Nanoscale conductive niobium oxides made through low temperature phase transformation for electrocatalyst support†
Kan Huang,
Yunfeng Li,
Litao Yan and
Yangchuan Xing*
Department of Chemical Engineering, University of Missouri, Columbia, MO 65211, USA. E-mail: xingy@missouri.edu; Fax: +1 573 884 4049; Tel: +1 573 884 1067
First published on 29th January 2014
Abstract
We report an effective approach to synthesize nanoscale Nb2O5 coated on carbon nanotubes (CNTs) and transform it at low temperatures to the conductive form of NbO2. The latter, when used as a Pt electrocatalyst support, shows significant enhancement in catalyst activity and durability in the oxygen reduction reaction (ORR). Direct phase transformation of Nb2O5 to NbO2 often requires temperatures above 1000 °C. Here we show that this can be achieved at a much lower temperature (e.g. 700 °C) if the niobium oxide is first activated with carbon. Low temperature processing allows retaining nanostructures of the oxide without sintering, keeping its high surface areas needed for being a catalyst support. We further show that Pt supported on the conductive oxides on CNTs has two times higher mass activity for the ORR than on bare CNTs. The electrochemical stability of Pt was also outstanding, with only ca. 5% loss in electrochemical surface areas and insignificant reduction in half-wave potential in ORR after 5000 potential cycles.
1 Introduction
Lithium–air batteries (LABs) have attracted intensive investigation due to their high theoretical energy densities, comparable to that of gasoline.1–3 The development of an aqueous version of LABs has opened an alternative way to aprotic LABs.4 Such hybrid LABs have been demonstrated in aqueous neutral,5 acidic6–8 and alkaline9,10 electrolytes in the cathodes. When batteries are discharged, oxygen is reduced to water through O2 + 4H+ + 4e− → 2H2O in acidic media or to OH− through O2 + 2H2O + 4e− → 4OH− in neutral/alkaline electrolytes, similar to the oxygen reduction reaction (ORR) taken place at cathode side in proton exchange membrane fuel cells (PEMFCs). Li et al.11 found Pt supported on carbon nanotubes (CNTs) served as cathode material has superior Li–air battery performance than Pt on carbon black. They found that the novel structure played the major role, in which CNTs intertwined in a horizontal direction and formed large open channels for air diffusion, while carbon black only generated a compact layer with less porosity.One persistent conundrum is the sluggish kinetics of ORR, even for the most active Pt and its alloys that are commonly utilized as catalysts for ORR.12 The practical onset potential of ORR is usually 0.3–0.4 V below its reversible thermodynamic potential (1.23 V vs. SHE), which comes from the strong adsorption of O and OH on Pt.13 To maintain practical current densities, more electrochemical active sites are required, leading to the high loading of the costly noble metal. Depositing a Pt monolayer on a metal support was proposed to increase the ORR activity and reduce Pt usage.14–17 However, the large scale production is a challenge due to the sophisticated procedure. Another critical challenge is to alleviate the dissolution and degradation of Pt for long-term application. Pt on carbon black is commonly used, but its instability has been reported.18,19 More stable Pt supported on CNTs was developed, but it is still less than desired stability requirements.20–22
Extensive attention has been paid to explore metal oxides as durable Pt support, among which TiO2 has been investigated.23–25 Several methods have been proposed, e.g., reducing TiO2 into a Magnéli phase (TinO2n−1) (ref. 26–29) or doping TiO2 with other elements (e.g. Nb).30–32 Comparable ORR activities but significantly improved catalyst stabilities were reported.23,30,33 Similarly, n-type semiconductor NbO2 was singled out as the next most stable material,34 which can be obtained from reducing Nb2O5 between 900 and 1300 °C.35 Sasaki et al.36 reported Pt on nano- to submicron-sized niobium oxide powders (NbO2 or Nb2O5) as electrocatalyst for ORR. The Pt/NbO2 mixed with carbon black has three times higher Pt mass activity than Pt on carbon black and exhibits extraordinary stability. Zhang et al.37 used a magnetron sputter to make NbO2 on carbon nanotube arrays on Inconel substrates, with niobium oxide layers ranging from 0.2–1 nm in thickness. When Pt was sputtered on the resulting materials, it displayed improved corrosion resistance but at the sacrifice of ORR activity.
In this study we developed a simple sol–gel procedure to make Nb2O5 nanocoatings on powder form CNTs (Nb2O5/CNTs), in which the micron long CNTs provide a backbone to support the nanocoatings. The Nb2O5 was further converted to the conductive form of NbO2 at a low temperature, which was made possible by carbon activation and post-annealing. The low temperature phase conversion allows maintaining the nanostructures. It was demonstrated that Pt on such supports has much higher ORR mass activity than on bare CNTs. The catalyst also showed extraordinary stability on the oxide supports.
2 Experimental
2.1 Synthesis of catalysts
CNTs (Pyrograf Products, Inc., 60–150 nm in diameter and 30–100 microns in length) were treated with 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) : 1 H2SO4–HNO3 (volume ratio) in an ultrasonic bath at 60 °C for 2 hours for surface functionalization,33 followed by filtration and thorough washing with deionized water. 10 mg treated CNTs were dispersed in a solution containing 8 mL ethanol, 2 mL benzyl alcohol plus excess water with aid of ultrasonication and stirring. 20 μL niobium ethoxide (Sigma-Aldrich) was dissolved in N2-saturated ethanol and slowly dipped into CNTs suspension and stirred for 2 h, followed by vacuum filtration and drying in 80 °C in a vacuum oven overnight.
: 1 H2SO4–HNO3 (volume ratio) in an ultrasonic bath at 60 °C for 2 hours for surface functionalization,33 followed by filtration and thorough washing with deionized water. 10 mg treated CNTs were dispersed in a solution containing 8 mL ethanol, 2 mL benzyl alcohol plus excess water with aid of ultrasonication and stirring. 20 μL niobium ethoxide (Sigma-Aldrich) was dissolved in N2-saturated ethanol and slowly dipped into CNTs suspension and stirred for 2 h, followed by vacuum filtration and drying in 80 °C in a vacuum oven overnight.
The synthesized Nb2O5/CNTs were thermally activated with carbon under a gas mixture of 10% acetylene in N2 at 700 °C for 20 min (sample designated as c-Nb2O5/CNTs). The c-Nb2O5/CNTs was further annealed under 10% H2 in N2 at 700–800 °C for 3 h, denoted as c-Nb2O5/CNTs-700 and c-Nb2O5/CNTs-800, respectively. Loading 10 wt% Pt on c-Nb2O5/CNTs-700 or c-Nb2O5/CNTs-800 was achieved by a polyol reduction method,38 and the obtained Pt catalysts were further heat treated under 10% H2 in N2 for 1 hour at 500 °C for stabilization.33 For comparison, the same amount of Pt was also deposited on Nb2O5/CNTs (Pt/Nb2O5/CNTs) and bare CNTs (Pt/CNTs).
2.2 Catalyst characterization
Morphologies were examined by a transmission electron microscope (TEM) (FEI Tecnai F20) operating at 300 kV and a scanning electron microscope (SEM) (Helios NanoLab 600) operating at 30 kV. Crystalline structures of the catalysts were analyzed by X-ray diffractometer (XRD) (X-Pert Philips) equipped with Cu K(α); XRD patterns were collected from 20 to 60° at a scanning rate of 0.026° s−1.2.3 Electrochemical testing
All electrochemical experiments were performed with an Electrochemical Workstation (Bioanalytical Sciences, BAS 100). The working electrode was a glassy carbon rotating disk electrode (RDE) with a disk diameter at 5 mm. A Pt wire was used as the counter electrode and 3 M Ag/AgCl as a reference electrode. The catalyst powder was dispersed in 0.05 wt% Nafion (Alfa Aesar) in ethanol (Alfa Aesar) by sonication for 20 min to form 5 mg mL−1 catalyst suspension. 20 μL of the suspension was pipetted onto the disk and dried in air, keeping the Pt loading at 0.01 mg. Cyclic voltammetry (CV) was carried out in N2 purged 0.1 M HClO4 at a potential range of 0–1.2 V (vs. RHE; the same hereafter unless otherwise noted). ORR measurement was taken in an O2-saturared 0.1 M HClO4 at different rotating speeds and the potential was swept in the positive direction from 0.2 to 1.0 V. To study the durability of Pt catalysts on the oxide support, potential cycling was conducted in air saturated 0.1 M HClO4 in the potential range between 0.6 and 1.0 V up to 5000 cycles. The initial and final CV and ORR performances were recorded.3 Results and discussion
3.1 Characterization of niobium oxide–CNTs and Pt electrocatalysts
Fig. 1(a) shows typical SEM images of Nb2O5 coated CNTs. The thin Nb2O5 coating was found to uniformly cover the CNTs surface with insignificant aggregation. EDX analysis shows the presence of Nb element in a Nb2O5/CNTs sample (Fig. S1†). The Nb2O5/CNTs obtained by the sol–gel method lack discernible diffraction peaks except the graphite (002) plane, suggesting an amorphous structure, as shown in Fig. 2. Annealing Nb2O5/CNTs at 600 °C in N2, a Nb2O5 pseudo-hexagonal crystalline structure was obtained and typical diffraction peaks at (001), (100) and (101) planes emerged.39 In a separate experiment, Nb2O5/CNTs was annealed in air at 600 °C, which also transforms the pseudo-hexagonal structure. However, the graphitic structure of the CNTs identified was no longer observed, indicating complete oxidation of the CNTs (Fig. 2). The morphology of oxides after removal of CNTs exhibits a nanotube structure as shown in Fig. 1(b). Smooth Nb2O5 nanotubes exhibited as several microns long with a wall thickness of ca. 10 nm; the hollow structure can be maintained during thermal oxidation. In situ thermal analysis was conducted which revealed that there is about 50 wt% of Nb2O5 in the Nb2O5/CNTs composite (Fig. S2†).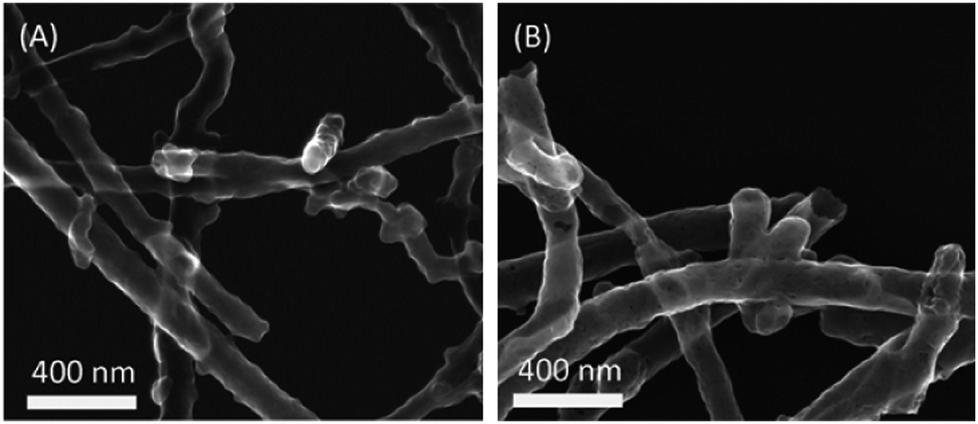 | ||
| Fig. 1 SEM images of (a) carbon nanotubes coated with amorphous Nb2O5, and (b) thin hexagonal Nb2O5 nanotubes after removing the carbon nanotubes template. | ||
 | ||
| Fig. 2 XRD patterns of (a) Nb2O5/CNTs obtained directly after the sol–gel process, (b) amorphous Nb2O5/CNTs annealed in N2 at 600 °C (resulting in hexagonal Nb2O5), and (c) amorphous Nb2O5/CNTs annealed in O2 at 600 °C (removal of the carbon nanotube templates and completion of transformation to hexagonal Nb2O5). | ||
Crystalline phase information of carbon activated Nb2O5/CNTs was illustrated in Fig. 3(a). After carbon activation, the XRD patterns of c-Nb2O5/CNTs still exhibited as Nb2O5/CNTs, suggesting that the bulk physical property was not changed. Hahn et al.40 suggested that at low temperatures, carbon doping TiO2 is mostly a surface deposition process and only distorts the surface lattice rather than the bulk. Our previous studies observed that carbon doping in TiO2 nanocoatings at 700 °C resulted in a shift of Ti 2p and formation of suboxide, instead of a generation of a new phase.33,41 We speculate that carbon activation in Nb2O5 would have a similar effect. Indeed, X-ray photoelectron spectroscopy (XPS) analysis showed Nb 3d in c-Nb2O5/CNTs shifted negatively at 0.8 eV as compared to that of Nb2O5/CNTs (Fig. S3†), which confirms our speculation.
 | ||
| Fig. 3 XRD patterns of (a) c-Nb2O5/CNTs, c-Nb2O5/CNTs-700, and c-Nb2O5/CNTs-800, (b) c-Nb2O5/CNTs and Nb2O5/CNTs further annealed in H2/N2 at 700 °C, (c) c-Nb2O5/CNTs and Nb2O5/CNTs further annealed in H2/N2 at 800 °C, and (d) Pt electrocatalysts after deposition of Pt on the niobium oxide-CNTs (e.g. Pt/Nb2O5/CNTs). | ||
Further annealing under H2/N2 at 700 °C (Fig. 3(a)), the characteristic peaks of NbO2, such as (400) and (222), started to appear, indicative of partial phase transformation from Nb2O5 to NbO2. At 700 °C, the niobium oxide was a mixture of Nb2O5 and NbO2. Raising the annealing temperature to 800 °C, however, the Nb2O5 phase almost disappeared and the oxide was transformed to NbO2. In a separate experiment, non-activated Nb2O5/CNTs were treated under the same conditions. Their XRD patterns were given in Fig. 3(b) for that treated at 700 °C and Fig. 3(c) treated for 800 °C along with their respected activated samples. It can be seen that phase transformation did not occur in these non-activated samples, indicative of that carbon activation has facilitated the phase transformation at a much lower temperature.
It was previously reported that such direct phase conversion requires a high calcination temperature up to 1300 °C under H2 atomosphere.35 But doping with other elements may lower phase transformation energies and allows low temperature processing. This has observed in studies of TiO2. Toyoda et al.42 annealed a mixture of TiO2 and carbon source under N2 instead of directly reducing TiO2 under H2 atmosphere, and they found formation of titanium suboxide (e.g. as Ti4O7) at a lower temperature (800 °C), which may imply that carbon activation helped in the transformation. Koc and Folmer43 thermally treated carbon coated TiO2 in argon and observed formation of titanium suboxides. They suggested that carbon was involved in partial substitution of the oxygen atoms in TiO2. Huang et al.33 reported that carbon atoms can be interstitially incorporated into the TiO2 lattice, resulting in a lattice expansion. It was speculated in this study that the doped carbon may also partially react with oxygen in Nb2O5 during the thermal treatment and form oxygen vacancies. Carbon may have incorporated into Nb2O5 lattice, similar to that in TiO2. An expanded and relaxed Nb2O5 lattice would favor the phase transformation to NbO2 in the further annealing, as observed in the study. Finally, the obtained Pt nanoparticles are metallic,38 as indicated by its characteristic peaks in Fig. 3(d).
Fig. 4 shows the TEM images of Pt nanoparticles decorated on c-Nb2O5/CNTs-700, c-Nb2O5/CNTs-800, and CNTs. As can be seen form Fig. 4(a), the Pt particles were not perfectly dispersed on the CNTs, and minor aggregation was observed, despite that the CNTs was pre-treated in acid to improve their wetting behavior.38,44 In the case of Pt on c-Nb2O5/CNTs −700 or −800 (see Fig. 4(b) and (c)), aggregation of Pt particles is very minimal, even after the as-synthesized Pt particles were post annealed at 500 °C for their anchoring on the support. Individual Pt particles were distinguishable and uniformly dispersed on the c-Nb2O5/CNTs −700 or −800 surfaces, suggesting an improved interaction between Pt and the oxide supports. In addition, the thin Nb2O5 film was found to be well retained during the thermal treatment.
 | ||
| Fig. 4 TEM images of (a) Pt/CNTs, (b) Pt/c-Nb2O5/CNTs-700, (c) Pt/c-Nb2O5/CNTs-800, and (d) c-Nb2O5/CNTs-800. The inset in (d) shows the indexed selected area diffraction (SAD) pattern, corresponding to the characteristic basal planes of NbO2. | ||
NbO2 has a much stronger interfacial interaction with CNTs than Pt with CNTs due to the stronger adsorption energy of NbO2 on a graphite surface.37 Zhang et al.45 suggested adding a Ti buffer layer between CNTs and metal particles can significantly improve the wetting due to stronger metal–metal bonds than simple metal–CNTs van der Waals force. Incorporating a titanium oxide and niobium oxide interlayer between CNTs and Pt was also reported to improve the wetting and interfacial interaction.33,37,46 Thus, the better dispersion of Pt on the niobium oxides than on bare CNTs may be attributed to the stronger metal–support interaction. The average Pt particle sizes are estimated, based on TEM image processing, from over 100 randomly picked particles. The particle sizes on CNTs, c-Nb2O5/CNTs-700, and c-Nb2O5/CNTs-800 were measured to be 5.45 ± 1.15 nm, 7.16 ± 2.80 nm, and 5.24 ± 1.17 nm, respectively. The size differences in Pt on c-Nb2O5/CNTs-700 and c-Nb2O5/CNTs-800 indicate a variation in the degree of wetting between the Pt particles and the metal oxide supports. Zhang et al.46 suggested that the chemical interaction between Pt with NbO is the strongest, followed by NbO2, and Nb2O5 is the weakest. Since the XRD showed that c-Nb2O5/CNTs-700 is a mixture of Nb2O5 and NbO2, and NbO2 is the main component in c-Nb2O5/CNTs-800, our observation is in agreement with the previous study. The differences of Pt particle sizes can be attributed to the wetting differences of the supports. Further confirmation of the NbO2 phase was obtained through selected area diffraction (SAD) of c-Nb2O5/CNTs-800, as shown in Fig. 4(d).
3.2 Electrochemical activities of the catalysts
Fig. 5(a) shows the CV curves of Nb2O5/CNTs, c-Nb2O5/CNTs-700, and c-Nb2O5/CNTs-800, with the amount of material on each disk kept the same. No specific oxidation and reduction current peaks were observed at the scanning potentials from 0–1.2 V, except for very small ramps above 1.1 V. Among them, Nb2O5/CNTs has the biggest double layer capacity, followed by c-Nb2O5/CNTs-700. c-Nb2O5/CNTs-800 has the smallest double layer capacity. Nb2O5 is an insulator (10−4 S cm−1 at room temperature) and therefore showed the largest capacitance,35 whereas NbO2 is an n-type semiconductor (∼10−3 S cm−1 at room temperature) with consequently lowest capacitance.47 Fig. 5(b) exhibits the ORR polarization curves of three supports; all possess negligible catalytic activity towards ORR. Fig. 5(c) presents the CV curves of niobium oxide supported Pt, including CNTs supported Pt for comparison. Due to the high electrical resistance of Nb2O5, its CV curve fundamentally differs from the others. The Pt oxide reduction peak shifts negatively and only hydrogen adsorption peak appears; this is presumably due to the poor conductivity of the Nb2O5 which separates Pt from CNTs and inhibits the electron flow.46 While the delay of reduction of Pt-oxide was not observed for Pt on c-Nb2O5/CNTs-700 and c-Nb2O5/CNTs-800, both exhibit similar CV behaviors as Pt on CNTs, suggesting carbon activation and annealing indeed increase the electronic conductivity of Nb2O5/CNTs and enable successful electron transfer.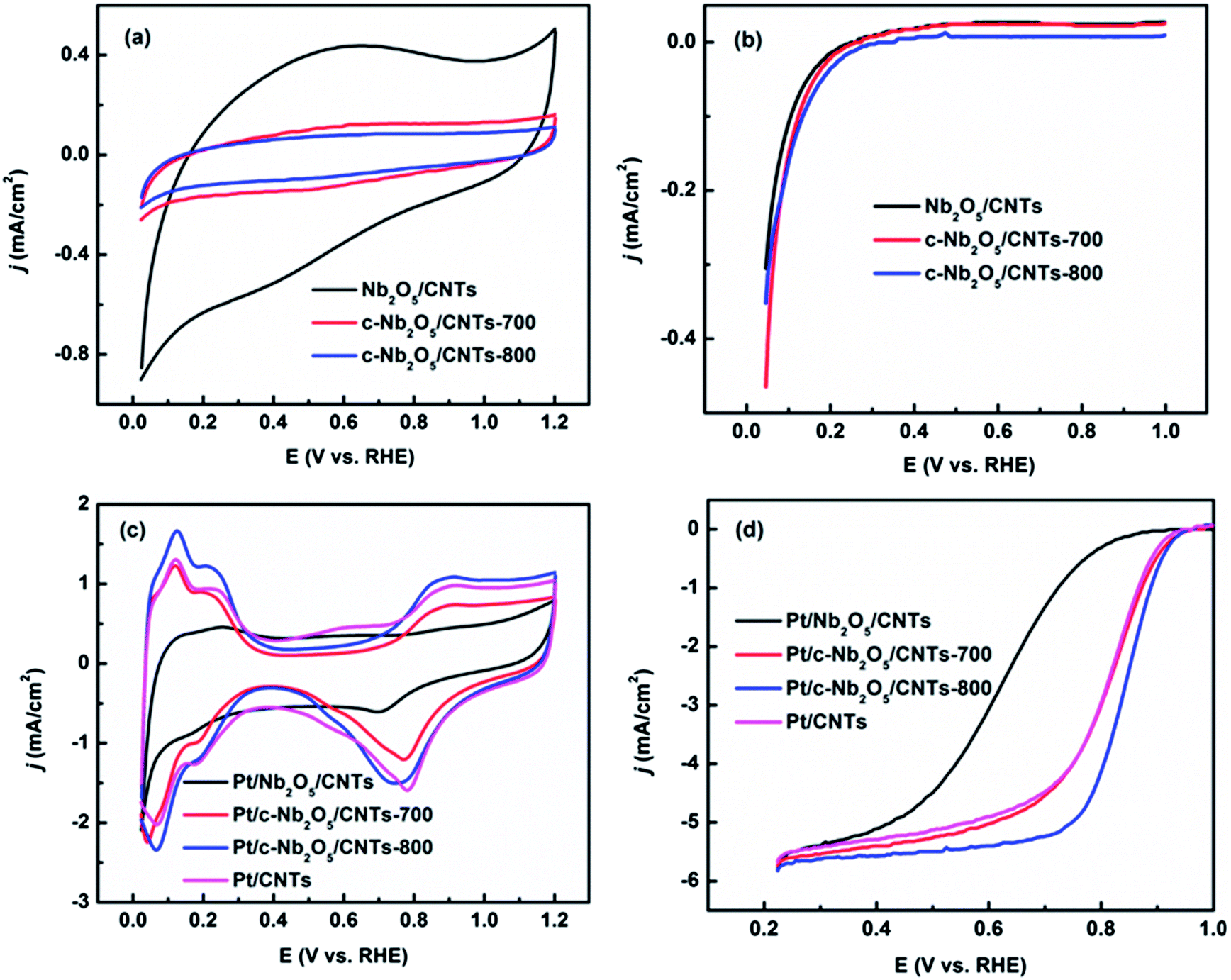 | ||
| Fig. 5 Electrochemical characterizations of catalysts: (a) cyclic voltammograms of Nb2O5/CNTs, c-Nb2O5/CNTs-700, and c-Nb2O5/CNTs-800. The scan rate: 50 mV s−1, (b) polarization curves for ORR of Nb2O5/CNTs, c-Nb2O5/CNTs-700, and c-Nb2O5/CNTs-800. The scan rate: 5 mV s−1, (c) cyclic voltammograms of Pt/Nb2O5/CNTs, Pt/c-Nb2O5/CNTs-700, Pt/c-Nb2O5/CNTs-800, and Pt/CNTs. The scan rate: 50 mV s−1, and (d) polarization curves for ORR of Pt/Nb2O5/CNTs, Pt/c-Nb2O5/CNTs-700, Pt/c-Nb2O5/CNTs-800, and Pt/CNTs. The scan rate: 5 mV s−1. | ||
The electrochemical surface area (ESA) of Pt/c-Nb2O5/CNTs-700, Pt/c-Nb2O5/CNTs-800, and Pt/CNTs were obtained by integrating the area of the hydrogen adsorption region (∼0.05–0.3 V)38 and calculated to be 468, 582 and 417 cm2 mgPt−1, respectively. The measurements of ORR activities were illustrated on Fig. 5(d). Similar Pt particle size based on TEM investigation makes Pt/CNTs and Pt/c-Nb2O5/CNTs-800 suitable for comparison. The half-wave potentials for Pt on Nb2O5/CNTs, c-Nb2O5/CNTs-700, c-Nb2O5/CNTs-800, and CNTs are 0.611 V, 0.804 V, 0.829 V and 0.802 V, respectively. Considering the same Pt mass loading, Pt/c-Nb2O5/CNTs-800 exhibited the best ORR mass activity. The insulating nature of Nb2O5 takes the main responsibility for the lowest observed activity of Pt/Nb2O5/CNTs. The better performance of Pt/c-Nb2O5/CNTs-800 than Pt/c-Nb2O5/CNTs-700 may come from the higher conductivity of oxide support36 and larger ESA (smaller catalyst size). In addition, Pt/CNTs was found to have similar ORR performance as Pt/c-Nb2O5/CNTs-700.
The RDE data was further analyzed using the Koutecky–Levich (K–L) equation:48
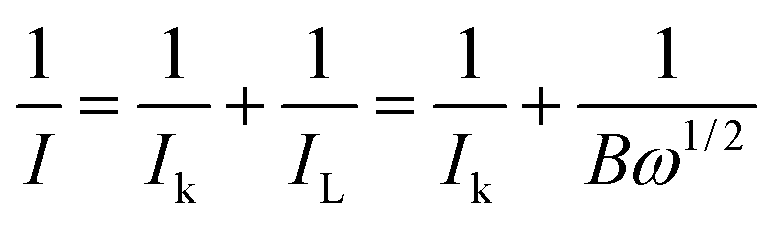
 | ||
| Fig. 6 (a) Polarization curve obtained with a rotating disk electrode for the ORR on Pt/c-Nb2O5/CNTs-800, and (b) Koutecky–Levich plots at different potentials using the data obtained from (a). | ||
The intrinsic activities of Pt catalysts were normalized ESA to obtain the so called specific activity (jk = Ik/ESA), and the mass activity was obtained by the mass loading (jk = Ik/mass of Pt loading). The comparisons were provided in Table 1. The results demonstrated that Pt/c-Nb2O5/CNTs-800 exhibited 2.1 times higher Pt mass activity and about 1.5 times specific activity over those of Pt/CNTs; but Pt/c-Nb2O5/CNTs-700 has only comparable activities.
| Catalysts | Mass activity (mA mgPt−1) | Specific activity (mA cmPt−2) |
|---|---|---|
| Pt/c-Nb2O5/CNTs-700 | 43.3 | 0.093 |
| Pt/c-Nb2O5/CNTs-800 | 80.5 | 0.138 |
| Pt/CNTs | 38.5 | 0.093 |
The high adsorption of OH and O on Pt surface which blocks the further access of oxygen molecules is a major cause of ORR loss.13 A four electron ORR must involve both the O2 dissociation (breaking O–O bond) and O–H formation (adding proton to O).49 The structure and occupancy of metal d-band, especially the d-band center (εd), plays an important role in the catalyst surface reactivity.50,51 According to those studies, the most active platinum monolayer should have an intermediate value of εd; a higher-lying εd tends to bind adsorbates more strongly and enhance the kinetics of dissociation reactions. On the other hand, a lower-lying εd tends to bind adsorbates more weakly and favor the formation of bonds among them.49,52 Computationally, Pt/NbO2 was found to bind atomic oxygen weakly and exhibit enhanced ORR activity.46 Experimentally, Sasaki et al.36 observed, through in situ XANES, and concluded that oxidation of Pt on NbO2 supports requires higher potential than that of Pt on carbon, which was attributed to the lateral repulsion between OH and O with the oxide's surface. The suppressed formation of Pt-oxide partly was attributed to the enhanced ORR activity. The suppressed adsorption of OH or O on Pt appears to explain the better ORR activity of Pt/c-Nb2O5/CNTs-800 in this study.
The electronic structure of substrates that affects the catalytic activity of deposited Pt is also an important factor.46,50 Zhang et al.37 examined the electronic structure of supported Pt and found the difference in Fermi energies between graphene and NbO2; the former forms a net positive charge and the latter a net negative charge. Based on the equilibrium of the Fermi levels to attain thermodynamic equilibrium, at Pt/NbO2 interface the electronic charge will flow from NbO2 to Pt and generate a negative charge on Pt, which is believed to benefit ORR reaction.36,46 For Pt/CNTs, the electron flow reverses, leaving a positive charge on the Pt. This could be another cause of the difference in ORR activity. Surface strain in a Pt monolayer is known to alter electronic configuration and change ORR activity.15,16,51 The interaction between Pt and niobium oxide is different from Pt and CNTs, and reflects in the different wetting behavior from TEM investigation. Strain-induced effect may also have contributed to the observed ORR activity.
Nb2O5 was reported to have a similar repulsion effect with OH or O as NbO2 and inhibits the oxidation of Pt supported on Nb2O5.36 However, the insulator nature of Nb2O5 should be responsible for the much lower ORR activity of Pt/Nb2O5, as compared with Pt/NbO2.36 The difference between c-Nb2O5/CNTs-700 and c-Nb2O5/CNTs-800 is likely to be one of possible causes of the difference in ORR activity observed for Pt/c-Nb2O5/CNTs-700 and Pt/c-Nb2O5/CNTs-800. In addition, Pt/Nb2O5 interface would render a net positive charge on Pt, opposite to the negative charge in Pt/NbO2, which may account for another cause of the differences in ORR activity.
The electrochemical stability of Pt/c-Nb2O5/CNTs-800 was tested by cycling it in air-saturated 0.1 M HClO4 in the potential range of 0.6–1.0 V for 5000 cycles. The initial and after-cycling ORR activities were recorded and shown in Fig. 7. The inset in Fig. 7 shows no significant change in CV in the hydrogen region before and after the cycling, verified by a small ESA loss of ca. 5%. However, there is an apparent change in the oxide reduction peak which shows a small shift to the positive. This change is attributed to Pt size increase (to 6.13 ± 1.34 nm) induced in the cycling process, since the Pt–O formation is size dependent as reported previously.53,54
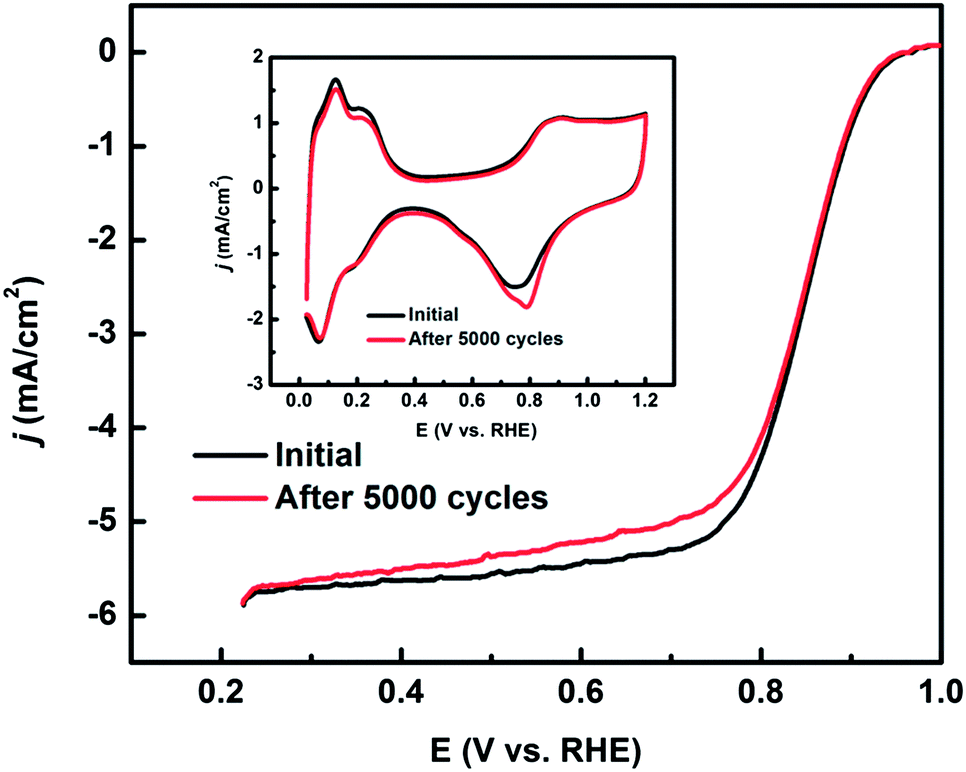 | ||
| Fig. 7 Polarization curves of the ORR on Pt/c-Nb2O5/CNTs-800 before and after 5000 cycles. The potential cycling was performed from 0.6–1.0 V in air saturated 0.1 HClO4 at a scan rate of 50 mV s−1. The inset shows the CV curves before and after 5000 cycles. | ||
The half-wave potential suffered a tiny negative shift from 0.829 V to 0.825 V, demonstrating that the catalyst is fairly stable. Fig. 8 recorded the initial and final morphologies of the catalyst Pt/c-Nb2O5/CNTs-800. From the TEM images, it appears that after potential cycling Pt is still uniformly dispersed on c-Nb2O5/CNTs-800, despite the particle size growth, in agreement with the observations of electrochemical test results.
 | ||
| Fig. 8 TEM images of Pt/c-Nb2O5/CNTs-800 at (A) initial, and (B) after 5000 cycles, showing minimum changes in particle morphology. | ||
4 Conclusions
This paper reported a study on the preparation of nanoscale conductive niobium oxide used as electrocatalyst support. Nb2O5 was coated on CNTs in a sol–gel process, leading to a nanoscale thin Nb2O5 coating on the CNTs. The Nb2O5 nanocoatings on CNTs were transformed to the conductive form of NbO2 at low temperatures, which was attributed to the effect of carbon activation. The replacement of oxygen in Nb2O5 by carbon and the expanded Nb2O5 lattice may be responsible for the easier phase transformation. The low temperature process makes it possible to keep the nanocoatings intact without sintering.It was demonstrated that Pt supported on the conductive oxides on CNTs has two times higher mass activity for the ORR than on bare CNTs. The enhanced catalytic activity may be attributed to reduced OH adsorption on Pt and modification of electronic structure of supported Pt on the oxides. The incorporation of the niobium oxide interlayer between Pt and CNTs improved the Pt particles dispersion, which comes from stronger interaction between Pt and niobium oxides. Under oxidizing conditions of 5000 potential cycles from 0.6–1.0 V, the catalysts suffered only ca. 5% loss in ESA and almost no loss in ORR activity, despite some particle size increase. This work c-Nb2O5/CNTs-800 is an excellent electrocatalyst support for fuel cells and hybrid Li–air batteries in oxygen electrodes.
Acknowledgements
Partial financial support by the US Department of Energy ARPA-E grant DE-AR0000066 is gratefully acknowledged. We also thank Dr Eric Bohannan for his help in the XRD analysis, Dr Kai Song for taking TEM images, Mr Brian Porter for the XPS analysis.Notes and references
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef CAS.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- J. Christensen, P. Albertus, R. S. Sanchez-Carrera, T. Lohmann, B. Kozinsky, R. Liedtke, J. Ahmed and A. Kojic, J. Electrochem. Soc., 2012, 159, R1–R30 CrossRef CAS PubMed.
- S. J. Visco, B. D. Katz, Y. S. Nimon and L. C. D. Jonghe, U.S. Patent 7282295, 2007.
- T. Zhang, N. Imanishi, S. Hasegawa, A. Hirano, J. Xie, Y. Takeda, O. Yamamoto and N. Sammes, J. Electrochem. Soc., 2008, 155, A965–A969 CrossRef CAS PubMed.
- T. Zhang, N. Imanishi, Y. Shimonishi, A. Hirano, Y. Takeda, O. Yamamoto and N. Sammes, Chem. Commun., 2010, 46, 1661–1663 RSC.
- T. Zhang, N. Imanishi, Y. Shimonishi, A. Hirano, J. Xie, Y. Takeda, O. Yamamoto and N. Sammes, J. Electrochem. Soc., 2010, 157, A214–A218 CrossRef PubMed.
- K. Huang, Y. Li and Y. Xing, Electrochim. Acta, 2013, 103, 44–49 CrossRef CAS PubMed.
- P. He, Y. Wang and H. Zhou, Electrochem. Commun., 2010, 12, 1686–1689 CrossRef PubMed.
- Y. Wang and H. Zhou, J. Power Sources, 2010, 195, 358–361 CrossRef PubMed.
- Y. Li, K. Huang and Y. Xing, Electrochim. Acta, 2012, 81, 20–24 CrossRef PubMed.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. Zhang, M. B. Vukmirovic, K. Sasaki, A. U. Nilekar, M. Mavrikakis and R. R. Adzic, J. Am. Chem. Soc., 2005, 127, 12480–12481 CrossRef PubMed.
- Y. Xing, Y. Cai, M. B. Vukmirovic, W.-P. Zhou, H. Karan, J. X. Wang and R. R. Adzic, J. Phys. Chem. Lett., 2010, 1, 3238–3242 CrossRef CAS.
- R. Adzic, J. Zhang, K. Sasaki, M. Vukmirovic, M. Shao, J. Wang, A. Nilekar, M. Mavrikakis, J. Valerio and F. Uribe, Top. Catal., 2007, 46, 249–262 CrossRef CAS.
- S. W. Lee, S. Chen, J. Suntivich, K. Sasaki, R. R. Adzic and Y. Shao-Horn, J. Phys. Chem. Lett., 2010, 1, 1316–1320 CrossRef CAS.
- P. J. Ferreira, G. J. la O, Y. Shao-Horn, D. Morgan, R. Makharia, S. Kocha and H. A. Gasteiger, J. Electrochem. Soc., 2005, 152, A2256–A2271 CrossRef PubMed.
- K. . J. J. Mayrhofer, J. C. Meier, S. J. Ashton, G. K. H. Wiberg, F. Kraus, M. Hanzlik and M. Arenz, Electrochem. Commun., 2008, 10, 1144–1147 CrossRef CAS PubMed.
- S. K. Natarajan and J. Hamelin, J. Electrochem. Soc., 2009, 156, B210–B215 CrossRef CAS PubMed.
- D. Z. Mezalira and M. Bron, J. Power Sources, 2013, 231, 113–121 CrossRef CAS PubMed.
- L. Li and Y. Xing, J. Power Sources, 2008, 178, 75–79 CrossRef CAS PubMed.
- T. Ioroi, H. Senoh, S.-i. Yamazaki, Z. Siroma, N. Fujiwara and K. Yasuda, J. Electrochem. Soc., 2008, 155, B321–B326 CrossRef CAS PubMed.
- X. Li, A. L. Zhu, W. Qu, H. Wang, R. Hui, L. Zhang and J. Zhang, Electrochim. Acta, 2010, 55, 5891–5898 CrossRef CAS PubMed.
- S.-Y. Huang, P. Ganesan, S. Park and B. N. Popov, J. Am. Chem. Soc., 2009, 131, 13898–13899 Search PubMed.
- W.-Q. Han and Y. Zhang, Appl. Phys. Lett., 2008, 92, 203117–203113 CrossRef PubMed.
- C. D. A. Rodrigues, N. R. de Tacconi, W. Chanmanee and K. Rajeshwar, Electrochem. Solid-State Lett., 2010, 13, B69–B72 CrossRef PubMed.
- F. C. Walsh and R. G. A. Wills, Electrochim. Acta, 2010, 55, 6342–6351 CrossRef CAS PubMed.
- S. Tominaka, Chem. Commun., 2012, 48, 7949–7951 RSC.
- S.-Y. Huang, P. Ganesan and B. N. Popov, Appl. Catal., B, 2010, 96, 224–231 CrossRef CAS PubMed.
- Y. Liu, J. M. Szeifert, J. M. Feckl, B. Mandlmeier, J. Rathousky, O. Hayden, D. Fattakhova-Rohlfing and T. Bein, ACS Nano, 2010, 4, 5373–5381 CrossRef CAS PubMed.
- N. R. Elezovic, B. M. Babic, V. R. Radmilovic, L. M. Vracar and N. V. Krstajic, Electrochim. Acta, 2011, 56, 9020–9026 CrossRef CAS PubMed.
- K. Huang, K. Sasaki, R. R. Adzic and Y. Xing, J. Mater. Chem., 2012, 22, 16824–16832 RSC.
- J. I. Martínez, H. A. Hansen, J. Rossmeisl and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 045120 CrossRef.
- C. M. Reich, A. Kaiser and J. T. S. Irvine, Fuel Cells, 2001, 1, 249–255 CrossRef CAS.
- K. Sasaki, L. Zhang and R. R. Adzic, Phys. Chem. Chem. Phys., 2008, 10, 159–167 RSC.
- L. Zhang, L. Wang, C. M. B. Holt, B. Zahiri, Z. Li, K. Malek, T. Navessin, M. H. Eikerling and D. Mitlin, Energy Environ. Sci., 2012, 5, 6156–6172 CAS.
- Y. Xing, J. Phys. Chem. B, 2004, 108, 19255–19259 CrossRef CAS.
- C. Yan and D. Xue, Adv. Mater., 2008, 20, 1055–1058 CrossRef CAS.
- R. Hahn, A. Ghicov, J. Salonen, V.-P. Lehto and P. Schmuki, Nanotechnology, 2007, 18, 105604 CrossRef.
- K. Huang, Y. Li and Y. Xing, J. Mater. Res., 2013, 28, 454–460 CrossRef CAS.
- M. Toyoda, T. Yano, B. Tryba, S. Mozia, T. Tsumura and M. Inagaki, Appl. Catal., B, 2009, 88, 160–164 CrossRef CAS PubMed.
- R. Koc and J. S. Folmer, J. Mater. Sci., 1997, 32, 3101–3111 CrossRef CAS.
- Y. Xing, L. Li, C. C. Chusuei and R. V. Hull, Langmuir, 2005, 21, 4185–4190 CrossRef CAS PubMed.
- Y. Zhang and H. Dai, Appl. Phys. Lett., 2000, 77, 3015–3017 CrossRef CAS PubMed.
- L. Zhang, L. Wang, C. M. B. Holt, T. Navessin, K. Malek, M. H. Eikerling and D. Mitlin, J. Phys. Chem. C, 2010, 114, 16463–16474 CAS.
- G. Bélanger, J. Destry, G. Perluzzo and P. M. Raccah, Can. J. Phys., 1974, 52, 2272–2280 Search PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley &Sons, New York, 2001 Search PubMed.
- J. Zhang, M. B. Vukmirovic, Y. Xu, M. Mavrikakis and R. R. Adzic, Angew. Chem., 2005, 117, 2170–2173 CrossRef.
- B. Hammer and J. K. Nørskov, in Advances in Catalysis, ed. H. K. Bruce and C. Gates, Academic Press, 2000, pp. 71–129 Search PubMed.
- J. Greeley, J. K. Nørskov and M. Mavrikakis, Annu. Rev. Phys. Chem., 2002, 53, 319–348 CrossRef CAS PubMed.
- Y. Xu, A. V. Ruban and M. Mavrikakis, J. Am. Chem. Soc., 2004, 126, 4717–4725 CrossRef CAS PubMed.
- K. J. J. Mayrhofer, B. B. Blizanac, M. Arenz, V. R. Stamenkovic, P. N. Ross and N. M. Markovic, J. Phys. Chem. B, 2005, 109, 14433–14440 CrossRef CAS PubMed.
- B. E. Hayden, Acc. Chem. Res., 2013, 46, 1858–1866 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TEM image showing CNTs and oxide coating; EDX showing elements in the material; thermogravimetric analysis of the material; XPS study results of Nb 3d and O 1s. See DOI: 10.1039/c3ra47091a |
| This journal is © The Royal Society of Chemistry 2014 |