
GC-MS (r.t.) 8.19 min. LRMS (ESI+) m/z 299 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 8.44 (s, 1H), 8.01 (d, J = 5.0 Hz, 1H), 7.91 (d, J = 3.5 Hz, 1H), 7.31 (dd, J = 8.6, 3.7 Hz, 3H), 6.89 (d, J = 8.7 Hz, 2H), 4.51 (d, J = 5.8 Hz, 2H), 3.78 (s, 3H). 13C NMR (CDCl3, 101 MHz): δ 160.3, 159.1, 143.8, 137.2, 136.4, 134.1, 131.0, 129.1, 128.4, 116.3, 113.7, 101.8, 54.6, 43.1. IR (cm−1) 3333 (NH), 3106, 3074, 2836 (CH), 2220 (CN), 1655 (CO), 1615, 1534 (CC), 1512 (NH bend), 1415 (CH2), 1320 (CH3), 1249 (C–O), 1031 (C–O–C), 797 (p-C Ph).
LRMS (ESI+) m/z 283 (M + 1). 1H NMR CDCl3, 400 MHz: δ 8.46 (s, 1H), 7.74 (dd, J = 12.2, 4.4 Hz, 2H), 7.22–7.15 (m, 4H), 6.51 (s, 1H), 4.55 (d, J = 5.7 Hz, 2H), 2.35 (s, 2H), 1.56 (s, 3H). 13C NMR (CDCl3, 101 MHz): δ 160.4, 145.0, 137.7, 136.5, 136.4, 134.2, 134.1, 129.5, 128.5, 127.9, 117.0, 100.4, 44.3, 21.1. IR (cm−1) 3338 (NH), 3073, 3020 (CH), 2217 (CN), 1655 (CO), 1583, 1532 (CC), 1436 (CH2), 1360 (CH3), 808 (p-C Ph).
LRMS (ESI+) m/z 251 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 7.60–7.56 (m, 5H), 7.46–7.43 (m, 2H), 7.40–7.34 (m, 2H), 6.41 (s, 1H), 4.53 (d, J = 5.7 Hz, 2H), 3.43 (s, 2H). 13C NMR (CDCl3, 101 MHz): δ 160.7, 141.1, 140.5, 135.7, 128.9, 128.4, 127.7, 127.5, 127.1, 114.6, 44.1, 25.9. IR (cm−1) 3312 (NH), 3098, 3055, 3034, 2960, 2921 (CH), 2259 (CN), 1656 (CO), 1557 (CC), 1450 (CH2), 816 (p-C Ph).
Thiophene-2-carboxaldehyde (0.2 g, 1.8 mmol) and 3-fluorophenylacetonitrile (0.45 g, 1.8 mmol) were added together in EtOH (4 mL). Piperidine (cat.) was added and the reaction was irradiated with microwaves (120 °C, 200 W) for 20 min and then placed in the freezer for 60 min. The solution was filtered and washed with cold EtOH and dried under suction to afford 22g as an orange solid; 57%; mp 183–185 °C.
LRMS (ESI+) m/z 345 (M + 1). 1H NMR (DMSO-d6, 400 MHz): δ 8.98 (s, 1H), 8.47 (s, 1H), 8.11 (d, J = 5.0 Hz, 1H), 7.91 (d, J = 3.2 Hz, 1H), 7.64 (dd, J = 7.6, 6.1 Hz, 4H), 7.49–7.40 (m, 4H), 7.39–7.29 (m, 2H), 4.46 (d, J = 5.7 Hz, 2H). 13C NMR (DMSO-d6, 101 MHz): δ 161.5, 144.2, 140.4, 139.4, 138.7, 138.5, 136.3, 135.5, 129.4, 129.1, 128.5, 127.8, 127.1, 127.1, 117.0, 102.0, 43.4. IR (cm−1) 3323 (NH), 3104, 3086, 3025, 2971 (CH), 2221 (CN), 1655 (CO), 1583, 1537 (CC), 1414 (CH2), 810 (p-C Ph).
General procedure 4 – concurrent olefin and furan reduction
MS (ESI+) m/z 285 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 7.21–7.16 (m, 2H), 6.85–6.83 (m, 2H), 6.77 (br s, 1H), 4.43–4.30 (m, 2H), 4.05–3.86 (m, 1H), 3.83–3.78 (m, 1H), 3.77 (s, 3H), 3.75–3.66 (m, 1H), 3.55–3.53 (m, 1H), 2.24–2.17 (m, 1H), 2.09–1.95 (m, 2H), 1.92–1.81 (m, 2H), 1.55–1.54 (m, 1H). 13C NMR (CDCl3, 101 MHz): δ 164.7, 159.3, 129.4, 129.3, 118.2, 114.2, 75.8, 67.9, 55.4, 43.8, 36.3, 35.9, 31.4, 25.7. IR (cm−1) 3303, 3251 (NH), 3077, 2955, 2930, 2830 (CH), 2253 (CN), 1644 (CO), 1611 (CC), 1552 (NH bend), 1461, 1436 (CH2), 1351 (CH3), 1298 (C–O), 1244 (C–O), 1029 (C–O–C), 826 (p-C Ph).
General procedure 5 – selective olefin reduction
LRMS (ESI+) m/z 285 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 7.32 (d, J = 1.1 Hz, 1H), 7.15 (d, J = 8.6 Hz, 2H), 6.88–6.83 (m, 2H), 6.34 (br s, NH), 6.31 (dd, J = 3.1, 1.9 Hz, 1H), 6.22 (d, J = 3.2 Hz 1H), 4.47–4.32 (m, 2H), 3.80 (s, 3H), 3.70 (dd, J = 7.8, 5.6 Hz, 1H), 3.32 (m, 2H). 13C NMR (CDCl3, 101 MHz): δ 163.4, 159.5, 149.4, 142.6, 129.4, 129.0, 117.6, 114.4, 110.7, 108.6, 55.5, 44.1, 38.2, 28.8. IR (cm−1) 3305 (NH), 3121, 3020, 2931, 2834 (CH), 2251 (CN), 1642 (CO), 1615 (CC), 1555 (CC), 1461 (CH2), 1434 (CH2), 1247 (CH3), 1223 (C–O), 813 (p-C Ph).
General procedure 6 – selective nitrile and olefin reduction
LRMS (ESI+) m/z 289 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 7.27 (dd, J = 1.9, 0.8 Hz, 1H), 7.12 (d, J = 8.7 Hz, 2H), 6.82 (dd, J = 8.6, 1.9 Hz, 2H), 6.26 (dd, J = 3.1, 1.9 Hz, 1H), 6.09–5.96 (m, 1H), 4.38 (dd, J = 14.6, 5.9 Hz, 1H), 4.29 (dd, J = 14.6, 5.4 Hz, 1H), 3.78 (s, 3H), 3.02 (dd, J = 15.1, 7.5 Hz, 1H), 2.92 (dd, J = 12.7, 8.3 Hz, 1H), 2.88–2.80 (m, 1H), 2.76 (d, J = 15.0 Hz, 1H), 2.56 (dd, J = 17.4, 9.9 Hz, 1H), 1.46 (s, 2H). 13C NMR (CDCl3, 101 MHz): δ 173.8, 158.9, 153.3, 141.3, 130.6, 129.0, 114.0, 110.3, 106.6, 55.3, 48.4, 43.3, 42.8, 28.6. IR (cm−1) 3285 (NH2), 3065, 2933, 2836 (CH), 1643 (CO), 1612 (CC), 1548, 1511 (NH bend), 1463, 1440 (CH2), 1301 (CH3), 1243 (C–O), 1175 (C–O), 1030 (C–O–C), 808 (p-C Ph).
LRMS (ESI+) m/z 293 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 7.33–7.27 (m, 2H), 7.24 (d, J = 1.8 Hz, 1H), 7.11 (d, J = 8.4 Hz, 2H), 6.26 (dd, J = 3.0, 2.0 Hz, 1H), 6.04 (d, J = 2.9 Hz, 1H), 4.45 (dd, J = 9.6, 5.7 Hz, 1H), 4.29 (dd, J = 15.1, 5.4 Hz, 1H), 3.06–3.01 (m, 1H), 2.94 (dt, J = 21.5, 8.4 Hz, 2H), 2.80 (dd, J = 15.0, 7.2 Hz, 1H), 2.67 (dd, J = 7.8, 4.3 Hz, 1H), 2.39 (br s, 2H). 13C NMR (CDCl3, 101 MHz): δ 173.9, 152.9, 141.4, 137.0, 129.0, 128.7, 127.6, 110.4, 106.8, 47.5, 42.9, 42.6, 28.6. IR (cm−1) 3330 (NH), 3121, 3014, 2931, 2834 (CH), 16412 (CO), 1615 (CC), 1552 (CC), 1462, 1430 (CH2), 1223 (C–O), 813 (p-C Ph), 547 (Cl–C Ph).
LRMS (ESI+) m/z 273 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 7.28–7.26 (m, 1H), 7.19–7.09 (m, 4H), 6.27 (t, J = 2.2 Hz, 1H), 6.02 (d, J = 3.1 Hz, 1H), 4.43–4.31 (m, 2H), 3.04 (dd, J = 15.1, 7.4 Hz, 1H), 2.92 (dd, J = 12.7, 8.4 Hz, 1H), 2.85 (dd, J = 7.8, 4.8 Hz, 1H), 2.77 (dd, J = 15.1, 7.4 Hz, 1H), 2.56 (qd, J = 7.6, 4.1 Hz, 1H), 2.32 (s, 3H), 1.35 (br s, NH). 13C NMR (CDCl3, 101 MHz): δ 173.9, 153.3, 141.3, 136.9, 135.4, 129.26, 127.6, 110.3, 106.6, 48.4, 43.3, 43.1, 28.6, 21.1. IR (cm−1) 3305 (NH), 3121, 3020, 2931, 2834 (CH), 2251 (CN), 1642 (CO), 1615 (CC), 1555 (CC), 1461 (CH2), 1434 (CH2), 1247 (CH3), 1223 (C–O), 813 (p-C Ph) 550 (Cl–C Ph).
LRMS (ESI+) m/z 288 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 9.05 (s, NH), 7.76 (s, NH), 7.11 (d, J = 8.7 Hz, 2H), 6.86–6.80 (m, 2H), 6.61 (dd, J = 4.1, 2.6 Hz, 1H), 6.07–6.05 (m, 1H), 5.88 (s, 1H), 4.40–4.29 (m, 2H), 3.78 (s, 3H), 2.98–2.83 (m, 3H), 2.82–2.77 (m, 1H), 2.46–2.40 (m, 1H), 1.40 (s, 2H). 13C NMR (CDCl3, 101 MHz): δ 175.2, 158.9, 130.5, 129.5, 128.8, 116.9, 114.0, 107.6, 106.3, 55.3, 48.7, 43.9, 42.81, 27.9. IR (cm−1) 3310 (NH), 3101, 3025, 2899, 2835 (CH), 1642 (CO), 1618 (CC), 1532 (CC), 1463 (CH2), 1434 (CH2), 1249 (CH3), 1223 (C–O), 813 (p-C Ph).
LRMS (ESI+) m/z 292 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 8.97 (s, NH), 8.03 (s, NH), 7.26 (t, J = 3.2 Hz, 2H), 7.10 (d, J = 8.4 Hz, 2H), 6.62 (dd, J = 4.1, 2.6 Hz, 1H), 6.07 (dd, J = 5.6, 2.8 Hz, 1H), 5.89 (s, 1H), 4.38 (qd, J = 15.1, 5.9 Hz, 2H), 3.12–2.64 (m, 4H), 2.47 (td, J = 9.1, 4.5 Hz, 1H), 1.57 (s, 2H). 13C NMR (CDCl3, 101 MHz): δ 175.4, 137.0, 133.0, 128.8, 128.8, 117.0, 107.7, 106.4, 48.4, 43.9, 42.6, 27.9. IR (cm−1) 3287 (NH), 3084, 3035 2923, 2860 (CH), 1644 (CO), 1540, 1514 (CC), 1434 (CH2), 808 (p-C Ph), 519 (Cl–C Ph).
LRMS (ESI+) m/z 219 (M + 1). 1H NMR (Acetone, 400 MHz): δ 8.60 (s, 1H), 7.39–7.22 (m, 1H), 7.05–6.87 (m, 3H), 6.61 (dd, J = 3.9, 2.4 Hz, 1H), 6.10 (dd, J = 5.5, 2.8 Hz, 1H), 5.88 (s, 1H), 3.06–2.82 (m, 5H). 13C NMR (acetone, 101 MHz): δ 164.3, 146.0, 130.2, 129.7, 123.6, 116.6, 114.5, 113.6, 108.1, 106.4, 49.5, 46.9, 32.3. IR (cm−1) 3288 (NH), 3029, 2965 (CH), 1612, 1515 (CC), 1049 (F–C Ph), 759 (m-C Ph).
LRMS (ESI+) m/z 305 (M + 1). 1H NMR (acetone, 400 MHz): δ 7.59 (s, 1H), 7.23 (dd, J = 5.1, 1.0 Hz, 2H), 7.13 (d, J = 8.7 Hz, 2H), 6.91 (dd, J = 5.1, 3.4 Hz, 1H), 6.83–6.81 (m, 2H), 4.29 (dd, J = 25.4, 5.9 Hz, 2H), 3.76 (d, J = 1.2 Hz, 3H), 3.74 (d, J = 0.9 Hz, 1H), 3.41 (dd, J = 14.0, 7.9 Hz, 1H), 3.30 (d, J = 7.5 Hz, 1H), 3.21–3.16 (m, 1H), 3.04 (dd, J = 14.6, 6.2 Hz, 1H), 1.28 (s, 2H). 13C NMR (Acetone, 101 MHz): δ 173.2, 158.7, 142.6, 131.6, 128.6, 128.5, 126.6, 125.4, 123.43, 113.5, 54.6, 53.0, 50.1, 41.9, 30.3. IR (cm−1) 3285 (NH), 3106, 3056, 2834 (CH), 1645 (CO), 1612, 1534 (CC), 1415 (CH2), 1319 (CH3), 1250 (C–O), 1030 (C–O–C), 812 (p-C Ph).
LRMS (ESI+) m/z 289 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 7.12 (dd, J = 5.1, 1.0 Hz, 1H), 7.08 (d, J = 7.8 Hz, 2H), 7.02 (d, J = 7.9 Hz, 2H), 6.90 (dd, J = 5.1, 3.5 Hz, 1H), 6.79 (d, J = 3.0 Hz, 1H), 4.39–4.31 (m, 2H), 3.28–3.13 (m, 1H), 3.04–2.88 (m, 2H), 2.53–2.42 (m, 1H), 2.31 (s, 3H), 2.28 (dd, J = 11.7, 5.2 Hz, 1H), 1.38–1.17 (m, 2H). 13C NMR (CDCl3, 101 MHz): δ 173.7, 141.9, 135.3, 129.3, 127.6, 126.9, 125.8, 123.8, 114.1, 51.7, 43.3, 43.2, 30.3, 21.1. IR (cm−1) 3238 (NH), 3054, 3020 (CH), 1650 (CO), 1580, 1512 (CC), 1426 (CH2), 1360 (CH3), 808 (p-C Ph).
LRMS (ESI+) m/z 340 (M + 1). 1H NMR (CDCl3, 400 MHz): δ 7.56–7.48 (m, 3H), 7.45–7.32 (m, 5H), 7.21 (d, J = 8.1 Hz, 1H), 7.13 (dd, J = 3.8, 1.3 Hz, 1H), 6.98–6.86 (m, 1H), 6.82 (d, J = 3.0 Hz, 1H), 4.51–4.38 (m, 2H), 3.26 (dd, J = 9.3, 5.2 Hz, 1H), 3.02–2.91 (m, 2H), 2.83 (d, J = 12.0 Hz, 1H), 2.51 (dd, J = 7.4, 4.3 Hz, 1H). 13C NMR (CDCl3, 101 MHz): δ 173.8, 140.8, 140.3, 137.8, 137.4, 128.8, 128.1, 128.0, 127.4, 127.1, 123.8, 31.9, 30.2, 22.5, 14.0. IR (cm−1) 3287 (NH), 3060, 2930, 2858 (CH), 1643 (CO), 1612, 1511 (CC), 1462 (CH2), 1440 (CH2), 818 (p-C Ph).
General procedure 7 – reaction path B
MS (ESI−) m/z 455 (M − 1). 1H NMR (DMSO-d6, 400 MHz): δ 11.91 (br s, OH), 8.21 (t, J = 5.4 Hz, NH), 7.49 (d, J = 1.0 Hz, NH), 7.40 (t, J = 5.9 Hz, NH), 7.07 (d, J = 8.6 Hz, 2H), 6.83 (d, J = 8.6 Hz, 2H), 6.34 (dd, J = 3.0, 1.9 Hz, 1H), 6.08 (d, J = 2.9 Hz, 1H), 4.73 (d, J = 3.7 Hz, 1H), 4.40 (d, J = 3.8 Hz, 1H), 4.22 (dd, J = 14.9, 6.0 Hz, 1H), 4.11 (dd, J = 14.9, 5.6 Hz, 1H), 3.72 (s, 3H), 3.24–3.18 (m, 1H), 3.07–2.99 (m, 1H), 2.83 (s, 2H), 2.77–2.68 (m, 3H), 1.57–1.40 (m, 4H). 13C NMR (DMSO-d6, 101 MHz) δ 173.0, 172.7, 171.3, 158.6, 153.7, 141.9, 131.7, 128.9, 114.0, 110.8, 106.6, 79.3, 77.2, 55.5, 53.6, 52.0, 45.2, 42.0, 41.3, 29.2, 28.8, 28.6. IR (cm−1) 3295 (NH), 3091 (OH), 2985, 2937 (CH), 1692 (CO), 1651 (CC), 1562, 1514 (NH bend), 1302 (CH3), 1247 (C–O), 1032 (CH3), 818 (p-C Ph), 731 (CH2 bend).
:1 ratio); 0.3 g, overall yield 56%; mp 166 °C.MS (ESI−) m/z 459 (M − 1). 1H NMR (DMSO-d6, 400 MHz): δ 11.91 (br s, OH), 8.45 (t, J = 6.0 Hz, NH), 7.66 (t, J = 5.8 Hz, NH), 7.50 (d, J = 1.1 Hz, 1H), 7.32 (dd, J = 8.3, 1.5 Hz, 1H), 7.16 (t, J = 8.6 Hz, 2H), 6.35 (t, J = 11.7, 2.9 Hz, 1H), 6.09 (dd, J = 11.7, 2.9 Hz, 1H), 4.72 (dd, J = 3.5, 2.4 Hz, 1H), 4.40 (d, J = 3.4 Hz, 1H), 4.30–4.14 (m, 2H), 3.27–3.18 (m, 2H), 3.06–3.01 (m, 1H), 2.85–2.67 (m, 5H), 1.56–1.47 (m, 4H). 13C NMR (DMSO-d6, 101 MHz): δ 173.1, 173.0*, 171.3, 171.2*, 153.6, 153.6*, 142.0, 139.0, 138.9*, 131.6, 129.5, 129.4*, 128.5, 127.6, 110.8, 106.7, 106.6*, 79.3, 79.2*, 77.2, 77.2*, 53.6, 53.3*, 51.9, 51.6*, 45.3, 45.0*, 41.8, 41.3*, 29.4, 29.2*, 28.9, 28.8, 28.6*. IR (cm−1) 3309 (NH), 3063 (OH), 2984, 2967, 2920, 2879 (CH), 1729 (CO), 1692 (CO), 1647 (CC), 1537 (NH bend), 1247 (C–O), 1183 (C–O), 819 (p-C Ph), 737 (CH2 bend), 696 (C–Cl). *Diastereomers peaks.
:1 ratio); 0.42 g, overall yield 60%; mp 166–167 °C.MS (ESI−) m/z 439 (M − 1). 1H NMR (DMSO-d6, 400 MHz): δ 11.92 (br s, OH), 8.37 (t, J = 5.6 Hz, NH), 8.22 (t, J = 5.5 Hz, NH)*, 7.62 (t, J = 5.6 Hz, NH), 7.42 (t, J = 5.4 Hz, NH)*, 7.12–7.01 (m, 4H), 6.34 (s, 1H), 6.10 (dd, J = 12.0, 2.7 Hz, 1H), 4.72 (d, J = 10.0 Hz, 1H), 4.41 (s, 1H), 4.27–4.13 (m, 2H), 3.25–3.21 (m, 2H), 3.06–3.03 (m, 2H), 2.85–2.68 (m, 5H), 2.26 (s, 3H), 1.56–1.40 (m, 4H). 13C NMR (DMSO-d6, 101 MHz) δ 173.0, 172.9*, 172.9, 172.8*, 171.3, 171.2*, 153.7, 153.7*, 141.9, 141.9*, 136.8, 136.7*, 136.1, 136.0*, 129.2, 127.6*, 110.8, 106.5, 106.6*, 79.3, 79.2*, 77.2, 77.2*, 53.6, 53.3*, 52.0, 51.7*, 45.2, 45.0*, 42.3, 41.3, 31.2, 29.4, 29.2*, 28.9, 28.8*, 28.8, 28.6*, 21.1. IR (cm−1) 3306 (NH), 3001 (OH), 2984, 2947, 2919, 2871 (CH), 1729 (CO), 1692 (CO), 1644 (CC), 1537 (NH bend), 1381 (CH3), 1251 (C–O), 1180 (C–O), 835 (p-Ar), 738 (CH2 bend). *Diastereomers peaks.
General procedure 8 – reaction path B, with selective nitrile and olefin reduction of thiophene and pyrrole based analogues
: EtoAc (1:1) (10 mL) to form a 0.05 M solution. This solution was hydrogenated using the H-Cube Pro™ with a RaNi catalyst at 1 mL min−1 flow rate, 70 °C and 70 bar H2 pressure. The solvent was removed in vacuo and to afford 2-((1H-pyrrol-2-yl)methyl)-3-amino-N-(4-methoxybenzyl)propanamide as a yellow oil, 0.11 g, 77%. The crude product and norcantharidin (2) (0.20 g, 1.8 mmol) were dissolved separately in acetone (10 mL × 2) and the solutions were added together and the reaction was left to stir at room temperature for 4 hours. The solution was filtered and washed with cold acetone and dried under suction to afford 24d as a white solid; 0.18 g, overall yield 26%; mp 166 °C.MS (ESI−) m/z 517 (M − 1). 1H NMR (DMSO-d6, 400 MHz): δ 11.89 (br s, OH), 10.39 (s, NH), 8.20 (t, J = 5.5 Hz, NH), 8.07 (t, J = 5.3 Hz, NH)*, 7.50 (t, J = 5.4 Hz, NH), 7.29 (t, J = 5.3 Hz, NH)*, 7.07 (d, J = 8.4 Hz, 2H), 6.82 (d, J = 8.5 Hz, 2H), 6.55 (s, 2H), 5.90–5.75 (m, 2H), 4.71 (s, 1H), 4.39 (s, 1H), 4.23–4.13 (m, 2H), 3.71 (s, 3H), 3.21–3.15 (m, 1H), 3.07 (dd, J = 12.0, 5.7 Hz, 1H), 2.86–2.61 (m, 5H), 1.55–1.39 (m, 4H). 13C NMR (DMSO-d6, 101 MHz): δ 174.0, 173.0, 172.8, 171.2, 142.1, 142.02, 140.5, 139.4, 139.1, 139.0, 129.4, 128.4, 128.3, 128.2, 127.8, 127.2, 127.0, 126.9, 126.0, 124.5, 79.3, 79.2, 77.2, 53.7, 53.3, 51.8, 48.4, 48.1, 42.2, 41.4, 31.7, 30.4, 29.4, 29.2, 28.9, 26.8, 22.5, 14.3. IR (cm−1) 3432 (NH), 3306 (NH), 3074 (OH), 2992, 2937, 2882, 2834 (CH), 1685 (CO), 1653 (CO), 1635 (CC), 1538, 1513 (NH bend), 1302 (CH3), 1245, 1223 (C–O), 806 (p-C Ph). *Diasteromer peaks.
:0.7 ratio); 0.1 g, overall yield 27%; mp 129–131 °C.MS (ESI−) m/z 459 (M − 1). 1H NMR (DMSO-d6, 400 MHz): δ 11.94 (br s, OH), 8.45 (t, J = 6.1 Hz, NH), 8.30 (t, J = 5.8 Hz, NH), 7.67 (t, J = 5.6 Hz, NH), 7.43 (t, J = 5.7 Hz, NH), 7.32 (d, J = 8.3 Hz, 2H), 7.17 (t, J = 8.5 Hz, 2H), 6.14 (d, J = 2.9 Hz, 1H), 6.01 (dd, J = 12.6, 2.9 Hz, 1H), 5.11 (t, J = 5.5 Hz, 1H), 4.74 (d, J = 3.1 Hz, 1H), 4.71 (s, 1H), 4.41 (d, J = 12.9 Hz, 1H), 4.31 (d, J = 5.2 Hz, 2H), 4.29–4.14 (m, 2H), 3.26–3.20 (m, 1H), 3.06 (d, J = 5.4 Hz, 1H), 2.87–2.67 (m, 5H), 1.57–1.41 (m, 4H). 13C NMR (DMSO-d6, 101 MHz): δ 173.5, 173.1*, 173.0, 172.9*, 171.3, 171.2*, 154.4, 152.9, 152.8*, 139.0, 138.9*, 131.6, 129.5, 129.4*, 128.6, 128.5*, 127.6, 108.0, 107.1*, 80.1, 79.3, 79.2*, 77.2, 56.1, 53.6, 53.2*, 51.9, 51.6*, 51.2, 45.1, 44.9*, 41.9, 41.3*, 31.2, 29.2*, 28.9, 28.8*, 28.8, 27.9*. IR (cm−1) 3292 (NH), 3087 (OH), 2987, 2984, 2920, 2875 (CH), 1702 (CO), 1646 (CC), 1545, 1494 (NH bend), 1240, 1200 (C–O), 1170 (C–O), 799 (p-C Ph). *Diastereomers peaks.
MS (ESI−) m/z 385 (M − 1). 1H NMR (CDCl3, 400 MHz): δ 8.35 (br s, NH), 7.27–7.24 (m, 1H), 6.94 (td, J = 8.1, 2.2 Hz, 2H), 6.90–6.85 (m, 1H), 6.63 (dd, J = 4.0, 2.8 Hz, 1H), 6.06 (dd, J = 6.0, 2.8 Hz, 1H), 5.77 (s, 1H), 4.83 (dt, J = 5.2, 2.8 Hz, 2H), 3.77 (dd, J = 13.4, 8.4 Hz, 1H), 3.66 (dd, J = 13.4, 6.4 Hz, 1H), 3.42–3.33 (m, 1H), 2.91 (d, J = 6.9 Hz, 2H), 2.76 (d, J = 7.3 Hz, 2H), 1.90–1.83 (m, 2H), 1.60 (q, J = 6.4 Hz, 2H). 13C NMR (CDCl3, 101 MHz): δ 177.4, 177.4, 162.8 (d, J = 246 Hz), 143.9, 130.0, 128.8, 123.5, 116.7, 114.7, 114.1, 108.3, 106.9, 79.1, 77.2, 49.8, 49.8, 43.6, 31.4, 28.6, 28.5.
MS (ESI−) m/z 471 (M − 1).1 H NMR (DMSO-d6, 400 MHz): δ 11.89 (br s, 1H), 8.33 (t, J = 4.7 Hz, NH), 8.20 (t, J = 5.6 Hz, NH)*, 7.58 (t, J = 5.9 Hz, NH), 7.40 (t, J = 6.2 Hz, NH)*, 7.30 (d, J = 5.2 Hz, 1H), 7.25 (t, J = 6.0 Hz, 1H)*, 7.17 (d, J = 8.5 Hz, 2H), 7.03 (t, J = 8.2 Hz, 2H), 6.98–6.87 (m, 1H), 6.90–6.74 (m, 3H), 4.72 (d, J = 9.9 Hz, 1H), 4.41 (d, J = 2.8 Hz, 1H)*, 4.37 (d, J = 4.0 Hz, 1H)*, 4.23–4.09 (m, 2H), 3.71 (s, 3H), 3.26–3.17 (m, 1H), 3.05 (dd, J = 12.8, 7.5 Hz, 1H), 3.01–2.93 (m, 1H), 2.93–2.66 (m, 4H), 1.55–1.38 (m, 4H). 13C NMR (DMSO-d6, 101 MHz): 172.9, 172.8, 171.2, 158.5, 142.1, 142.0*, 131.8, 129.0*, 128.9, 127.2, 126.0, 124.4, 114.0, 79.2, 77.3, 55.5, 53.4, 48.0, 42.0, 41.3, 30.4, 29.4, 28.9, 22.5. IR (cm−1) 3308 (NH), 3078 (OH), 2984, 2964, 2921, 2874 (CH), 1694 (CO), 1646 (CC), 1545, (NH bend), 1382 (CH3), 1242 (C–O), 817 (p-C Ph). * Diastereomers peaks.
:1 ratio); 0.13 g, overall yield 38%; mp 161–163 °C.MS (ESI−) m/z 455 (M − 1). 1H NMR (DMSO-d6, 400 MHz): δ 11.91 (br s, OH), 8.33 (t, J = 5.8 Hz, NH)*, 8.2 (t, J = 5.8 Hz, NH), 7.61 (t, J = 5.7 Hz, NH)*, 7.41 (t, J = 5.8 Hz, NH), 7.32 (dd, J = 5.1, 0.8 Hz, 1H), 7.05 (d, J = 7.9 Hz, 2H), 6.98 (t, J = 8.5 Hz, 2H), 6.94–6.90 (m, 1H), 6.83 (dd, J = 8.9, 2.9 Hz, 1H), 4.75–4.69 (m, 1H)*, 4.42 (d, J = 3.3 Hz, 1H), 4.27–4.11 (m, 2H), 3.27–3.19 (m, 1H), 3.09–2.95 (m, 2H), 2.89 (dd, J = 12.3, 6.9 Hz, 1H), 2.84–2.67 (m, 2H), 2.51–2.49 (m, 2H), 2.25 (s, 3H), 1.57–1.40 (m, 4H). 13C NMR (DMSO-d6, 101 MHz): δ 172.9, 172.8*, 172.7, 171.2*, 171.1, 142.1, 142.0*, 136.8, 136.6*, 136.1, 136.0*, 129.1, 127.6, 127.2*, 126.0, 124.4, 80.1, 79.3, 79.2*, 77.2, 77.2*, 53.6, 53.3*, 51.9, 51.7*, 51.2, 48.3, 48.0*, 42.3, 41.3*, 30.4, 30.2*, 29.4, 29.2*, 28.9, 28.8*, 21.1. IR (cm−1) 3310 (NH), 3062 (OH), 2986, 2963, 2920, 2874 (CH), 1695 (CO), 1646 (CC), 1545, (NH bend), 1382 (CH3), 1244 (C–O), 816 (p-C Ph). *Diastereomers peaks.
:1 ratio); 0.23 g, overall yield 46%; mp 171–174 °C.1H NMR (DMSO-d6, 400 MHz): δ 11.89 (br s, OH), 8.44 (t, J = 5.3 Hz, NH), 8.31 (t, J = 5.6 Hz, NH)*, 7.63 (d, J = 7.9 Hz, 2H), 7.61 (d, J = 2.1 Hz, 1H)*, 7.59 (d, J = 2.3 Hz, 1H), 7.54 (d, J = 8.2 Hz, 2H), 7.46 (d, J = 7.6 Hz, 2H), 7.35 (dd, J = 8.8, 6.2 Hz, 2H), 7.18 (t, J = 8.6 Hz, 2H), 6.97–6.91 (m, 1H), 6.85 (dd, J = 8.7, 3.2 Hz, 1H), 4.73 (d, J = 11.1 Hz, 1H), 4.50–4.40 (m, 1H), 4.37–4.09 (m, 2H). 3.25 (d, J = 7.0 Hz, 1H), 3.08 (dd, J = 8.0, 5.2 Hz, 1H), 2.96–2.71 (m, 5H), 1.46–1.37 (m, 2H), 1.27–1.17 (m, 2H). 13C NMR (DMSO-d6, 101 MHz): δ 174.0, 173.0, 172.8, 171.2, 142.1, 142.02, 140.5, 139.4, 139.1, 139.0, 129.4, 128.4, 128.3, 128.2, 127.8, 127.2, 127.0, 126.9, 126.0, 124.5, 79.3, 79.2, 77.2, 53.7, 53.3, 51.8, 48.4, 48.1, 42.2, 41.4, 31.7, 30.4, 29.4, 29.2, 28.9, 26.8, 22.5, 14.3. IR (cm−1) 3316 (NH), 3070 (OH), 3033, 2985, 2923, 2876 (CH), 1691 (CO), 1646 (CC), 1534, 1487 (NH bend), 1242, (C–O), 819 (p-C Ph). *Diastereomers peaks.
Acknowledgements
This project as supported by the Australian Cancer Research Foundation, Ramaciotti Foundation and the Australian Research Council. CPG is the recipient of an ARC DECRA fellowship. LH acknowledges the receipt of a Postgraduate Scholarship from the University of Newcastle.Notes and references
- L. Moed, T. A. Shwayder and M. W. Chang, Arch. Dermatol. Res., 2001, 137, 1357–1360 CAS.
- T. Eisner, S. R. Smedley, D. K. Young, M. Eisner and B. Roach, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 6499–6503 CrossRef CAS.
- G.-S. Wang, J. Ethnopharmacol., 1989, 26, 147 CrossRef CAS.
- S. S. Taylor and A. P. Kornev, Trends Biochem. Sci., 2011, 36, 65 CrossRef CAS PubMed.
- Y. Shi, Cell, 2009, 139, 468 CrossRef CAS PubMed.
- X. Li, S.-L. Zheng, X. Li, J.-L. Li, O. Qiang, R. Liu and L. He, Eur. J. Med. Chem., 2012, 54, 42 CrossRef CAS PubMed.
- R. S. de Jong, E. G. E. de Vries, S. Meijer, P. E. de Jong and N. H. Mulder, Cancer Chemother. Pharmacol., 1998, 42, 160–164 CrossRef CAS.
- F. Massicot, H. Dutertre-Catella, C. Pham-Huy, X.-H. Liu, H. T. Duc and J.-M. Warnet, Basic Clin. Pharmacol. Toxicol., 2005, 96, 26 CrossRef CAS PubMed.
- J. H. Cao, B. Xu, D. Z. Wu, W. Huang and J. R. Cui, Ai Zheng., 2007, 26, 361 CAS.
- D. Liu and Z. Chen, Anti-Cancer Agents Med. Chem., 2009, 9, 1871 CrossRef.
- L. P. Deng, J. Dong, H. Cai and W. Wang, Curr. Med. Chem., 2013, 20, 159 CrossRef CAS.
- L. Deng, J. Dong and W. Wang, Mini-Rev. Med. Chem., 2013, 13, 1166–1176 CrossRef CAS.
- J. A. Sakoff, S. P. Ackland, M. L. Baldwin, M. A. Keane and A. McCluskey, Invest. New Drugs, 2010, 11, 1 Search PubMed.
- J. A. Sakoff and A. McCluskey, Curr. Pharm. Des., 2004, 10, 1139 CrossRef CAS.
- A. McCluskey, A. T. R. Sim and J. A. Sakoff, J. Med. Chem., 2002, 45, 1151–1175 CrossRef CAS.
- L. H. Zheng, Y. L. Bao, Y. Wu, C. L. Yu, X. Meng and Y. X. Li, Cancer Lett., 2008, 272, 102–109 CrossRef CAS PubMed.
- C.-H. Hsieh, K. S. C. Chao, H.-F. Liao and Y.-J. Chen, J. Evidence-Based Complementary Altern. Med., 2013, 838651 Search PubMed.
- J. Bajsa, A. McCluskey, C. P. Gordon, S. G. Stewart, T. A. Hill, R. Sahu, S. O. Duke and B. L. Tekwani, Bioorg. Med. Chem. Lett., 2010, 20, 6688–6695 CrossRef CAS PubMed.
- B. E. Campbell, M. Tarleton, C. P. Gordon, J. A. Sakoff, J. Gilbert, A. McCluskey and R. B. Gasser, Bioorg. Med. Chem. Lett., 2011, 21, 3277–3281 CrossRef CAS PubMed.
- Y. Baba, N. Hirukawa, N. Tanohira and M. Sodeoka, J. Am. Chem. Soc., 2003, 125, 9740 CrossRef CAS PubMed.
- C. E. Puerto Galvis, L. Y. Vargas Méndez and V. V. Kouznetsov, Chem. Biol. Drug Des., 2013, 82, 477 CAS.
- T. A. Hill, S. G. Stewart, C. P. Gordon, S. P. Ackland, J. Gilbert, B. Sauer, J. A. Sakoff and A. McCluskey, ChemMedChem, 2008, 3, 1878 CrossRef CAS PubMed.
- S. G. Stewart, T. A. Hill, J. Gilbert, S. P. Ackland, J. A. Sakoff and A. McCluskey, Bioorg. Med. Chem., 2007, 15, 7301 CrossRef CAS PubMed.
- M. Tarleton, J. Gilbert, J. A. Sakoff and A. McCluskey, Eur. J. Med. Chem., 2012, 54, 573 CrossRef CAS PubMed.
- B. Sauer, J. Gilbert, J. A. Sakoff and A. McCluskey, Lett. Drug Des. Discovery, 2009, 6, 1 CrossRef CAS.
- L. Deng, Z. Yong, W. Tao, J. Shen and W. Wang, J. Heterocycl. Chem., 2010, 48, 158 CrossRef.
- A. Thaqi, J. L. Scott, J. Gilbert, J. A. Sakoff and A. McCluskey, Eur. J. Med. Chem., 2010, 45, 1717 CrossRef CAS PubMed.
- A. McCluskey, M. A. Keane, L.-M. Mudgee, A. T. R. Sim and J. Sakoff, Eur. J. Med. Chem., 2000, 35, 957 CrossRef CAS.
- C. P. Gordon, N. Byrne and A. McCluskey, Green Chem., 2010, 12, 1000 RSC.
- K. Paulvannan and J. W. Jacobs, Tetrahedron, 1999, 55, 7433 CrossRef CAS.
- K. Paulvannan, Tetrahedron Lett., 1999, 40, 1851 CrossRef CAS.
- P. S. Sarang, A. A. Yadav, P. S. Patil, U. Murali Krishna, G. K. Trivedi and M. M. Salunkhe, Synthesis, 2007, 1091 CAS.
- C. W. Laidley, W. G. Dauben, Z. R. Guo, J. Y. L. Lam and J. E. Casida, Bioorg. Med. Chem., 1999, 7, 2937 CrossRef CAS.
- M. Tarleton, J. Gilbert, M. J. Robertson, A. McCluskey and J. A. Sakoff, MedChemComm, 2011, 2, 31 RSC.
- M. Tarleton, L. Dyson, J. Gilbert, J. A. Sakoff and A. McCluskey, Bioorg. Med. Chem., 2013, 21, 333 CrossRef CAS PubMed.
- M. Tarleton, J. Gilbert, J. A. Sakoff and A. McCluskey, Eur. J. Med. Chem., 2012, 57, 65 CrossRef CAS PubMed.
- A. McCluskey and J. A. Sakoff, Mini-Rev. Med. Chem., 2001, 1, 43 CrossRef CAS.
- B. H. Wojcik, Ind. Eng. Chem., 1948, 40, 210 CrossRef CAS.
- J. Kijeński, P. Winiarek, T. Paryjczak, A. Lewicki and A. Mikołajska, Appl. Catal., A, 2002, 233, 171 CrossRef.
- M. Maris, T. Bürgi, T. Mallat and A. Baiker, J. Catal., 2004, 226, 393 CrossRef CAS PubMed.
- P. Feiertag, M. Albert, U. Nettekoven and F. Spindler, Org. Lett., 2006, 8, 4133–4135 CrossRef CAS PubMed.
- M. Maris, T. Mallat, E. Orglmeister and A. Baiker, J. Mol. Catal., 2004, 219, 371 CrossRef CAS PubMed.
- M. Maris, W. R. Huck, T. Mallat and A. Baiker, J. Catal., 2003, 219, 52–58 CrossRef CAS.
- M. Sebek, J. Holz, A. Börner and K. Jähnisch, Synlett, 2009, 461 CAS.
- R. Kuwano, Heterocycles, 2008, 76, 909 CrossRef CAS PubMed.
- R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854 RSC.
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133 CrossRef CAS.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301 RSC.
- J. H. Clark, Green Chem., 1999, 1, 1 RSC.
- C. P. Gordon, B. Venn-Brown, M. J. Robertson, K. A. Young, N. Chau, A. Mariana, A. Whiting, M. Chircop, P. J. Robinson and A. McCluskey, J. Med. Chem., 2013, 56, 46 CrossRef CAS PubMed.
- M. C. Bryan, D. Wernick, C. D. Hein, J. V. Petersen, J. W. Eschelbach and E. M. Doherty, Beilstein J. Org. Chem., 2011, 7, 114 Search PubMed.
- M. Tarleton and A. McCluskey, Tetrahedron Lett., 2011, 52, 1583 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: GCMS chromatograms, 1H and 13C NMR spectra. See DOI: 10.1039/c3ra47657j |
| ‡ ThalesNano provide details of standard reducing conditions for a variety of functional groups: http://www.thalesNano.com. |
| This journal is © The Royal Society of Chemistry 2014 |