
Synthesis and photovoltaic properties of solution-processable star-shaped small molecules with triphenylamine as the core and alkyl cyanoacetate or 3-ethylrhodanine as the end-group
Abstract
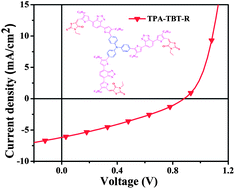
Two novel star-shaped donor–acceptor small molecules, TPA–TBT–CN and TPA–TBT–R, with triphenylamine (TPA) as the core, benzothiadiazole (BT) as the arm, and alkyl cyanoacetate or 3-ethylrhodanine as the end-group are synthesized for application as donor materials in OSCs. The two small molecule films show broad absorption bands from 300 nm to 850 nm, narrow optical band gaps (1.5–1.7 eV), deep HOMO energy levels (−5.0 to −5.1 eV) and moderate hole mobilities. OSCs based on blends of the two donors and PC70BM acceptors exhibit power conversion efficiencies of 1.34% and 1.79%, respectively. Notably, TPA–TBT–R with 3-ethylrhodanine as the end-group displays a broader solar spectral coverage, a lower HOMO level, a higher hole mobility and higher photovoltaic properties. Our results indicate that 3-ethylrhodanine as the acceptor and end-group is a promising linker in constructing donor materials for high efficiency OSCs.
 Please wait while we load your content...
Please wait while we load your content...

