Enhanced thermochemical CO2 splitting over Mg- and Ca-doped ceria/zirconia solid solutions
Min Kangab,
Xiaomin Wuab,
Jun Zhang*d,
Ning Zhao*a,
Wei Wei*ac and
Yuhan Sunad
aState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, South Taoyuan Road 27#, Taiyuan, 030001, China. E-mail: zhaoning@sxicc.ac.cn; weiwei@sxicc.ac.cn; Fax: +86-351-041153; Tel: +86-351-4049612
bUniversity of Chinese Academy of Sciences, Beijing 100049, PR China
cCenter for Greenhouse Gas and Environmental Engineering, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201210, PR China
dCAS Key Lab of Low-Carbon Conversion Science and Engineering, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201210, PR China. E-mail: zhangj@sari.ac.cn
First published on 17th December 2013
Abstract
Thermochemical CO2 splitting was carried out over ceria/zirconia solid solutions prepared via a P123-added hydrothermal method in the temperature range of 1100–1400 °C. XRD, Raman and TPR characterization indicated that the introduction of Mg and Ca into ceria/zirconia could produce lattice defects in the fluorite lattice, and then strongly modify the mobility of oxygen as well as the thermal stability of the samples. As compared to Mg-doped samples, faster reaction rates and higher CO2 splitting reactivity were obtained over Ca-doped samples, because of the faster oxygen mobility in Ca–Ce–Zr–O ternary solid solutions. Moreover, the porous structure with small particle size favoured the thermal reduction and the mass diffusion. As a result, fast reaction rates and relatively high fuel productivity were obtained at the moderate thermal reduction temperature (1200 °C).
1. Introduction
Thermochemical CO2 splitting cycles using concentrated solar energy or nuclear heat to drive the high temperature endothermic chemical reactions were proposed to recycle CO2 into fuels, which has the potential of reducing CO2 emission associated to the challenging problems of climate change and the energy crisis.1–3 The product, CO and/or H2 (via H2O splitting cycles), can be then used as fuels directly, or valuable raw materials to synthesize liquid fuels or other chemicals via water-gas shift, hydrogenation and/or Fischer–Tropsch synthesis.4 The liquid fuels can be used on the existing technologies, and thus bypass the problems associated with CO2 geological storage that do not meet all the expectations in terms of costs and long term CO2 emission management.The feasibility of direct thermal dissociation of CO2 had also been investigated,5,6 and the main challenges of the process are the requirement of extremely high temperatures (>2400 °C) in order to achieve reasonable degree of CO2 dissociation and the following high temperature separation of CO and O2. As an alternative method for CO2 reduction, the CO2 splitting reaction in thermochemical cycles is decomposed into two steps in order to avoid the problems of the extremely high operating temperatures and the high temperature CO/O2 separation. Typically, thermochemical CO2 splitting cycles contain a metal oxide thermal reduction (T-R, eqn (1)) step which usually proceeds at high temperatures followed by low temperature carbon dioxide splitting (CD-S, eqn (2)) step, in which the thermally reduced metal oxide is re-oxidized by CO2 and then recycled into the T-R step.1,7 Generally, the reaction can be represented as follows:
| MOx → MOx−y + y/2O2 | (1) |
| MOx−y + yCO2 → MOx + yCO | (2) |
Due to its high melting point, cerium based material showed to be relatively stable in thermochemical cycles at temperatures of 800–1500 °C,18,19 and is one of the promising candidates for thermochemical CO2 and/or H2O splitting reactions. However, the reduction temperature of pure CeO2 is very high. This would lead to a loss in material partial sublimation and reactivity during cycling. It was reported that ceria doped with a metallic cation, such as Zr, Mn, Fe and Ni etc., could decrease the thermal reduction temperature by inducing structural defects or oxygen vacancies and improve its thermal stability while maintaining the cubic fluorite structure.22–24 Zr-doped CeO2 solid solutions could be thermally reduced under high oxygen partial pressure, even in air at 1500 °C.22,23 As a result, compared to pure CeO2, doped samples showed improved cycling reactivity due to the high thermal stability as well as the high O2 conductivity of the solid solutions.
Recently, Gal and Abanades investigated the thermochemical H2O splitting reaction using trivalent cations doped ceria/zirconia solid solutions.25 They found that doped samples showed high and stable reactivity during repeated cycles due to their high thermal resistance and enhanced redox properties.25,26 It was also reported that the simultaneous presence of CeO2, ZrO2 and some alkaline earth metal could create strain in the fluorite-type lattice and then strongly modify surface and bulk oxygen handling properties.27–29 CaO has the optimum ionic radius for divalent cations in ceria-based material (0.111 nm) and gives the minimum association enthalpy between dopant ion and oxygen vacancy, thus the oxygen handling properties could be strongly modified by the introduction of Ca into ceria/zirconia lattice.28 Consequently, Ca-doped ceria/zirconia sample is expected to show a fast bulk oxygen mobility and improved thermal stability and thus enhance the thermochemical CO2 splitting reactivity.
In our previous work, the introduction of Mg was observed to improve the CO productivity and the cycling stability due to a relatively high thermal stability.30 However, Mg2+ and Ca2+ have different ionic radius with the same electronic shell structure, which would give rise to the different effect on the microstructure and oxygen storage capacity (OSC).28,31 For the above purpose, Mg- and Ca-doped ceria/zirconia samples were prepared via a P123-added hydrothermal treatment, and then characterized for CO2 splitting reaction via thermogravimetric analysis (TGA) in the temperature range of 1100–1400 °C. Furthermore, in order to alleviate the unfavorable structure change caused by the high cycling temperature (1400 °C) and investigate the CO2 splitting reactivity under relatively low T-R temperatures, the cyclic reaction was also conducted at 1000–1200 °C using the porous doped samples.
2. Experimental
2.1. Material preparation
Mg- and Ca-doped ceria/zirconia solid solutions were prepared via a P123-added hydrothermal synthesis.30 Ce(NO3)3·6H2O, Zr(NO3)4·5H2O, Mg(NO3)2·6H2O and Ca(NO3)2·4H2O were used as starting precursor salts. All chemicals used were analytical reagents and were used as received without further purification.Mg- and Ca-doped ceria/zirconia samples with the compositions of Mg0.05Ce0.75Zr0.2O1.95, Mg0.1Ce0.7Zr0.2O1.9, Ca0.05Ce0.75Zr0.2O1.95 and Ca0.1Ce0.7Zr0.2O1.9 were prepared via the hydrothermal treatment, and then denoted as H-CZMg5, H-CZMg10, H-CZCa5 and H-CZCa10, respectively. Appropriate amounts of nitrates and P123 were dissolved into deionized water under stirring. NaOH solution was used as precipitate agent. The pH during precipitation was kept at approximately 10. After stirred at 80 °C for 3 h, the product was filtered and washed thoroughly with deionized water. The obtained suspension was dispersed in deionized water again and then transferred into an autoclave filled with deionized water up to 80% of the total volume. Hydrothermal synthesis was then carried out at 140 °C for 24 h. After cooling to room temperature naturally, the product was collected, dried at 80 °C and calcined at 600 °C in air for 4 h.
For comparison, Ce0.8Zr0.2O2, Mg0.05Ce0.75Zr0.2O1.95 and Ca0.05Ce0.75Zr0.2O1.95 were also prepared via conventional co-precipitation method,30 and denoted as c-CZ, c-CZMg5 and c-CZCa5, respectively.
2.2. Characterization
Thermochemical CO2 splitting reactions are performed at high temperatures, usually higher than 1400 °C, which is significantly higher than the calcination temperature during the sample preparation process. As a result, some unfavorable changes on the structure characteristics and oxygen handling properties of the cycled materials would certainly happen as compared to fresh samples, and then further change the redox properties. Thus, in order to investigate the structure, thermal stability and oxygen mobility of the samples, the as-prepared samples as well as that thermally treated at 1400 °C (10 °C min−1) in air for 2 h were characterized by XRD, Raman, N2 adsorption, TPR and SEM.X-ray diffraction (XRD) patterns of the samples were recorded on Rigaku D/max-2200/PC diffractometer equipped with Cu Kα radiation (λ = 0.15418 nm) at the tube current of 20 mA and tube potential of 40 kV.
Raman spectra measurements were performed on a confocal microprobe Raman system (LabRam II, Dilor, France). A liquid nitrogen-cooled 1024 × 800 pixels charge-coupled device was used as a detector and an exciting line of 632.8 nm was supplied by a He–Ne laser using a power of ca. 5 mW. Calibration was done by using the 519 cm−1 line of silicon as a reference.
Temperature-programmed reduction (TPR) was carried out in a quartz tube reactor; TCD was used for the collection of the H2 consumption signal. Prior to reduction, 50 mg of sample was heated in Ar (30 mL min−1) from room temperature to 300 °C in order to remove surface contaminants. After cooled down to room temperature, the reduction process was started using 5% H2/Ar mixture with a flow rate of 30 mL min−1 and recorded from 100 °C to 1000 °C with a heating rate of 10 °C min−1.
The cycled samples were pulverized using a mortar and pestle, and the morphology of the samples was analyzed by a Scanning Electron Microscope (SEM, JSM-7001F). The specific surface area (BET) and pore volume of the samples were measured via N2 adsorption–desorption isotherms using Micromeritics ASAP-2000.
2.3. Two step CO2 splitting reactions
The thermochemical CO2 splitting reactivity of the materials was studied on Netzsch STA 449 F3 Jupiter via thermogravimetric analysis. Approximate 50 mg of the sample was used in each TGA experiment. Three successive cycles were performed for each sample according to the following temperature program: the temperature was firstly increased to 1400 °C under argon (99.99%) to perform the T-R step, after heating at 1400 °C for 40 min the temperature was decreased to 1100 °C; pure CO2 was then introduced to perform the CD-S step, which was also performed for 40 min. After the CD-S step, the temperature was increased to 1400 °C under argon again, and thus the T-R and CD-S reactions were cyclically performed. The heating and cooling rates were set at 20 °C min−1 and the flow rate of Ar and CO2 in the T-R and CD-S steps were 30 mL min−1. The mass variation due to oxygen release during T-R step and oxygen uptake during CD-S step was recorded continuously, and consequently the amounts of O2 and CO produced in the T-R and CD-S step, respectively, can be calculated from the mass variation.For the successive cycles performed at lower temperature, a T-R temperature of 1200 °C and a CD-S temperature of 1000 °C were used and the duration of the two steps were 60 and 40 min, respectively. Two successive cycles were performed for each sample with the rest reaction conditions the same as that mentioned above.
The volume of O2 released (mL g−1) in the T-R step is calculated according to eqn (3).
| VO2 = VmΔmloss/(MO2ms) | (3) |
The CO production (mL g−1) in the CD-S step is calculated using eqn (4).
| VCO = VmΔmgain/(MOms) | (4) |
3. Results and discussion
3.1. Physicochemical–chemical properties
Fig. 1 shows the XRD patterns of the as-prepared Mg- and Ca-doped samples. Clearly, all samples showed the single fluorite-like cubic phase, indicating that the introduction of Mg and Ca did not change the phase structure of ceria/zirconia. No extra peaks ascribed to ZrO2, MgO and CaO were observed in the diffraction patterns, which was due to the incorporation of ZrO2, MgO and CaO into the CeO2 lattice and then the formation of M–Ce–Zr–O ternary solid solutions. Compared to co-precipitated samples, diffraction peaks for hydrothermal-treated materials were broadened to some degree, indicating their presence in small particles. Besides, the diffraction peaks slightly shifted toward the higher angles for Mg-doped samples, suggesting the lattice shrinkage caused by the introduction of Mg2+ with smaller ionic radius (0.066 nm) than that of Ce4+ (0.097 nm).28 Similarly, lattice expansion upon the substitution of Ca was expected due to larger ionic radius of Ca2+ (0.111 nm). However, diffraction patterns of Ca-doped samples also shifted toward the same higher angles, which were also observed over some other Ca-doped ceria and ceria/zirconia samples.32,33 This might be related to the fact that the structural parameter of Ca–Ce–Zr–O solid solution is strongly influenced by the variations in the local order, which is a function of the preparation method.32,33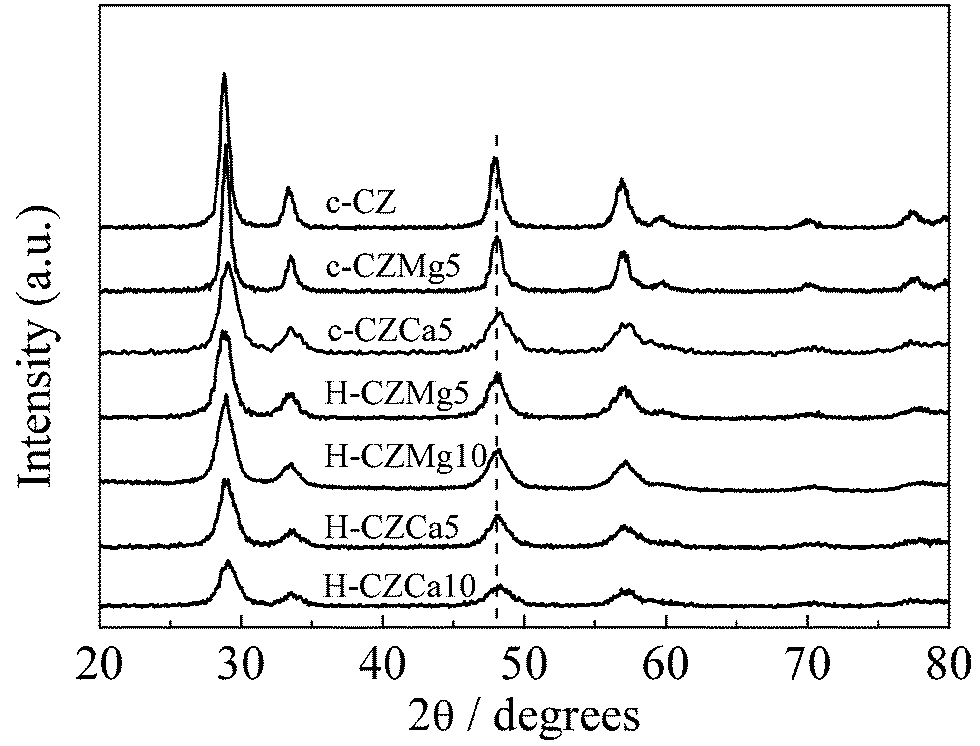 | ||
| Fig. 1 XRD patterns of the as-prepared samples. | ||
In order to further determine the exact phases and lattice defects of those solid solutions, the as-prepared samples were also analyzed by Raman spectra, an effective tool to analyze the M–O bond arrangement especially for ceria-based materials. As shown in Fig. 2, all samples were dominated by a strong band at ca. 470 cm−1, which could be attributed to the F2g vibration of the fluorite-structured phase.32,34 Two weak bands centered at about 304 and 622 cm−1 could also be observed due to tetragonal displacement of oxygen atoms from their ideal fluorite lattice positions, suggesting the t′′ or t′ phase.32 Furthermore, it can also be noticed that the intensity of the F2g vibration peaks for the doped samples were lower than that of the undoped sample. Considering about the Raman spectra sensitivity towards the crystalline symmetry,34 the intensity decrease of the F2g peaks suggested that the presence of Mg2+ and Ca2+ could cause the deformation of ceria/zirconia lattice structure, especially for the Ca-doped ones. The much lower intensity of F2g vibration for Ca-doped samples than that of Mg-doped samples significantly indicated a higher disorder on the anion sublattice.
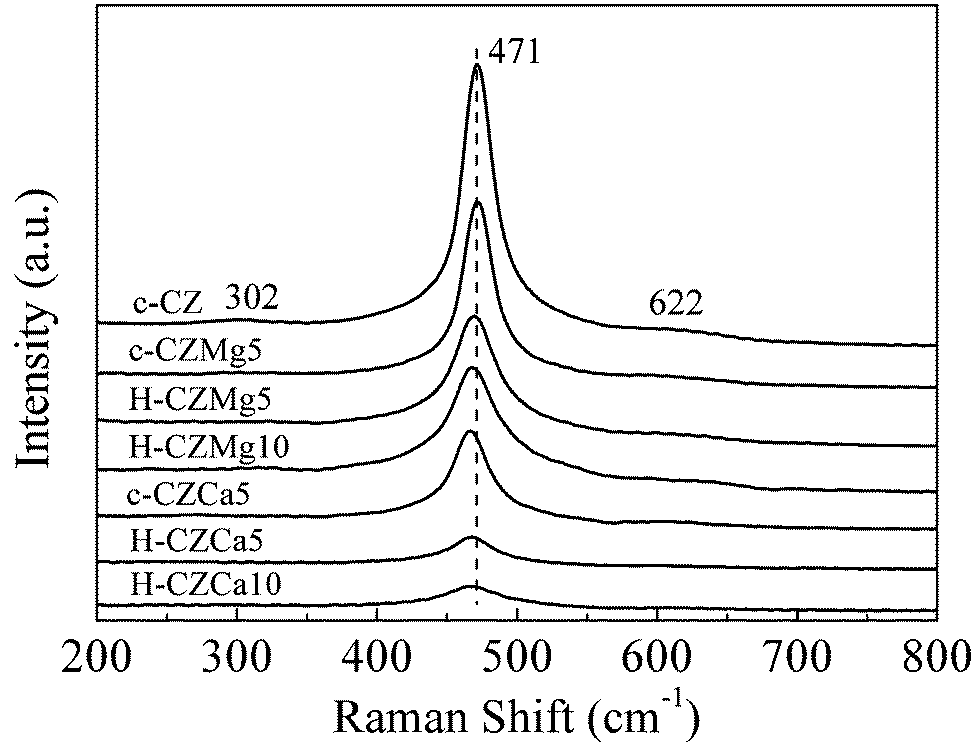 | ||
| Fig. 2 Raman spectra of the as-prepared samples. | ||
As mentioned above, both XRD and Raman spectra indicated that Mg and Ca were incorporated into the ceria/zirconia lattice, forming M–Ce–Zr–O ternary solid solutions. The incorporation of Mg/Ca with different ionic radius could lead to a shrink/expansion of the cell lattice and then the generation of lattice defects and oxygen vacancies, which was reported to favour the mobility of oxygen and then influence the redox performance of the materials.34
Fig. 3 shows the TPR profiles of the fresh samples. It can be seen that all samples exhibited one broad peak centered at ca. 570–610 °C and several minor peaks at lower temperatures, indicated that the reduction of ceria located at the surface and the bulk occurred almost concurrently. It can also be observed that, compared to the co-precipitated samples, the reduction temperature of the hydrothermal treated materials decreased to lower temperatures (ca. 30 °C). And the area of the reduction peaks for the hydrothermal treated materials was also higher than that of the co-precipitated samples due to the smaller particles, indicating the higher reduction extent. Moreover, the main reduction peaks of H-CZCa5 and H-CZCa10 were significantly stronger and sharper than that of the co-precipitated and Mg-doped samples, suggested that most of ceria was reduced almost at the same time, which is the characteristic of better oxygen mobility. As a result, it can be concluded that the templated Mg- and Ca-doped samples had an enhanced reduction performance compared to co-precipitated samples, which was due to both small particle size and relative good oxygen mobility.
 | ||
| Fig. 3 Temperature-programmed reduction (TPR) profiles of the fresh samples. | ||
However, significant change was observed over the reduction profiles of all the samples after a high temperature thermal treatment at 1400 °C. And the sintered Mg- and Ca-doped samples showed different reduction behavior, as shown in Fig. 4. As compared to fresh samples, the main reduction peaks of all the sintered samples significantly shifted towards higher temperature due to the severely particle aggregation. Sintered c-CZ only had a bulk reduction peak at about 742 °C, but for c-CZMg5, another main reduction peak centered at 658 °C could also be observed. According to the previous studies, the lower temperature reduction peak could be ascribed to the reduction of small particles and surface/sub-surface of the samples, and accordingly, the reduction peak at the higher temperature could be assigned to the reduction of bulk crystallites.35,36 This implied that sintered c-CZMg5 possessed higher surface area than sintered c-CZ, i.e. the agglomeration for c-CZ was more serious than c-CZMg5. The intensity of the surface reduction peak of H-CZMg5 was stronger than c-CZMg5; meanwhile, the surface and bulk reduction temperatures of H-CZMg5 were somewhat lower than that of c-CZMg5. And H-CZMg10 even showed a main reduction peak at about 640 °C with a minor bulk reduction peak at high temperature. This suggested that the presence of Mg enhanced the sample thermal stability and the hydrothermal-treated samples possessed relative higher thermal stability than co-precipitated materials.
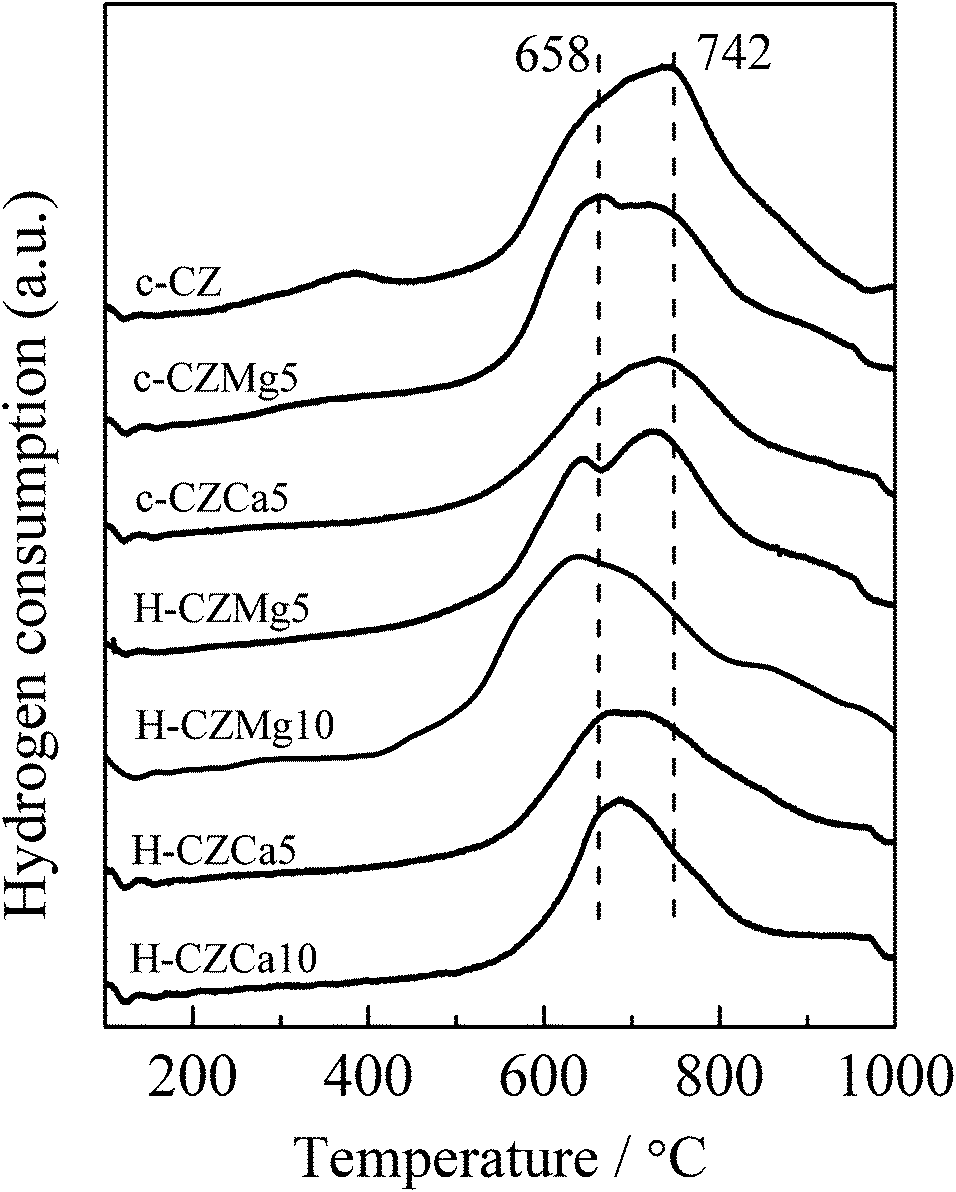 | ||
| Fig. 4 TPR profiles of the samples sintered at 1400 °C for 2 h. | ||
Different from sintered Mg-doped samples, all the Ca-doped samples still showed only one main reduction peak even after the high temperature thermal treatment, which was centered between the surface (ca. 660 °C) and the bulk (ca. 740 °C) reduction peaks of the sintered Mg-doped samples, suggesting the concurrent reduction of ceria located at the surface and the bulk.27,36 It means that oxygen located in the bulk would fast diffuse to the surface as surface oxygen was consumed, which is the indicator of fast oxygen mobility. As a result, Ca-doped samples may possess both higher oxygen mobility and better homogeneity than Mg-doped samples after the severely thermal treatment at 1400 °C.
Obviously, the introduction of Mg and Ca into ceria/zirconia solid solution could improve oxygen mobility and more importantly enhance the thermal stability, which are the key factors to improve the cycling performance of the samples in high temperature thermochemical cycles.
3.2. Thermochemical CO2 splitting reactivity
Fig. 5 shows the TGA curves of the thermochemical CO2 splitting cycles using the hydrothermal-treated samples. Three consecutive cycles were carried out for each sample and the mass variation was recorded during the cyclic experiment. It could be found from all the TGA curves that a fast mass loss beginning at ca. 1100 °C was observed due to the thermal reduction of samples, and then the weight loss proceeded slowly at the temperature up to 1400 °C, which suggested a two stage T-R process, the initial fast surface reduction at low temperature and the subsequent bulk reduction at high temperature.17,20 For the CO2 splitting reaction, CO could be produced via the regeneration of reduced samples by the re-oxidation of CO2, and in the first few minutes after the injection of CO2, the mass recovery proceeded very fast and then slowly afterwards. As a result, the rapid surface reaction upon CO2 injection was then followed by a slow re-oxidation step, which was mainly controlled by the diffusion gas-phase inside the powder.20 Meanwhile, the fast surface reaction in CD-S step tended to disappear and then showed a main slow step after the first cycle for H-CZMg5, which was also observed over some other ceria based materials.17,20 This mainly originated from the significant sintering, which strongly restricted the diffusion of CO2 in the particle bed. As a result, the CD-S reaction would transform to the diffusion controlled regime after the limited amount of ceria located at the surface of particles was re-oxidized by CO2, and then a relatively long duration of the CD-S reaction was desirable to have a complete re-oxidation. As can be seen from the TGA curve, the weight of H-CZMg5 increased in the whole CD-S step. While H-CZMg10 showed more stable cycling reactivity, due to the higher thermal stability (see Fig. 6). | ||
| Fig. 5 TGA of CO2 splitting experiments during the three successive cycles using the hydrothermal treated samples. | ||
 | ||
| Fig. 6 SEM images of (a) H-CZMg5, (b) H-CZMg10, (c) H-CZCa5 and (d) H-CZCa10 after three cyclic reactions in the temperature range of 1100–1400 °C. | ||
On the other hand, it can be noticed that all the Ca-doped samples had higher mass loss in the T-R step, suggesting the higher thermal reduction extent of ceria due to the enhanced oxygen diffusion rate. Because of the increased reduction yield, which means higher oxygen vacancy concentration, the Ca-doped samples also showed higher CO2 splitting reactivity as compared to Mg-doped samples during the three cyclic reactions. And the fast CO2 re-oxidation step could be observed in all the three cyclic reactions for the two Ca-doped samples without the apparent rate-limiting transition between the surface reaction and the bulk reaction. The weight of H-CZCa5 slightly increased after the first cyclic reaction, while that of H-CZCa10 kept decreasing during all the three cycles. Although within error, this might be mainly originated from the relative high re-oxidation extent of the reduced sample (the CO/O2 ratio was 1.56) in the first cycle and the low reduction extent of Ce4+ in the second cyclic reaction. However, in comparison to the fresh sample, the weight of H-CZCa5 still decreased after the three cyclic reactions because of the incomplete re-oxidation of the reduced sample, as it also happened in the case of H-CZCa10.
The amounts of O2 and CO produced in the TGA experiments with respect to each cycle were estimated from the mass change of the samples during the three cyclic reactions (see Table 1). With the increase of cyclic number, the reactivity of all the samples somewhat decreased because of the significant particle aggregation. It can also be seen that the Ca-doped samples released more oxygen than Mg-doped samples, suggesting that the presence of Ca could more effectively enhance the thermal reduction extent of ceria compared to Mg. Meanwhile, Ca-doped samples also produced much higher amounts of CO than Mg-doped samples and the results obtained over some other ceria and ceria/zirconia solid solutions.17,20 This might originate from the synergism caused by co-presence of Zr and Ca, which could more effectively improve both bulk oxygen mobility and thermal stability as compared to Mg dopant. As a result, the bulk can also participate in the cyclic reactions leading to the higher CO2 splitting cycling reactivity of the Ca-doped samples.
| O2 released (mL g−1) | CO produced (mL g−1) | CO/O2 ratio | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 1st cycle | 2nd cycle | 3rd cycle | 1st cycle | 2nd cycle | 3rd cycle | 1st cycle | 2nd cycle | 3rd cycle | |
| a It is taken from our previous results.30 | |||||||||
| c-CZa | 2.60 | 2.60 | — | 4.65 | 3.97 | — | 1.79 | 1.53 | — |
| H-CZMg5 | 3.41 | 2.53 | 2.53 | 4.34 | 4.78 | 3.89 | 1.27 | 1.89 | 1.54 |
| H-CZMg10 | 3.36 | 3.03 | 2.73 | 4.77 | 5.44 | 5.41 | 1.42 | 1.80 | 1.98 |
| H-CZCa5 | 4.90 | 3.08 | 2.80 | 7.64 | 6.16 | 5.18 | 1.56 | 2.00 | 1.85 |
| H-CZCa10 | 4.83 | 3.41 | 3.27 | 6.03 | 5.57 | 5.53 | 1.25 | 1.63 | 1.69 |
The P123-templated hydrothermal treated samples were composed by nanosized particles (not shown here).30 Fig. 6 shows the SEM images of the samples obtained after the three cyclic reactions. As compared to the fresh samples, significant particle aggregation was observed over all the samples due to the successive high temperature thermal treatment (1400 °C). As a general trend, the particle size of the cycled samples decreased with the increase of Mg and Ca doping content, indicating that the presence of Mg and Ca can effectively enhance the sample thermal stability. The sample thermal stability is essential for the cycling rate and CO2 splitting reactivity, especially for Mg-doped samples, which could achieve lower ceria reduction extent due to the relative slower oxygen diffusion rate. For example, H-CZMg5, which suffered more significant sintering, showed slower re-oxidization rate in the CD-S step than H-CZMg10.
3.3. Thermochemical CO2 splitting at relatively low T-R temperature
The present P123-added hydrothermal-treated Mg- and Ca-doped samples had a high surface area and porous structure, and these samples also possessed relative high oxygen mobility. Fig. 5 indicated that a large amount of mass loss took place before the temperature was increased up to 1400 °C, which indicated that the cyclic reaction could probably be carried out at a lower T-R temperature. Thus, the CO2 splitting reaction was then conducted using H-CZMg5 and H-CZCa5 at the T-R temperature of 1200 °C and a CD-S temperature of 1000 °C, respectively.Fig. 7 gives the mass variation of the two materials during the two cyclic reactions. The mass loss occurred below 900 °C could be ascribed to the desorption of those surface contaminants (such as H2O and CO2) and the decomposition of carbonates, and the thermal reduction of ceria took place afterwards. A relatively high reduction degree of ceria was observed at the T-R temperature of 1200 °C with the mass recovery in the subsequent CD-S step. But the TGA curves appeared remarkably different from that at a relatively high T-R temperature (e.g. 1400 °C). Except the first T-R reaction, a linear change mass in both T-R and CD-S reactions was observed for the two samples in both the two cycles, i.e. it nearly completed in the first few minutes (<10 min) and hardly changed in the subsequent time. This suggested that the dwell time of T-R and CD-S steps would be greatly shortened, which is important for the increase in fuel productivity.
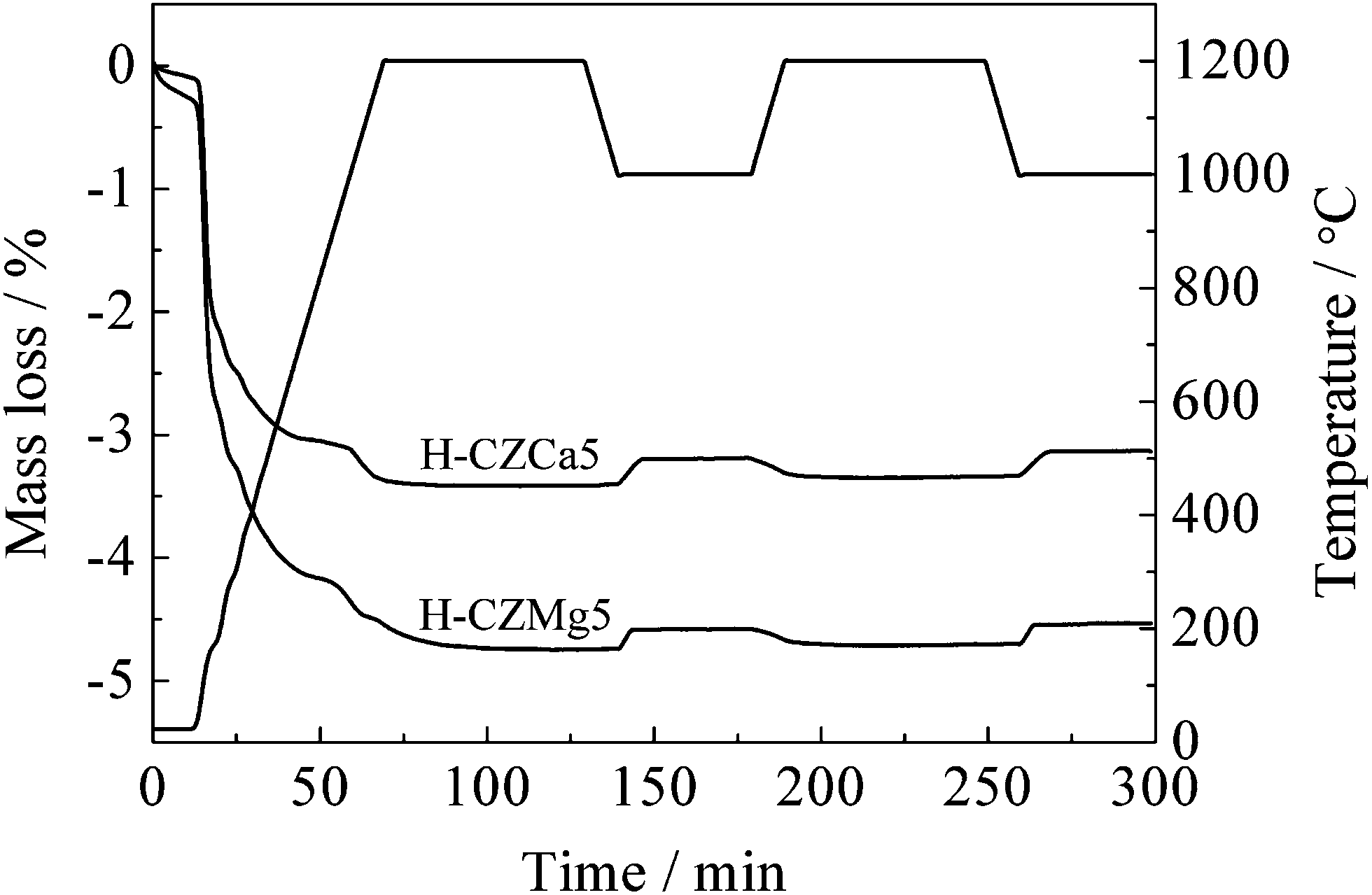 | ||
| Fig. 7 TGA of CO2 splitting experiments performed at 1000–1200 °C using H-CZMg5, and H-CZCa5. | ||
The BET surface areas of the fresh hydrothermal treated samples were in the range of 80–100 m2 g−1, which decreased to near zero after successive cyclic CO2 splitting reactions under 1100–1400 °C. However, the BET surface areas of H-CZMg5 and H-CZCa5 cycled in the temperature range of 1000–1200 °C were about 1.2 m2 g−1. Fig. 8 shows the SEM images of H-CZMg5 and H-CZCa5 obtained after the two cyclic reactions. Obviously, both crystallite growth and sintering still took place over the two samples. However, both of the two cycled samples were porous with the particle size significantly smaller than those obtained at the T-R temperature of 1400 °C. Clearly, the small particle size and pore structure were important for CO productivity and reaction kinetics. The small particle size allow the fast diffusion of oxygen between bulk and surface during the reduction and re-oxidation reactions; and the porosity facilitated the diffusion of reactant (CO2) and the evolution of the product gases (O2 and CO) during repeated cycles. And thus both the T-R and CD-S steps only showed the fast reaction step without the presence of the gas-phase diffusion controlled step. As a result, rapid cycling rates were observed in the cycling experiments, which was extraordinarily different from that obtained at 1100–1400 °C.
 | ||
| Fig. 8 SEM images of (a) H-CZMg5 and (b) H-CZCa5 after the two cyclic reactions in the temperature range of 1000–1200 °C. | ||
The amounts of oxygen released over H-CZMg5 were 1.86 and 0.86 mL g−1 in the first and the second cycle, corresponding to CO of 2.44 and 2.35 mL g−1, respectively, and for H-CZCa5, the volumes of oxygen released in the two cycles were 2.1 and 1.04 mL g−1 with the amounts of produced CO of 3.24 and 2.89 mL g−1, respectively. Rudisill et al. reported a more rapid cycling rates with the duration of each step less than 2 min over three-dimensionally ordered and nonordered macroporous CeO2 in the temperature range of ∼850–1200 °C,21 and the average O2 and CO production values over the macroporous CeO2 were about 0.5 and 1 mL g−1, respectively, which were lower than our results. The better thermochemical cycling behavior in the present work could be ascribed to both higher thermal stability and better oxygen handling properties of ternary solid solutions than pure CeO2.
The present results indicated that the cyclic thermochemical reactions could be performed at moderate T-R temperatures while maintaining relative high fuel productivity, by using samples with proper morphology and more importantly fast oxygen diffusion rate. Less unfavorable change on the structure characteristics of the samples would happen under lower temperatures and then decrease the reactivity loss due to sample aggregation caused by high operating temperatures. Moreover, this kind of materials could be cycled under a modest temperature swing between T-R and CD-S steps (1100–1200 °C) and even an isothermal redox cycle (1200–1200 °C), which has the advantage of reaching significantly higher energy conversion efficiency.37,38
In addition, it should be mentioned that the conditions in a TGA setup will be greatly different from those in solar reactor, in which more larger amount of redox samples would be used as compared to TGA experiment. Then, temperature gradients would exist in solar reactor because of the low effective conductivity of the porous sample beds and the high heat fluxes required for rapid heating and cooling, especially for the experiments performed under low T-R temperatures (the samples would be far different from dense samples).21 In contrast, less or even no temperature gradient would exist in TGA experiment, because of the relative slower heating and cooling rates and the lower amount of loaded samples. Meanwhile, it also can be anticipated that the flow of gas in solar reactor would be different from that of the TGA experiment. Obviously, both the temperature and gas diffusion have significant influence on the cycling performance of the materials. Considering these aspects, the reactivity as well as the long term stability of the samples will be investigated in our future work using solar reactor and/or packed bed reactor.
4. Conclusions
With the aim of recycling CO2 into valuable CO feedstock, the thermochemical fuel production via CO2 splitting reaction was carried out by using Mg- and Ca-doped ceria/zirconia solid solutions in the temperature range of 1100–1400 °C. The presence of Mg and Ca led to the lattice defects in the fluorite lattice and then the strongly modification of oxygen handling properties. More importantly, thermal stability of the doped samples was greatly enhanced by the doping of Mg and Ca. The addition of Ca could more effectively enhance the mobility of oxygen in ceria/zirconia lattice than Mg, and thus faster reaction rates and higher CO2 splitting reactivity could be observed over Ca-doped samples compared to Mg-doped samples.The samples modified by addition of P123 template were composed of small particles and then allowed the fast diffusion of oxygen in the short diffusion route. Greatly different from the samples cycled at 1100–1400 °C, the samples cycled at the T-R temperature of 1200 °C were of porosity with greatly smaller particle size. As a result, fast reaction rates and relative stable cycling performance were observed at 1000–1200 °C.
Acknowledgements
This work was supported by the Natural Science Foundation of China (21301186); “Strategic Priority Research Program-Climate Change: Carbon Budget and Related Issues” of the Chinese Academy of Sciences, Grant no. XDA05010109, XDA05010110 and XDA05010204.Notes and references
- G. Centi and S. Perathoner, ChemSusChem, 2010, 3, 195–208 CrossRef CAS PubMed.
- T. Kodama and N. Gokon, Chem. Rev., 2007, 107, 4048–4077 CrossRef CAS PubMed.
- A. Meier and A. Steinfeld, Adv. Sci. Technol., 2010, 74, 303–312 CrossRef CAS.
- J. Kim, T. A. Johnson, J. E. Miller, E. B. Stechel and C. T. Maravelias, Energy Environ. Sci., 2012, 5, 8417–8429 CAS.
- R. J. Price, D. A. Morse, S. L. Hardy, T. H. Fletcher, S. C. Hill and R. J. Jensen, Ind. Eng. Chem. Res., 2004, 43, 2446–2453 CrossRef CAS.
- A. J. Traynor and R. J. Jensen, Ind. Eng. Chem. Res., 2002, 41, 1935–1939 CrossRef CAS.
- G. P. Smestad and A. Steinfeld, Ind. Eng. Chem. Res., 2012, 51, 11828–11840 CrossRef CAS.
- A. H. McDanie, E. C. Miller, D. Arifin, A. Ambrosini, E. N. Coker, R. O'Hayre, W. C. Chueh and J. Tong, Energy Environ. Sci., 2013, 6, 2424–2428 Search PubMed.
- D. Arifin, V. J. Aston, X. Liang, A. H. McDaniel and A. W. Weimer, Energy Environ. Sci., 2012, 5, 9438–9443 CAS.
- E. N. Coker, A. Ambrosini, M. A. Rodriguez and J. E. Miller, J. Mater. Chem., 2011, 21, 10767–10776 RSC.
- E. N. Coker, J. A. Ohlhausen, A. Ambrosini and J. E. Miller, J. Mater. Chem., 2012, 22, 6726–6732 RSC.
- S. Abanades and H. I. Villafan-Vidales, Chem. Eng. J., 2011, 175, 368–375 CrossRef CAS PubMed.
- P. G. Loutzenhiser, M. E. Galvez, I. Hischier, A. Graf and A. Steinfeld, Chem. Eng. Sci., 2010, 65, 1855–1864 CrossRef CAS PubMed.
- S. Abanades and M. Chambon, Energy Fuels, 2010, 24, 6667–6674 CrossRef CAS.
- W. C. Chueh and S. M. Haile, ChemSusChem, 2009, 2, 735–739 CrossRef CAS PubMed.
- P. Furler, J. R. Scheffe and A. Steinfeld, Energy Environ. Sci., 2012, 5, 6098–6103 CAS.
- A. L. Gal, S. Abanades and G. Flamant, Energy Fuels, 2011, 25, 4836–4845 CrossRef.
- W. C. Chueh and S. M. Haile, Philos. Trans. R. Soc., A, 2010, 368, 3269–3294 CrossRef CAS PubMed.
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797–1801 CrossRef CAS PubMed.
- A. L. Gal, S. Abanades, N. Bion, T. L. Mercier, and V. Harle, Energy Fuels, 2013, 27, 6068–6078 Search PubMed.
- S. G. Rudisill, L. J. Venstrom, N. D. Petkovich, T. Quan, N. Hein, D. B. Boman, J. H. Davidson and A. Stein, J. Phys. Chem. C, 2013, 117, 1692–1700 CAS.
- H. Kaneko, S. Taku, Y. Naganuma, T. Ishihara, N. Hasegawa and Y. Tamaura, J. Sol. Energy Eng., 2010, 132, 21202–21206 CrossRef.
- H. Kaneko, S. Taku and Y. Tamaura, Sol. Energy, 2011, 85, 2321–2330 CrossRef CAS PubMed.
- H. Kaneko, T. Miura, H. Ishihara, S. Taku, T. Yokoyama, H. Nakajima and Y. Tamaura, Energy, 2007, 32, 656–663 CrossRef CAS PubMed.
- A. L. Gal and S. Abanades, J. Phys. Chem. C, 2012, 116, 13516–13523 Search PubMed.
- P. Vidmar, P. Fornasiero, J. Kaspar, G. Gubitosa and M. Graziani, J. Catal., 1997, 171, 160–168 CrossRef CAS.
- J. Fan, D. Weng, X. Wu and R. Ran, J. Catal., 2008, 258, 177–186 CrossRef CAS PubMed.
- Y. An, M. Shen and J. Wang, J. Alloys Compd., 2007, 441, 305–310 CrossRef CAS PubMed.
- M. Fernandez-Garcia, A. Martinez-Arias, A. Guerrero-Ruiz, J. C. Conesa and J. Soria, J. Catal., 2002, 211, 326–334 CAS.
- M. Kang, J. Zhang, C. Wang, F. Wang, N. Zhao, F. Xiao, W. Wei and Y. Sun, RSC Adv., 2013, 3, 18878–18885 RSC.
- Y. Yao, D. Li, T. Xu and T. Fu, Seventh International Conference on Thin Film Physics and Applications, 2011 Search PubMed.
- M. Fernández-García, X. Wang, C. Belver, A. Iglesias-Juez, J. C. Hanson and J. A. Rodriguez, Chem. Mater., 2005, 17, 4181–4193 CrossRef.
- J. A. Rodriguez, X. Wang, J. C. Hanson, G. Liu, A. Iglesias-Juez and M. Fernández-García, J. Chem. Phys., 2003, 119, 5659–5669 CrossRef CAS PubMed.
- I. Atribak, A. Buenolopez and A. Garciagarcia, J. Catal., 2008, 259, 123–132 CrossRef CAS PubMed.
- Q. Wang, G. Li, B. Zhao, M. Shen and R. Zhou, Appl. Catal., B, 2010, 101, 150–159 CrossRef CAS PubMed.
- F. Fally, V. Perrichon, H. Vidal, J. Kaspar, G. Blanco, J. M. Pintado, S. Bernal, G. Colon, M. Daturi and J. C. Lavalley, Catal. Today, 2000, 59, 373–386 CrossRef CAS.
- R. Bader, L. J. Venstrom, J. H. Davidson and W. Lipiński, Energy Fuels, 2013, 27, 5533–5544 CAS.
- C. L. Muhich, B. W. Evanko, K. C. Weston, P. Lichty, X. Liang, J. Martinek, C. B. Musgrave and A. W. Weimer, Science, 2013, 341, 540–542 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |