
A facile bi-phase synthesis of Fe3O4@SiO2 core–shell nanoparticles with tunable film thicknesses†
Abstract
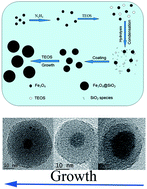
Monodisperse Fe3O4@SiO2 nanoparticles are prepared using hydrazine as a catalyst via a biphase approach without any alcohols or surfactants. Fe3O4 seeds can be dispersed well in this system. The sizes of Fe3O4@SiO2 nanoparticles with a single core could be regulated from 20 nm to 50 nm corresponding to SiO2 shell thickness from 3 nm to 17 nm. Core-free SiO2 nanoparticles are not observed in this system. The coating process can be implemented at a temperature greater than 90 °C, which results in a short coating duration from 2 h to 8 h for different shell thicknesses. Hydrazine can prevent the Fe3O4 core from oxidization during coating at this temperature. Fe3O4@SiO2 nanoparticles have high chemical stability and magnetic saturation. A plausible formation mechanism of these nanoparticles is also presented.
 Please wait while we load your content...
Please wait while we load your content...

