
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen adsorption and dissociation on AunY (n = 1–12) nanoclusters: a DFT investigation†
Nguyen Van Danga,
Ngo Thi Lan *a,
Nguyen Thi Maib,
Son Tung Ngocd,
Phung Thi Thue and
Nguyen Thanh Tungb
*a,
Nguyen Thi Maib,
Son Tung Ngocd,
Phung Thi Thue and
Nguyen Thanh Tungb
aInstitute of Science and Technology, TNU-University of Sciences, Tan Thinh Ward, Thai Nguyen City, Vietnam. E-mail: lannt@tnus.edu.vn
bInstitute of Materials Science, Vietnam Academy of Science and Technology, Hanoi, Vietnam
cLaboratory of Biophysics, Institute for Advanced Study in Technology, Ton Duc Thang University, Ho Chi Minh City, Vietnam
dFaculty of Pharmacy, Ton Duc Thang University, Ho Chi Minh City, Vietnam
eUniversity of Science and Technology of Hanoi, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Hanoi, Vietnam
First published on 14th July 2025
Abstract
The interaction between nanomaterial systems and hydrogen has recently emerged as a compelling study model, offering valuable insights for designing materials with applications in nanotechnology, catalysis, and energy storage. Among these, transition metal-doped gold clusters exhibits intriguing stability and electronic properties, making them promising candidates for hydrogen related processes. In this study, we employ density functional theory (DFT) to investigate the interaction between H2 molecules and small-sized gold clusters doped with yttrium. Our finding reveals that most of the bare cluster structures remain intact during the H2 adsorption, regardless of whether the process occurs molecularly or dissociatively. A comprehensive analysis indicates that the preferred adsorption configuration is governed by multiple factors, including adsorption site (surface vs. encapsulated), relative electronegativity, and the atomic coordination number. The calculations demonstrate that dissociative adsorption of H2 on Au6Y and Au11Y clusters is both thermodynamically and kinetically favorable. However, for AunY (n = 1, 4–5, 8, 10) dissociative adsorption is hindered by a significant energy barrier before reaching the final state, while for species with n = 2, 3, 7, and 9 molecular adsorption is more favorable due to intrinsic energy preferences. This study provides fundamental insights into the adsorption sites and detailed analysis of adsorption kinetics on AunY clusters, laying the groundwork for further theoretical an ed experimental investigations into the hydrogenation process in nanostructured materials.
1. Introduction
Nowadays, hydrogen has been envisioned as a key energy carrier for sustainable development of the future economy, meeting essential environmental and energy requirements.1–3 Compared to traditional fuels like gasoline (44 MJ kg−1) and diesel (45.6 MJ kg−1),4,5 hydrogen offers the highest energy density per unit mass (∼120 MJ kg−1) and conversion efficiency and can be produced through water-splitting reactions without CO2 emissions. Moreover, it can be conveniently stored in various forms, including gaseous, liquid, or metal hydrides.6–8 However, the practical implementation of hydrogen energy storage after generation and during transport faces significant challenges, primarily due to its kinetic stability under ambient conditions and the low energy density per unit volume of solid storage materials.To address this bottleneck, various strategies have been explored.9–12 Among them, metal hydride materials have garnered special attention as model solid-state hydrogen storage systems, offering a more compact and secure alternative. They provide the possibility of preventing critical operating conditions like high pressures and elevated temperatures, which are typical of conventional compressed gas and liquefied hydrogen storage.13 To enhance the practical prospect of metal hydride, a deeper understanding of the interaction between hydrogen and potential surface at the molecule level is essential. Therefore, nanoclusters consisting of a few atoms have emerged as promising candidates for providing fundamental insights into potentially reactive sites, given significant variation in physical and chemical properties with size and composition.
Among various metal nanoclusters, gold nanoclusters (Aun) are considered particularly advantageous due to their superior stability, abundance, and cost-effectiveness, even surpassing palladium and platinum in many aspects. While gold is chemically inert in its bulk form, it exhibits remarkable catalytic activity at the nano scale. For example, gold nanoclusters have shown promising performance in catalytic processes such as water-splitting reaction14 and CO oxidation.15 Interestingly, this catalytic behavior is highly dependent on their geometric structure. Bootharaju and co-workers16 investigated the Au12Ag32/TiO2, system, which features a gold icosahedral core encapsulated by a silver dodecahedral shell. Their finding revealed a substantial enhancement in solar H2 production efficiency, approximately 6.2 and 37.8 times higher than those of Ag44/TiO2 and bare TiO2, respectively. Furthermore, key factors influencing the reactivity and catalytic potential of gold nanoclusters include cluster size, atomic composition, and charge state.17–19 By tailoring these parameters, such as through doping or precise control of cluster size, researchers can design tunable materials optimized for specific catalytic processes. For instance, both experimental and theoretical studies have explored O2 activation on small-sized anionic Aun− clusters.20 S. Pal and D. Manzoor have compared the catalytic efficiency of Au8, Pd8, Au8−n Pdn (n = 1–7), Au7Si,21,22 Au18 and endohedral Au18M (M = Na, K, Mg, Ca, Al, Ga) cages,23 and Al-, Hf-, and Ge-doped Au20 cages.24 Similarly, Zeng and co-workers25 using DFT calculation studied the catalytic behavior of sub-nanometer-sized gold nanoclusters supported on TiO2. They observed that cluster shape and size significantly influenced catalytic performance, with adsorption energies for O2 and CO decreasing as the cluster size increased within the range of 16–35 atoms.15 Recent studies have also highlighted the emerging role of hydrogen atoms as dopants in gold nanoclusters.26 Beyond their electronegativity, hydrogen atoms can induce chemisorption and activation on previously inert or closed-shell gold clusters,27 modulated by cluster shape,28,29 and charge state.30 This, in turn, enhances the adsorption and activation of O2 and CO molecules, opening new directions for designing advanced gold-based catalysts.
Research efforts have expanded beyond the studies of O2 adsorption and CO oxidation to include investigations of metal nanoclusters and their alloys in order to deepen our understanding of the kinetics and dynamics of hydrogenation reactions. Numerous density functional theory (DFT)31–34 studies have investigated the adsorption and dissociation of hydrogen on metal nanoclusters. The H2 absorption process has been found to strongly depend on the physical and chemical properties, structural, size, and composition of nanoclusters, as demonstrated in 3d transition metal (Sc–Zn)-doped Cu clusters35 and Au24M, Au36M (M = Pt and Pd).36 Theoretical studies have demonstrated that the incorporation of a Pt dopant can significantly alter the electronic structure of gold nanoclusters Aun+1 (n = 1–12), thereby enhancing their reactivity toward H2 molecules.37 Li and coworkers investigated the effect of adsorbed hydrogen atoms on the geometric and electronic structures of alkali-doped gold nanoclusters, providing insights into hydrogen-cluster interaction.38 Additionally, the adsorption and dissociation behavior of TiMgn (n = 1–12) clusters has been examined DFT, revealing key factors that govern their hydrogen activity.39 Furthermore, mass spectrometry and infrared multiple photon dissociation spectroscopy combined with theoretical calculations, have provided insights into the competition between molecular and dissociative hydrogen chemisorption, which is highly dependent on the size of cationic aluminum-doped Rh clusters.40,41 Recently, Lan et al. conducted comparative studies on the influence of 3d transition metal atoms on hydrogen adsorption and dissociation by examining size-dependent AgnCr (n = 2–12) clusters and composition-dependent Au9M2+ (M = Sc–Ni) clusters.42,43 Their research has revealed that Ag3Cr, Ag6Cr, and Au9Ti2+, clusters exhibit the ability to adsorb and dissociate H2, leading to the formation of metal hydrides even at room temperature.
Additionally, 4d transition metal atoms have shown significant potential for hydrogen storage, attributed to their enhanced stability, fully filled electron configurations across multiple energy levels, diverse oxidation states, and distinct chemical properties compared to their 3d counterparts.44,45 Moreover, the lower electronegativity of 4d transition metals relative to 3d elements influences their interaction with hydrogen, raising several intriguing questions, such as how charge transfer occurs in clusters and the nature of interactions between dopant clusters and hydrogen molecules. To address these questions, the effects of boron and 4d transition metal (Y–Mo) on hydrogen adsorption and storage properties have been systematically investigated.45–47 Notably, the impact of charge transfer, electronegativity, and the atomic radius of the dopant elements on the catalytic mechanism and hydrogen storage capacity of MgH2 clusters has been clarified.48,49 C.S. Sergio et al. have clarified the correlation between hydrogen storage capacity and the surface binding energy of 4d transition metal-doped carbon nanoflakes.50 Recent work by J. Barabás et al., combining gas-phase reaction investigations and DFT computations, has demonstrated that the catalytic activity of gold cation clusters (Aun+) and yttrium-doped gold (Aun−1Y+, n = 4–20) clusters can be effectively tuned by cluster size, charge state, and dopant introduction.51 While initial studies have explored the influence of 4d transition metals on cluster stability and reactivity, a systematic investigation of the hydrogen storage mechanism in noble metal clusters doped with 4d transition metals remains lacking. A deeper understanding of the kinetics and dynamics of hydrogenation reactions in precious metal species and their alloys could pave the way for new research directions in hydrogen storage materials.52,53
Driven by the pursuit of nanocluster materials for efficient hydrogen storage, this study aims to theoretically explore the critical role of yttrium doping in H2 adsorption and dissociation on small gold clusters. Using DFT calculations,54 we systematically analyze the geometrical, relative stability, bonding characteristics, electronic properties, and adsorption energies of AunY (n = 1–12) clusters. The insights gained from this study provide a deeper understanding of the influence of 4d transition metal dopants on hydrogen adsorption and dissociation mechanisms. Furthermore, these findings offer valuable guidelines for future experimental studies, aiding the rational design of advanced nanomaterials for hydrogen storage applications.
2. Computational method
This work uses the DFT calculations implemented in the Gaussian 09 program55,56 in combination with GaussView, to predict the geometric and electronic structures of AunY (n = 1–12) clusters in their ground state, along with their molecularly and dissociatively adsorbed variants AunY–H2 and AunY–2H. To generate initial cluster structures, two complementary approaches were employed: (1) a stochastic algorithm was used to explore a broad range of feasible AunY (n = 1–12) configurations;57 (2) manual structure construction, in which a Y atom systematically replaced an Au atom at all possible positions within the cluster. Subsequently, hydrogen adsorption configurations were investigated based on the lowest-energy AunY (n = 1–12) structures, where a single H2 molecule or two dissociated H atoms were attached at various adsorption sites.Computationally, predicted structures were first optimized by the TPSSTPSS functionals in combination with the cc-pVDZ-pp basis set for Au and Y atoms, and LanL2DZ for H atoms. For each cluster size, various structural and spin configurations were explored and subjected to the convergence criteria. Nevertheless, only species with relative energies less than 2.0 eV were selected for single-point energy recalculations at the same functional level but with an enhanced basis set, specifically cc-pVTZ-pp for Au and Y atoms and LanL2DZ for H atoms. This approach represents a balanced trade-off between computational accuracy and efficiency and has been shown to be reliable for 4d transition metal-doped noble metal systems.58 To validate the accuracy and reliability of our computational methodology, we compared binding energies (BE, eV) of Au2, AuY, AuH, and YH dimers across different functionals, including TPSSTPSS, BP86, BPW91, BLYP, and PBEPBE. Tables 1 and S1† summarize the results and benchmarks them against available experimental data, confirming that the TPSSTPSS/cc-pVTZ-pp (for Au and Y) and LanL2DZ (for H) combination yields the closest agreement with experimental values. The self-consistent field (SCF)59 calculations were conducted with a convergence threshold of 2.0 × 10−5 Hartree per Å for gradients and 5.0 × 10−3 Å for displacements. The stability of the lowest-energy structures was further confirmed by calculating harmonic vibrational frequencies, ensuring they correspond to true minima on the potential energy surface. To reinforce the robustness of the ground-state optimization, multiple spin multiplicities were examined for each structure. The electronic configurations of all lowest-energy structures were analyzed using the natural bond orbital (NBO) method.60 To explore the reaction mechanisms, transition-state (TS) geometries were identified using the quadratic synchronous transit (QST) method,61,62 with relaxed potential energy surface (PES) scans along appropriate internal coordinates. To further verify the transition states, intrinsic reaction coordinate (IRC) calculations were performed, ensuring a continuous and valid connection between reactants and products.
| Dimer | BP86 | PW91 | BLYP | PBEPBE | TPSSTPSS | Experiment63 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| LanL2DZ | SDD | LanL2DZ | SDD | LanL2DZ | SDD | LanL2DZ | SDD | LanL2DZ | SDD | ||
| AuH | 3.07 | 3.11 | 3.02 | 3.00 | 2.94 | 2.97 | 2.94 | 2.98 | 3.00 | 3.02 | 3.03 ± 0.08 |
| YH | 2.90 | 3.12 | 2.74 | 2.82 | 2.88 | 3.02 | 2.79 | 2.85 | 2.94 | 3.23 | — |
3. Optimized geometrical structures
We first optimized the lowest-energy structures of bare AunY clusters and then examined both molecular and dissociative adsorption of hydrogen, leading to the formation of AunY–H2 and AunY–2H clusters, respectively. To ensure the stability of the obtained geometries, we conducted frequency calculations, confirming the absence of imaginary frequencies in all optimized structures. While multiple geometrical and spin isomers exist for each stoichiometry, only the lowest-energy structures of AunY, AunY–H2, and AunY–2H clusters were selected for further discussion of hydrogen adsorption behavior, as shown in Fig. 1. Other low-lying isomers for AunY–H2 and AunY–2H clusters are provided in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 46
46
 | ||
| Fig. 1 Optimized structures of AunY, AunY–H2, and AunY–2H clusters (n = 1–12). The yellow, purple, and red spheres represent Au, Y, and H atoms, respectively. | ||
Our results indicate that the stable spin configurations of AunY (n = 1–12) clusters follow an alternating pattern with a singlet spin state for odd n and a doublet for even n. The lowest-energy AunY structures tend to adopt a cage-like form as the cluster size increases, where the Y atom prefers a highly coordinated position. Interestingly, the structural framework of AunY (n = 1–12) clusters undergoes significant reconstruction upon yttrium doping, deviating from the planar configuration of pure Aun clusters (n ≤ 11).64 Unlike pure gold clusters, where the first 3D structure appears at n = 12, the introduction of a Y atom stabilizes 3D geometries at much smaller sizes, starting from n = 4, where four Au atoms coordinate around the Y dopant. The lowest-lying isomers of AunY (n = 5–8) retain a 2D morphology, whereas larger clusters (n = 9–12) exhibit 3D structural preferences. These findings align well with theoretical predictions and far-infrared spectral studies of yttrium-doped gold clusters.65
Our calculations suggest that hydrogen adsorption does not significantly alter the geometric structure of bare AunY clusters, except for Au5Y, where H2 preferentially attaches to energetically unstable bridge sites (Au–Y and Au–Au). Similar behavior has been observed in transition metal-doped clusters.41–43,66 For AunY (n = 1–5, 7, 9, and 12), the hydrogen molecule favors direct attachment to the Y dopant, attributed to differences in electronegativity χ between Y (1.22), H (2.2), and Au (2.54). Fundamentally, hydrogen bonding arises from electrostatic attraction, facilitated by charge transfer between atoms with significant electronegativity differences. Consequently, bonds between the low-electronegativity Y atom and high-electronegativity H atom are more favorable, in agreement with previous reports, where surface impurities are among the most favored adsorption sites for a hydrogen molecule on transition metal doped clusters.41–43,67 Furthermore, hydrogen adsorption is not solely governed by electronegativity differences but also strongly influenced by atomic coordination number (N). Lower coordination numbers correspond to higher surface activity, facilitating stronger H2 adsorption.68 Consequently, atomic clusters with low-coordination sites exhibit enhanced hydrogen interaction capability. For example, experimental and theoretical studies on singly rhodium-doped cationic aluminum clusters (AlnRh+) using time-of-flight mass spectrometry and infrared multiple photon dissociation, in combination with DFT calculations, have demonstrated that hydrogen preferentially binds to the Al atom rather than the Rh impurity in cluster sizes where Rh is coordination-saturated or encapsulated.41 This principle helps explain the behavior observed in Au6Y–H2, Au8Y–H2, Au10Y–H2, and Au11Y–H2 clusters, where a competition between electronegativity and coordination occurs as H2 preferentially attaches to a low-coordination Au vertex (NAu = 2 or 3) instead of the highly coordinated Y impurity (NY = 6, 8, or 9). A similar hydrogen adsorption trend has been reported for the clusters AlnCr, AgnCr, Au9M2+, AlnRh2+, and MgnCo, further reinforcing the critical role of coordination number in determining the preferred hydrogen adsorption site.41–43,66,69
Upon molecular adsorption, the hydrogen molecule may dissociate, triggering a structural transformation of the bare clusters.70 However, the ground-state geometry of AunY clusters generally remains intact upon hydrogen adsorption, with only minor distortions observed in Au2Y–2H, Au5Y–2H, and Au8Y–2H. As illustrated in Fig. 1, following H2 dissociation one Au atom shifts closer to another Au atom in Au2Y–2H, while in Au5Y–2H and Au8Y–2H, a single Au atom is slightly displaced from two-membered and three-membered Au chains, respectively. Notably, both cluster size and the position of the Y dopant significantly influence H2 dissociation and adsorption behavior, which can be categorized into two distinct trends. For small clusters (n = 2–4), individual hydrogen atoms preferentially bind to the surface Y atom, typically at the Au–Y bridge site. As the cluster size increases (n = 5–12), the Y atom becomes progressively encapsulated by surrounding Au atoms, leading to H atoms favoring Au–Au bridge adsorption rather than direct interaction with Y. This trend is in good agreement with previous investigations.43 In the case of AuY–2H, both H atoms preferentially adsorb on top of the Y surface site, highlighting the role of dopant coordination and atomic arrangement in governing hydrogen interaction dynamics.
4. Relative stability
The relative stability of a clusters is a key parameter that provides insight into its bonding or dissociation energy, which are critical in determining reactivity and stability in chemical reactions. This stability is typically assessed through average binding energies (BE, in eV) calculations. The BE is defined as the mean energy difference between the sum of the total energies of all isolated constituent atoms and the total energy of the cluster. The calculation follows the equations below:
 | (1) |
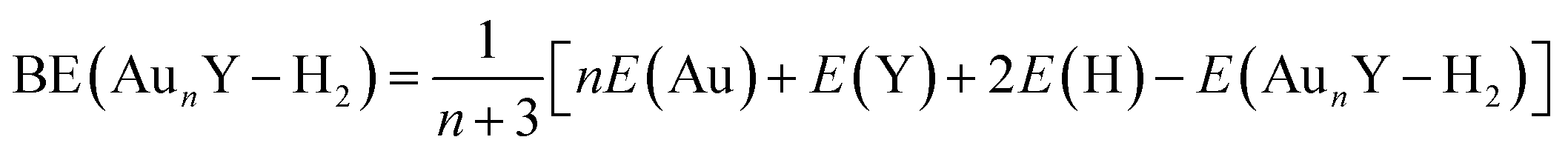 | (2) |
 | (3) |
 | ||
| Fig. 2 Comparison of per-atom binding energies (BE, in eV) for AunY, AunY–H2, and AunY–2H cluster. | ||
| n | BE/eV | Eads/eV | dH−H/Å | ||||
|---|---|---|---|---|---|---|---|
| AunY | AunY–H2 | AunY–2H | AunY–H2 | AunY–2H | AunY–H2 | AunY–2H | |
| 1 | 1.59 | 1.96 | 2.38 | 0.12 | 1.81 | 0.81 | 3.36 |
| 2 | 2.19 | 2.28 | 2.14 | 0.29 | −0.39 | 0.85 | 4.16 |
| 3 | 2.55 | 2.48 | 2.08 | 0.15 | −2.27 | 0.75 | 2.40 |
| 4 | 2.43 | 2.40 | 2.42 | 0.16 | 0.27 | 0.76 | 3.35 |
| 5 | 2.51 | 2.45 | 2.51 | 0.05 | 0.52 | 0.83 | 3.44 |
| 6 | 2.50 | 2.46 | 2.51 | 0.13 | 0.52 | 0.76 | 3.44 |
| 7 | 2.57 | 2.53 | 2.51 | 0.19 | 0.00 | 0.75 | 3.46 |
| 8 | 2.50 | 2.47 | 2.51 | 0.12 | 0.53 | 0.74 | 3.44 |
| 9 | 2.57 | 2.53 | 2.51 | 0.15 | −0.11 | 0.76 | 3.45 |
| 10 | 2.53 | 2.50 | 2.53 | 0.14 | 0.56 | 0.74 | 3.46 |
| 11 | 2.57 | 2.53 | 2.55 | 0.13 | 0.40 | 0.74 | 3.51 |
| 12 | 2.58 | 2.55 | 2.53 | 0.27 | −0.11 | 0.77 | 5.54 |
5. Hydrogen adsorption process
The accumulation of small molecules on a surface is known as adsorption, which can be classified as either physisorption or chemisorption, depending on the nature of the interaction between the adsorbate and the surface. Physisorption is governed by weak van der Waals forces, whereas chemisorption involves the formation of chemical bonds. The adsorption process is influenced by several key factors, including the host cluster, the type of adsorption, and the energy barrier between the adsorbate and the surface. A comprehensive understanding of hydrogen adsorption on AunY (n = 2–12) clusters can be obtained through detailed analysis of bonding characteristics, particularly examining the interaction of a hydrogen molecule or two dissociated hydrogen atoms within a specific chemical equilibrium. To gain fundamental insights into the adsorption processes of H2 on AunY clusters, we calculate the adsorption energy and H–H bond length for both molecular and dissociative adsorption configurations using the following equations:| Eads(AunY−H2) = E(AunY) + E(H2) − E(AunY−H2) | (4) |
| Eads(AunY−2H) = E(AunY) + E(H2) − E(AunY−2H) | (5) |
The adsorption energies (Eads) were calculated as the energy difference between the total energy of the reactants (AunY + H2) and the products (AunY–H2 or AunY–2H), as expressed in eqn (4) and (5). In essence, Eads represents the minimum energy required to desorb hydrogen from the clusters surface, as listed in Table 2. Comparing the adsorption energy of molecular and dissociative adsorption configurations, we find that all computed Eads values for the molecular adsorption (AunY–H2) are positive, ranging from 0.05 to 0.29 eV, indicating that the formation of AunY–H2 is thermodynamically favorable. For dissociative adsorption (AunY–2H), Eads values range from −0.39 to 1.81 eV. Notably, the Eads values for Au2Y–2H, Au3Y–2H, Au7Y–2H, Au9Y–2H, and Au12Y–2H clusters are negative or zero (Au7Y–2H), suggesting that dissociative adsorption on these clusters is endothermic and unlikely to occur spontaneously. This phenomenon can be attributed to the decrease in average binding energy for these clusters compared to their bare counterparts (Au2Y, Au3Y, Au7Y, Au9Y, and Au12Y). In contrast, H2 dissociation on AunY–2H (n = 1, 4–6, 8, 10 and 11) clusters is exothermic and thermodynamically favorable, as evidenced by the higher computed Eads values for dissociative adsorption AunY–2H compared to molecular adsorption AunY–H2. It is worth mentioning that molecular adsorption is more likely to occur for AunY with n = 2, 3, 7, 9, and 12. On the other hand, when H2 approaches the surface of AunY (n = 1, 4–6, 8, 10, and 11), it first undergoes molecular adsorption. Subsequently, the hydrogen molecule may either desorb from the cluster surface or eventually undergo dissociation, forming strong atomic bonds with the cluster. The detachment of adsorbed H2 molecules may dominate in certain cases, as it is influenced not only by adsorption energy but also by the activation barrier of the transition state and multiple intermediate states before reaching a stable dissociative configuration. A more detailed discussion on this topic will be provided in the following sections.
To better understand the binding nature of adsorbed hydrogen molecules on AunY (n = 1–12) clusters, we analyzed the densities of electronic states (DOS) for both molecular and dissociative adsorption configurations. The DOS results for several thermodynamically preferred configurations are illustrated in Fig. 3, while data for other species are provided in the ESI.†46 For each DOS graph in Fig. 3, both partial and total DOS are depicted. Solid lines represent spin-up (α) states, whereas dashed lines correspond to spin-down (β) states. As shown in Fig. 3, for species where physisorption is energetically feasible (AunY–H2 with n = 3, 7), most of electronic states associated with adsorbed hydrogen (red line) appear on the low-energy side (below −13.0 eV), indicating a weak interaction between hydrogen and the cluster surface. These states are clearly separated from the electronic states of the cluster, which extend beyond −8.5 eV for n = 3 and −10.5 eV for n = 7, respectively. This indicates that the chemical bonding contribution between hydrogen and these clusters is relatively weak or potentially negligible. This observation aligns well with previous studies on molecular hydrogen adsorption in transition metal-doped Agn (n = 2–12), Au102+ or even [Mo13S13]2− clusters, where van der Waals forces and/or electrostatic interactions between charge-polarized hydrogen molecules and cluster surfaces were identified as the dominant bonding mechanisms.42,43,67
 | ||
| Fig. 3 Density of electronic states (DOS) for the most stable configuration of hydorgen adsorbed on AunY (n = 3–4, 6–8, and 11) clusters. | ||
The DOS for dissociative configurations of AunY–2H (n = 4, 6, 8, 11) in Fig. 3, along with additional results for n = 1, 5, and 10 provided in the ESI,†46 exhibit a distinctly different behavior compared to their molecular adsorption counterparts. Remarkably, an analysis of the DOS spectra reveals an intriguing observation: the expected low-energy contribution of s-H2 states, typically associated with molecular adsorption, disappears entirely in the dissociative configurations. Instead, the electronic states of hydrogen extend significantly toward higher energies, generally exceeding −10.5 eV, with the exception of n = 1 and 4, where they remain above −8.5 eV. This shift is attributed to the strong hybridization of valence s-electrons from hydrogen atoms with those of the cluster, leading to the formation of stable chemical bonds within these systems. Additionally, in most cases, the highest occupied molecular orbital (HOMO) states are predominantly contributed by s-Au orbitals, further reinforcing the role of gold in hydrogen dissociation mechanisms. It is important to note that bonding interactions are often accompanied by electron transfer processes. Therefore, these findings support the earlier conclusion that hydrogen adsorption preferentially occurs at Au surface sites.
To gain deeper insight into the dissociative hydrogen adsorption mechanism in AunY–2H (n = 1, 4–6, 8, 10 and 11) clusters, we computed possible reaction pathways linking the reactants to their final dissociated states for single H2 adsorption and dissociation. Fig. 4 presents the hydrogen dissociation pathways for Au4Y, Au6Y, Au8Y, and Au11Y clusters, while additional data for AuY, Au5Y, and Au10Y are provided in the ESI.†46 The activation energy for hydrogen dissociation was determined by incorporating zero-point energy corrections and electronic energy values of the reactants and transition states (TS). Specifically, the activation barrier is quantified as the total energy difference between the reactants and the TS, offering insights into the feasibility of hydrogen dissociation on AunY clusters.
 | ||
| Fig. 4 Computed reaction pathways and relative energies (in eV) for H2 dissociative adsorption on Au4Y, Au6Y, Au8Y, and Au11Y clusters. Intermediates and transition states are denoted as Ii and TSi, respectively. Yellow, purple, and red spheres represent Au, Y, and H atoms, respectively. | ||
As shown in Fig. 4, the interaction of H2 with Au4Y clusters follows a stepwise dissociative adsorption mechanism. Initially, molecular hydrogen preferentially binds to the transition metal atom Y with an adsorption energy of −0.16 eV. However, to activate the adsorbed H2, the Au4Y clusters must overcome two activation barriers (TS1 and TS2) associated with the dissociation process. The first activation barrier is 0.78 eV (+0.62 eV relative to the reactants at −0.16 eV), while the second barrier is 0.73 eV (+0.60 eV relative to the intermediate state I1 at −0.13 eV). Before reaching the final dissociated product (Au4Y–2H), the system passes through another intermediate state (I2) at 0.44 eV. The final state, where the H2 molecule is fully dissociated, has an energy of −0.27 eV, which is 0.11 eV lower than the reactant state (−0.16 eV), indicating a thermodynamically favorable process. A similar dissociative adsorption mechanism is observed for AunY clusters with n = 1, 5, 6, 8, 10 and 11). As detailed in the ESI,† the Au5Y cluster follows a comparable pathway. The activation barrier for H2 dissociation (TS1) is 0.75 eV (+0.70 eV relative to the reactant state), followed by a significant energy decrease through the intermediate state I1 (−0.04 eV). Ultimately, the system reaches a fully dissociated state, forming Au5Y–2H with an energy of −0.52 eV, which is 0.47 eV lower than both the reactants and the H2 adsorption state (−0.05 eV).
Similarly, for Au6Y cluster, H2 activation requires overcoming an energy barrier of 0.68 eV for the first transition state (TS1, +0.55 eV relative to the reactant state). The system then proceeds through an intermediate state (I1) with a slight energy drop of 0.21 eV before reaching the final dissociated state, which is 0.39 eV lower than the initial molecular adsorption state (−0.13 eV). A similar dissociative adsorption pattern is observed for AuY, Au8Y and Au10Y clusters. The activation barriers for the first transition state are 0.48 eV, 0.74 eV and 0.80 eV for the transition state TS1 (0.36 eV, 0.62 eV and 0.66 eV relative to the reactant state), respectively. Before reaching their final stable states, Au8Y and Au10Y pass through intermediate states with energy drops of +0.56 eV and −0.28 eV, respectively. In contrast, the AuY cluster reaches its final state at −1.81 eV without any intermediate states. Interestingly, the Au11Y cluster exhibits the lowest activation energy for H2 dissociation among the studied clusters, with a barrier of 0.63 eV for TS1 (+0.50 eV). The system transitions through an intermediate state (I1, +0.46 eV) before fully dissociating into the most stable state, Au11Y–2H, at −0.40 eV (0.27 eV lower than the reactants and the H2 adsorption state, −0.13 eV). Comparing the activation barriers for H2 dissociation across Au5Y (0.75 eV), Au6Y (0.68 eV), Au8Y (0.74 eV), Au10Y (0.80 eV), and Au11Y (0.63 eV), it is evident that the Au10Y cluster exhibits the highest energy barrier at 0.8 eV, suggesting a more challenging dissociative adsorption process compared to other clusters.
Although the final products of H2 dissociation on the Au4Y, Au5Y, Au8Y, and Au10Y clusters exhibit relatively low energies of −0.27 eV, −0.52 eV, −0.53 eV, and −0.56 eV, respectively, the activation energy barriers for H2 dissociation remain relatively high at 0.78 eV, 0.75 eV, 0.74 eV, and 0.80 eV. This suggests that while H2 adsorption is energetically favorable for these clusters, dissociative adsorption is unlikely to occur without external stimuli, particularly for the Au4Y and Au10Y clusters. For the AuY cluster, the activation barrier for adsorption-dissociation is relatively low at 0.48 eV, and the final product has a significantly low energy of −1.81 eV. However, as previously mentioned, this cluster is known to be unstable, limiting its potential for real-world catalytic applications. In contrast, the Au6Y and Au11Y clusters exhibit the lowest activation energy barriers of 0.68 eV and 0.63 eV, respectively. This indicates that Au6Y and Au11Y could serve as promising superatoms for hydrogen storage applications.
6. Conclusion
In summary, we have systematically investigated the interaction of H2 with small-sized AunY clusters (n = 1–12) using DFT calculations. Our findings highlight the crucial role of transition metal dopants in stabilizing cluster geometries and enhancing reactivity toward hydrogen adsorption and dissociation. We identified that the geometric structure, relative electronegativity of the dopant atom, and atomic coordination number (N) are key factors governing the preferred adsorption configurations. The Y atom exhibits a strong preference for high-coordination sites, favoring surface positions in smaller clusters (n = 1–5) before becoming gradually encapsulated by Au atoms in larger clusters (n = 6–12). Due to its relatively lower electronegativity (χY = 1.22) compared to gold (χAu = 2.54), H2 adsorption initially favors direct interaction with Y. However, as cluster size increases (n = 7–12) and the Y dopant attains higher coordination numbers, hydrogen adsorption shifts toward Au surface sites or Au–Au bridge positions. Notably, our results also indicate that molecular hydrogen adsorption does not significantly alter the structure of bare AunY clusters (except for Au5Y). The adsorption of two dissociated hydrogen atoms preferentially occurs at Au–Au or Au–Y bridge sites, depending on cluster size and atomic arrangement. Average binding energy calculations reveal that the hydrogen adsorption on AunY (n = 4–12) generally maintains higher relative stability compared to their bare counterparts. Importantly, dissociative adsorption is unlikely to occur for AunY (n = 1, 4–5, 8, 10) clusters without external stimulation due to high energy barriers, while for n = 2, 3, 7, and 9 molecular adsorption is energetically preferred. Au6Y and Au11Y clusters exhibit remarkable stability and barrier-free dissociation pathways, making them promising candidates for hydrogen dissociation and storage applications. Our results provide a deeper insight into the interaction between H2 and AunY clusters, including the adsorption site preference, the stability of adsorption configuration, the effect of impurities, possible reaction pathways, and energy barriers involved with this process. This work offers valuable guidance for future experiments and contributes to the development of alloys metal clusters as fundamental components in the design of innovative nanostructured materials for hydrogen catalysis and storage applications.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Nguyen Van Dang: conceptualization, data curation, formal analysis, investigation, validation, writing – original draft, review and editing. Ngo Thi Lan: conceptualization, funding acquisition, data curation, validation, writing – original draft, review and editing. Nguyen Thi Mai: data curation, formal analysis, investigation, validation, review and editing. Son Tung Ngo: data curation, formal analysis, investigation, validation, review and editing. Phung Thi Thu: data curation, formal analysis, investigation, validation. Nguyen Thanh Tung: conceptualization, data curation, formal analysis, investigation, validation, writing – original draft, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Foundation of Science and Technology Development (NAFOSTED) under the grant number 103.01-2021.109.Notes and references
- L. Schlapbach and A. Züttel, Hydrogen-storage materials for mobile applications, Nature, 2001, 414(6861), 353–358 CrossRef CAS.
- L. Baetcke and M. Kaltschmitt, Hydrogen Storage for Mobile Application: Technologies and Their Assessment, Hydrogen Supply Chains, Elsevier, 2018, pp. 167–206 Search PubMed.
- A. Pareek, R. Dom, J. Gupta, J. Chandran, V. Adepu and P. H. Borse, Insights into renewable hydrogen energy: recent advances and prospects, Mater. Sci. Energy Technol., 2020, 3, 319–327 CAS.
- G. Nicoletti, N. Arcuri, G. Nicoletti and R. Bruno, A technical and environmental comparison between hydrogen and some fossil fuels, Energy Convers. Manage., 2015, 89, 205–213 CrossRef.
- M. Yue, H. Lambert, E. Pahon, R. Roche, S. Jemei and D. Hissel, Hydrogen energy systems: a critical review of technologies, applications, trends and challenges, Renew. Sustainable Energy Rev., 2021, 146, 111180 CrossRef.
- N. Klopčič, I. Grimmer, F. Winkler, M. Sartory and A. Trattner, A review on metal hydride materials for hydrogen storage, J. Energy Storage, 2023, 72, 108456 CrossRef.
- C. Drawer, J. Lange and M. Kaltschmitt, Metal hydrides for hydrogen storage–Identification and evaluation of stationary and transportation applications, J. Energy Storage, 2024, 77, 109988 CrossRef.
- F. J. Desai, M. N. Uddin, M. M. Rahman and R. Asmatulu, A critical review on improving hydrogen storage properties of metal hydride via nanostructuring and integrating carbonaceous materials, Int. J. Hydrogen Energy, 2023, 48(75), 29256–29294 CrossRef CAS.
- I. Hassan, H. S. Ramadan, M. A. Saleh and D. Hissel, Hydrogen storage technologies for stationary and mobile applications: review, analysis and perspectives, Renew. Sustainable Energy Rev., 2021, 149, 111311 CrossRef CAS.
- F. Schüth, Challenges in hydrogen storage, Eur. Phys. J. Special Top., 2009, 176, 155–166 CrossRef.
- W. L. L. Sun, Z. Li, M. Fujii, Y. Geng, L. Dong and T. Fujita, Trends and future challenges in hydrogen production and storage research, Environ. Sci. Pollut. Res., 2020, 27, 31092–31104 CrossRef PubMed.
- P. H. M. Babaie, F. Salek, N. Haque, R. Savage, S. Stevanovic, T. A. Bodisco and A. Zare, Advancements in hydrogen production, storage, distribution and refuelling for a sustainable transport sector: hydrogen fuel cell vehicles, Int. J. Hydrogen Energy, 2024, 52, 973–1004 Search PubMed.
- P. Nordlander, S. Holloway and J. Nørskov, Hydrogen adsorption on metal surfaces, Surf. Sci., 1984, 136(1), 59–81 CrossRef CAS.
- B. Kumar, T. Kawawaki, N. Shimizu, Y. Imai, D. Suzuki, S. Hossain, L. V. Nair and Y. Negishi, Gold nanoclusters as electrocatalysts: size, ligands, heteroatom doping, and charge dependences, Nanoscale, 2020, 12(18), 9969–9979 RSC.
- Y. Gao, N. Shao, Y. Pei, Z. Chen and X. C. Zeng, Catalytic activities of subnanometer gold clusters (Au16–Au18, Au20, and Au27–Au35) for CO oxidation, ACS Nano, 2011, 5(10), 7818–7829 CrossRef CAS PubMed.
- M. S. Bootharaju, C. W. Lee, G. Deng, H. Kim, K. Lee, S. Lee, H. Chang, S. Lee, Y. E. Sung, J. S. Yoo, N. Zheng and T. Hyeon, Atom-precise heteroatom core-tailoring of nanoclusters for enhanced solar hydrogen generation, Adv. Mater., 2023, 35(18), 2207765 CrossRef CAS.
- L. M. Wang, S. Bulusu, W. Huang, R. Pal, L.-S. Wang and X. C. Zeng, Doping the golden cage Au16− with Si, Ge, and Sn, J. Am. Chem. Soc., 2007, 129(49), 15136–15137 CrossRef CAS.
- R. Pal, L.-M. Wang, W. Huang, L.-S. Wang and X. C. Zeng, Structural evolution of doped gold clusters: MAux−(M = Si, Ge, Sn; x = 5− 8), J. Am. Chem. Soc., 2009, 131(9), 3396–3404 CrossRef CAS.
- L. M. Wang, S. Bulusu, H. J. Zhai, X. C. Zeng and L. S. Wang, Doping golden buckyballs: Cu@Au16− and Cu@Au17− cluster anions, Angew. Chem., Int. Ed., 2007, 46(16), 2915–2918 CrossRef CAS PubMed.
- R. Pal, L.-M. Wang, Y. Pei, L.-S. Wang and X. C. Zeng, Unraveling the mechanisms of O2 activation by size-selected gold clusters: transition from superoxo to peroxo chemisorption, J. Am. Chem. Soc., 2012, 134(22), 9438–9445 CrossRef CAS PubMed.
- M. A. Dar and S. Krishnamurty, Molecular and dissociative adsorption of oxygen on Au–Pd bimetallic clusters: role of composition and spin state of the cluster, ACS Omega, 2019, 4(7), 12687–12695 CrossRef CAS PubMed.
- D. Manzoor, S. Krishnamurty and S. Pal, Effect of silicon doping on the reactivity and catalytic activity of gold clusters, J. Phys. Chem. C, 2014, 118(14), 7501–7507 CrossRef CAS.
- D. Manzoor, S. Krishnamurty and S. Pal, Endohedrally doped gold nanocages: efficient catalysts for O2 activation and CO oxidation, Phys. Chem. Chem. Phys., 2016, 18(10), 7068–7074 RSC.
- D. Manzoor, S. Krishnamurty and S. Pal, Contriving a catalytically active structure from an inert conformation: a density functional investigation of Al, Hf, and Ge doping of Au20 tetrahedral clusters, J. Phys. Chem. C, 2016, 120(35), 19636–19641 CrossRef CAS.
- L. Li, Y. Gao, H. Li, Y. Zhao, Y. Pei, Z. Chen and Z. C. Zeng, CO oxidation on TiO2 (110) supported subnanometer gold clusters: size and shape effects, J. Am. Chem. Soc., 2013, 135(51), 19336–19346 CrossRef CAS PubMed.
- S. Maity, S. Takano, S. Masuda and T. Tsukuda, Bonding and electronic interactions of hydrogen with gold superatoms, J. Phys. Chem. C, 2023, 128(1), 19–30 CrossRef.
- D. Manzoor and S. Pal, Hydrogen atom chemisorbed gold clusters as highly active catalysts for oxygen activation and CO oxidation, J. Phys. Chem. C, 2014, 118(51), 30057–30062 CrossRef CAS.
- K. Mondal, S. Agrawal, D. Manna, A. Banerjee and T. K. Ghanty, Effect of hydrogen atom doping on the structure and electronic properties of 20-atom gold cluster, J. Phys. Chem. C, 2016, 120(33), 18588–18594 CrossRef CAS.
- Megha, C. Kamal, K. Mondal, T. K. Ghanty and A. Banerjee, Remarkable structural effect on the gold–hydrogen analogy in hydrogen-doped gold cluster, J. Phys. Chem. A, 2019, 123(10), 1973–1982 CrossRef CAS PubMed.
- S. Buckart, G. Ganteför, Y. D. Kim and P. Jena, Anomalous behavior of atomic hydrogen interacting with gold clusters, J. Am. Chem. Soc., 2003, 125(46), 14205–14209 CrossRef CAS.
- X. Ma, S. Liu and S. Huang, Hydrogen adsorption and dissociation on the TM-doped (TM= Ti, Nb) Mg55 nanoclusters: A DFT study, Int. J. Hydrogen Energy, 2017, 42(39), 24797–24810 CrossRef CAS.
- D. Shen, C.-P. Kong, R. Jia, P. Fu and H.-X. Zhang, Investigation of properties of Mg n clusters and their hydrogen storage mechanism: a study based on DFT and a global minimum optimization method, J. Phys. Chem. A, 2015, 119(15), 3636–3643 CrossRef CAS.
- D. Shi, Y. Ni, G. Li, Z. Yan, Q. Zhao, W. Zhao, W. Xie, X. Meng and J. Chen, A computational study of MgmHn nanoclusters with n: m ≥ 2:1 for efficient hydrogen storage, Int. J. Quantum Chem., 2023, 123(6), e27058 CrossRef CAS.
- M. Pozzo and D. Alfe, Hydrogen dissociation and diffusion on transition metal (=Ti, Zr, V, Fe, Ru, Co, Rh, Ni, Pd, Cu, Ag)-doped Mg (0001) surfaces, Int. J. Hydrogen Energy, 2009, 34(4), 1922–1930 CrossRef CAS.
- J. Li, Y. Liu, J. Zhang, X. Liang and H. Duan, Density functional theory study of the adsorption of hydrogen atoms on Cu2X (X = 3d) clusters, Chem. Phys. Lett., 2016, 651, 137–143 CrossRef CAS.
- W. C. G. Hu, K. Kwak, M. Kim, D. Jiang, J. Choi and D. Lee, Effects of metal-doping on hydrogen evolution reaction catalyzed by MAu24 and M2Au36 nanoclusters (M = Pt, Pd), ACS Appl. Mater. Interfaces, 2018, 10(51), 44645–44653 CrossRef.
- Z. Fang and X. Kuang, Hydrogen molecule adsorption on AunPt (n = 1–12) clusters in comparison with corresponding pure Aun+1 (n = 1–12) clusters, Phys. Status Solidi B, 2014, 251(2), 446–454 CrossRef CAS.
- Y. Li, Y.-F. Li, J.-J. Tan, B.-F. Jiang and Y.-Z. OuYang, Probing structure, electronic property, and hydrogen adsorption for the alkali auride series, Eur. Phys. J. Plus, 2017, 132(4), 159 CrossRef.
- D. Bandyopadhyay, S. Chatterjee, R. Trivedi and K. Dhaka, Insights into catalytic behavior of TiMgn (n = 1–12) nanoclusters in hydrogen storage and dissociation process: a DFT investigation, Int. J. Hydrogen Energy, 2022, 47(27), 13418–13429 CrossRef CAS.
- J. Vanbuel, M. Jia, P. Ferrari, S. Gewinner, W. Schöllkopf, M. T. Nguyen, A. Fielicke and E. Janssens, Competitive molecular and dissociative hydrogen chemisorption on size selected doubly rhodium doped aluminum clusters, Top. Catal., 2018, 61, 62–70 CrossRef CAS.
- M. Jia, J. Vanbuel, P. Ferrari, E. M. Fernández, S. Gewinner, W. Schöllkopf, M. T. Nguyen, A. Fielicke and E. Jansses, Size dependent H2 adsorption on AlnRh+ (n = 1–12) clusters, J. Phys. Chem. C, 2018, 122(32), 18247–18255 CrossRef CAS.
- N. T. Lan, N. T. Mai, N. T. Cuong, P. T. H. Van, D. D. La, N. M. Tam, S. T. Ngo and N. T. Tung, Exploring hydrogen adsorption on nanocluster systems: insights from DFT calculations of Au9M2+ (M = Sc-Ni), Chem. Phys. Lett., 2023, 831, 140838 CrossRef CAS.
- N. T. Lan, N. T. Mai, N. T. Cuong, P. T. H. Van, D. D. La, N. M. Tam, S. T. Ngo and N. T. Tung, Density functional study of size-dependent hydrogen adsorption on AgnCr (n = 1–12) clusters, ACS Omega, 2022, 7(42), 37379–37387 CrossRef CAS.
- A. S. Shajahan, N. Kalarikkal, N. Garg, Y. Kawazo and B. Chakraborty, A quest to high-capacity hydrogen storage in zirconium decorated pentagraphene: DFT perspectives, Int. J. Hydrogen Energy, 2022, 47(85), 36190–36203 CrossRef CAS.
- C. Guo and C. Wang, The hydrogen storage capacities of 4d transition metals in various boron systems, J. Energy Storage, 2023, 57, 106216 CrossRef.
- M. D. Esrafili and S. Sadeghi, Y decorated all-boron B38 nanocluster for reversible molecular hydrogen storage: a first-principles investigation, Int. J. Hydrogen Energy, 2022, 47(22), 11611–11621 CrossRef CAS.
- W. Liu, Y. Liu and R. Wang, Prediction of hydrogen storage on Y-decorated graphene: a density functional theory study, Appl. Surf. Sci., 2014, 296, 204–208 CrossRef CAS.
- M. El, K. M. Bhihi, S. Naji, H. Labrim, A. Benyoussef, A. El Kenz and M. Loulidi, Study of doping effects with 3d and 4d-transition metals on the hydrogen storage properties of MgH2, Int. J. Hydrogen Energy, 2016, 41(8), 4712–4718 CrossRef.
- J. Lyu, V. Kudiiarov, L. Svyatkin, A. Lider and K. Dai, On the catalytic mechanism of 3d and 4d transition-metal-based materials on the hydrogen sorption properties of Mg/MgH2, Catalysts, 2023, 13(3), 519 CrossRef CAS.
- C. Sergio, F. Pansini and M. de Campos, The relationship between hydrogen storage capacity and 4d transition metal–carbon surface binding energy, Chem. Phys. Lett., 2024, 846, 141338 CrossRef CAS.
- J. Barabás, P. Ferrari, V. Kaydashev, J. Vanbuel, E. Janssens and T. Höltzl, The effect of size, charge state and composition on the binding of propene to yttrium-doped gold clusters, RSC Adv., 2021, 11(47), 29186–29195 RSC.
- Q. L. M. Paskevicius, D. A. Sheppard, C. E. Buckley and A. W. Thornton, Hydrogen storage materials for mobile and stationary applications: current state of the art, ChemSusChem, 2015, 8(17), 2789–2825 CrossRef PubMed.
- M. L. S. Srivastava, T. Lakshmi, E. R. Sandadi, S. Gour, N. A. Thomas, S. S. Priya and K. Sudhakar, An overview of hydrogen storage technologies–Key challenges and opportunities, Mater. Chem. Phys., 2024, 325, 129710 CrossRef.
- M. D. Liptak and G. C. Shields, Comparison of density functional theory predictions of gas-phase deprotonation data, Int. J. Quantum Chem., 2005, 105(6), 580–587 CrossRef CAS.
- M. Frisch G. W. Trucks, H. B. Schlegel, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, Gaussian 09, Revision a. 02, 200, Gaussian, Inc., Wallingford, CT, 2009, p. 271 Search PubMed.
- P. Hohenberg and W. Kohn, Inhomogeneous electron gas, Phys. Rev., 1964, 136(3B), B864 CrossRef.
- T. B. Tai and M. T. Nguyen, A stochastic search for the structures of small germanium clusters and their anions: enhanced stability by spherical aromaticity of the Ge10 and Ge122− systems, J. Chem. Theory Comput., 2011, 7(4), 1119–1130 CrossRef CAS PubMed.
- Q. Du, X. Wu, P. Wang, D. Wu, L. Sai, R. B. King, S. J. Park and J. Zhao, Structure evolution of transition metal-doped gold clusters M@Au12 (M= 3d–5d): Across the periodic table, J. Phys. Chem. C, 2020, 124(13), 7449–7457 CrossRef CAS.
- R. McWeeny and B. T. Sutcliffe, Fundamentals of self-consistent-field (SCF), hartree-fock (HF), multi-configuration (MC) SCF and configuration interaction (CI) schemes, Comput. Phys. Rep., 1985, 2(5), 219–278 CrossRef.
- F. Weinhold, Discovering Chemistry with Natural Bond Orbitals, John Wiley & Sons, 2012 Search PubMed.
- C. Peng, P. Y. Ayala, H. B. Schlegel and M. J. Frisch, Using redundant internal coordinates to optimize equilibrium geometries and transition states, J. Comput. Chem., 1996, 17(1), 49–56 CrossRef CAS.
- N. Govind, M. Petersen, G. Fitzgerald, D. King-Smith and J. Andzelm, A generalized synchronous transit method for transition state location, Comput. Mater. Sci., 2003, 28(2), 250–258 CrossRef CAS.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC press, 2007 Search PubMed.
- L. Rincon, A. Hasmy, M. Marquez and C. Gonzalez, A perturbatively corrected tight-binding method with hybridization: application to gold nanoparticles, Chem. Phys. Lett., 2011, 503(1–3), 171–175 CrossRef CAS.
- L. Lin, P. Claes, P. Gruene, G. Mijer, A. Fielicke, M. T. Nguyen and P. Lievens, Far-infrared spectra of yttrium-doped gold clusters AunY (n = 1–9), Chem. Phys. Chem., 2010, 11(9), 1932–1943 CrossRef CAS PubMed.
- L. Guo, First-principles study of molecular hydrogen adsorption and dissociation on AlnCr (n = 1–13) clusters, J. Phys. Chem. A, 2013, 117(16), 3458–3466 CrossRef CAS PubMed.
- T. T. Phung, N. T. Huyen, N. T. Giang, N. M. Thu, N. T. Son, N. H. Tung, N. T. Lan, N. T. Mai and N. T. Tung, Unraveling hydrogen adsorption on transition metal-doped [Mo3S13]2− clusters: insights from density functional theory calculations, ACS Omega, 2024, 19, 18 Search PubMed.
- J. K. J. Greeley, N. A. Romero, V. A. Morozov, H. Falsig, A. H. Larsen, J. Lu, J. J. Mortensen, M. Dułak, K. S. Thygesen, J. K. Nørskov and K. W. Jacobse, Finite size effects in chemical bonding: from small clusters to solids, Catal. Lett., 2011, 141, 1067–1071 CrossRef.
- R. Trivedi and D. Bandyopadhyay, Hydrogen storage in small size MgnCo clusters: a density functional study, Int. J. Hydrogen Energy, 2015, 40(37), 12727–12735 CrossRef CAS.
- G. Ortega, E. Germán, M. J. Lopez and J. A. Alonso, Catalytic activity of Co–Ag nanoalloys to dissociate molecular hydrogen. New insights on the chemical environment, Int. J. Hydrogen Energy, 2022, 47(44), 19038–19050 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01901j |
| This journal is © The Royal Society of Chemistry 2025 |