DOI:
10.1039/D5RA01399B
(Paper)
RSC Adv., 2025,
15, 15550-15560
A comparative DFT study of different structures of ZnTe: for optoelectronic and thermoelectric applications
Received
27th February 2025
, Accepted 6th May 2025
First published on 12th May 2025
Abstract
In the present study, a comprehensive comparative investigation of ZnTe in its zinc-blende, wurtzite, and tetragonal structural phases was performed employing density functional theory to analyze their electronic, optical, and thermoelectric properties for potential optoelectronic and thermoelectric applications. All three phases possess a direct band gap at the Γ point, with computed energy gap values of 2.1 eV (zinc-blende), 1.5 eV (wurtzite), and 1.3 eV (tetragonal), revealing their semiconductivity. The density of states study found that Zn and Te orbitals made significant contributions across the valence and conduction bands, with substantial variations in bonding interactions between these phases. Optical investigation revealed that the tetragonal phase has the highest static dielectric constant (7.65) and refractive index (2.70), while the zinc-blende phase has superior absorption properties and the highest peak in the optical conductivity spectrum at 6.3 eV, demonstrating increased photon absorption efficiency. The energy loss function also shows that the tetragonal phase has greater energy dissipation potential. The Seebeck coefficient increased with temperature in all phases, with the tetragonal structure reaching the greatest value of 115 μV K−1 at 500 K. At high temperatures, the wurtzite phase had the maximum electrical conductivity, which can be associated with its favorable electronic band dispersion near the Fermi level. The figure of merit (ZT), an essential indicator for thermoelectric efficiency, is highest for the tetragonal phase (ZT = 0.387 at 500 K), overcoming both the wurtzite (ZT = 0.36) and zinc-blende structures. These results show that structural configuration has an essential effect on the multifunctional properties of ZnTe. The tetragonal phase is a very attractive candidate for thermoelectric applications, while the zinc-blende structure provides superior optical performance for optoelectronic devices.
1. Introduction
The II–VI group of semiconducting materials continues to be the main regime of research interest in condensed matter physics despite their high complexity and defective forms due to their wide band gap characteristics.1–3 One of the II–VI semiconductor material prototypes is zinc telluride.4 Due to their direct energy band gaps and ability to generate light at ambient temperature, these semiconductors have garnered a lot of attention in recent years.5–7 Additionally, as computer processing power increases, it becomes simpler to accurately calculate material properties such as solids' structural, electronic, and optical characteristics. These materials are useful for numerous technical applications, including THz emitters, detectors, imaging systems, thin-film transistors, photovoltaics, control systems, and solid-state lasers.8–10 Several properties of materials can now be computed because of advancements in ab initio approaches. The structural and mechanical characteristics of ZnX (X = O, S, Se, Te) have been the subject of numerous ab initio studies.11 Hattori et al.12 determined the refraction index of ZnTe in the visible spectrum. Recently, Yu et al.13 used first-principles ground-state and response-function computations to examine the elastic, dielectric, and thermodynamic properties of the B3 structure of ZnTe. The ZnTe band structure was studied by Walter et al.14 with the Wu–Cohen Generalized Gradient Approximation (WC-GGA) approximation. By utilizing DFT within the generalized gradient approximation (GGA) for exchange-correlation energy, Sahraoui et al.15 have calculated the elastic stiffness coefficients and bulk modulus for ZnS under hydrostatic pressure.
The zinc-blende ZnTe was studied by Ding et al.,16 whose space group was F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m and the lattice parameter was a = 6.1037 Å. The space group for wurtzite ZnTe is P63mc, and for this investigation, the initial structure parameters are used. The Zn and Te have orbitals 3d104s2 and 4d105s25p4, respectively, and these orbitals are regarded as being in the valence band. The experimental parameters serve as the first ones for the structural optimization.17 The ZnTe is significant for possible application in multi-junction tandem solar panels.18–22 At 300 K, ZnTe has a band gap energy of 2.26 eV, in the cubic phase, and has an experimental lattice parameter of 6.10372 Å, of the IIB–VIA family is ZnTe. It also exists in other phases, like as in the wurtzite with lattice constant (a = b = 4.27 Å, and c = 6.99 Å) and at a 10.5 GPa pressure with lattice constant of (a = b = 4.062 Å and c = 9.434 Å)23–26 discussed the experimental data for the electronic characteristics of ZnTe. Fig. 1 signifies the crystal structures for the investigated phases. The current research focuses on the electronic, optical, and thermoelectric properties of ZnTe in various structural forms, such as zinc-blende, wurtzite, and tetragonal configurations. Density functional theory is used in these analyses. Notably, the modified Beck–Johnson approximation is the most recent and precise approach for predicting the energy band gaps, with experimental precision nearing a 10% margin of error.27 This study is significant because there is a lack of knowledge about the structural, optoelectronic, and thermoelectric properties of ZnTe, which are vital for its usage in nonlinear optoelectronic and thermoelectric technologies.
3m and the lattice parameter was a = 6.1037 Å. The space group for wurtzite ZnTe is P63mc, and for this investigation, the initial structure parameters are used. The Zn and Te have orbitals 3d104s2 and 4d105s25p4, respectively, and these orbitals are regarded as being in the valence band. The experimental parameters serve as the first ones for the structural optimization.17 The ZnTe is significant for possible application in multi-junction tandem solar panels.18–22 At 300 K, ZnTe has a band gap energy of 2.26 eV, in the cubic phase, and has an experimental lattice parameter of 6.10372 Å, of the IIB–VIA family is ZnTe. It also exists in other phases, like as in the wurtzite with lattice constant (a = b = 4.27 Å, and c = 6.99 Å) and at a 10.5 GPa pressure with lattice constant of (a = b = 4.062 Å and c = 9.434 Å)23–26 discussed the experimental data for the electronic characteristics of ZnTe. Fig. 1 signifies the crystal structures for the investigated phases. The current research focuses on the electronic, optical, and thermoelectric properties of ZnTe in various structural forms, such as zinc-blende, wurtzite, and tetragonal configurations. Density functional theory is used in these analyses. Notably, the modified Beck–Johnson approximation is the most recent and precise approach for predicting the energy band gaps, with experimental precision nearing a 10% margin of error.27 This study is significant because there is a lack of knowledge about the structural, optoelectronic, and thermoelectric properties of ZnTe, which are vital for its usage in nonlinear optoelectronic and thermoelectric technologies.
 |
| | Fig. 1 The crystallographic structures and polyhedral model of the coordination for (a) and (b) zinc-blende, (c) and (d) wurtzite, and (e) and (f) tetragonal phases of the ZnTe system. | |
2. Computational details
The density functional theory was used in the present work, which employs both the WIEN2K code28 and the BoltzTrap package.29 For materials with localized electron states, such as transition metal complexes, modified Beck–Johnson exchange-correlation potential attempts to enhance the description of electronic properties, such as band gaps and optical properties.30 It modifies the exchange potential to more accurately predict the band gaps observed experimentally for different materials, such as semiconductors, insulators, and compounds. The Tran–Blaha modified Beck–Johnson (TB-mBJ) potential is employed for the calculation of the electronic characteristics of these structural phases. The modified Beck–Johnson potential was demonstrated as follows.| |
 | (1) |
here  is electronic density,
is electronic density,  is kinetic energy density, and the vBRx,σ shows Becke–Roussel potential.31 The real and imaginary components are probably presented separately.31
is kinetic energy density, and the vBRx,σ shows Becke–Roussel potential.31 The real and imaginary components are probably presented separately.31| |
 | (2) |
| |
 | (3) |
When creating the plane wave basis for the Brillouin zone of zinc-blende ZnTe, 256 k-points are used, whereas 550 k-points are used for the wurtzite structure. The first stage was to fine-tune the energy cutoff and the k-point mesh using self-consistent test computations. The stable structure's ground state volume is defined as the volume in which the system has the lowest total energy. For both Zn and Te atoms, the atomic muffin-tin radii (RMT) are set to 2. Where RMT represents the muffin tin's smallest radius and Kmax is the reciprocal lattice vector's greatest value, (RMT × Kmax) is set to 7, and the cutoff energy is 8.0 Ry. The k-point values are employed for both phases of their structural optimizations in the BZ integration and evaluated at 1000 K k-points while considering optical qualities.
3. Results and discussions
3.1 Structural properties
The ZnTe zinc-blende, in the cubic phase (see Fig. 1), has a space group F3m. The Zn2+ is attached to four equivalent Te2− atoms that form corner-sharing ZnTe4 tetrahedra. All Zn–Te bond lengths are 2.55 Å. The Te2− in the cubic phase was bonded to four equivalent Zn2+ atoms, and it forms a corner-sharing TeZn4 tetrahedra. The ZnTe in the wurtzite structure is in the hexagonal phase and has a space group P63mc. The Zn2+ in the wurtzite was bonded to its four equivalent Te2− atoms and formed a corner-sharing ZnTe4 tetrahedra. All Zn–Te bond lengths are 2.75 Å. The Te2− was bonded to the four corresponding Zn2+ and formed corner-sharing TeZn4 tetrahedra. Similarly, the zinc telluride structure crystallizes in a tetragonal structure with a space group P4/mmm. The Zn2+ in this phase was bonded in a linear geometry to four equivalent Te2− atoms. Both Zn–Te bond lengths are 3.30 Å. These materials' structural relaxation was performed using PBE-GGA approximations. The initially anticipated equilibrium volume remained constant while thirteen alternative volumes were created in the second stage. The structure was then relaxed by changing the atomic locations. By taking into account different exchange-correlation factors for structures across these atomic volumes, the cohesive energy was calculated. The lowest cohesive energy volume was identified as the ideal volume for the construction, from which the structural parameters were derived.32–34 We obtain significant ground-state characteristics, including the lattice constants, the bulk modulus B, and the optimized unit cell volume of these structures (see Table 1). The energies required for the formation of the various structural phases were determined, which were anticipated to be −1.47, −1.44, and −1.35 (eV per f.u.) for the zinc-blende, wurtzite, and tetragonal phases of ZnTe, respectively. The fact that these energies range from −1.3 eV per f.u. to −1.5 eV per f.u., suggesting that these materials are stable. The considerable connection between formation energy and the level of ionicity in Zn–Te bonds shows that lower formation energy is an indicator of stronger ionic bonds. A more dependable convex hull is associated with the reported formation energy values and is closest to the compositions of interest.32–34
Table 1 The atomic sites coordinate, lattice constants, formation energy, bulk modulus, and cohesive energy, for the investigated structural phases of ZnTe
| Systems |
PBE-GGA |
a (Å) |
b (Å) |
c (Å) |
Eform (eV per f.u.) |
B (GPa) |
Ecoh (eV per atom) |
| Atoms |
x |
y |
z |
| Cubic-ZnTe (F3m) |
|
|
|
|
6.14 |
6.14 |
6.14 |
−1.47 |
57.5 |
4.51 |
| 6.10 (ref. 16) |
|
|
|
52.0 (ref. 23) |
|
| 6.11 (ref. 26) |
|
|
|
|
|
| 6.19 (ref. 35) |
|
|
|
|
|
| 6.12 (ref. 36) |
|
|
|
|
|
| |
Zn |
0.500 |
0.500 |
0.000 |
|
|
|
|
|
|
| |
Te |
0.330 |
0.330 |
0.330 |
|
|
|
|
|
|
| Hexa-ZnTe (P63mc) |
|
|
|
|
4.41 |
4.41 |
7.22 |
−1.44 |
55.11 |
3.12 |
| 4.27 (ref. 17) |
4.27 (ref. 17) |
6.99 (ref. 17) |
|
51.0 (ref. 23) |
|
| 4.29 (ref. 25) |
4.29 (ref. 25) |
6.99 (ref. 25) |
|
|
|
| 4.43 (ref. 35) |
4.43 (ref. 36) |
7.17 (ref. 35) |
|
|
|
| 4.52 (ref. 36) |
4.52 (ref. 35) |
7.11 (ref. 36) |
|
|
|
| |
Zn |
0.500 |
0.250 |
0.003 |
|
|
|
|
|
|
| |
Te |
0.003 |
0.250 |
0.333 |
|
|
|
|
|
|
| Tetra-ZnTe (P4/mmm) |
|
|
|
|
|
|
|
|
|
|
| |
Zn |
0.250 |
0.356 |
0.777 |
3.34 |
3.34 |
6.73 |
−1.35 |
78.31 |
2.34 |
| |
Te |
0.500 |
0.350 |
0.666 |
|
|
|
|
|
|
3.2 Electronic band structures
By employing the Tran–Blaha modified Beck–Johnson (TB-mBJ) potential, the band structure in the zinc-blende, wurtzite, and tetragonal structure systems was computed along the chosen high-symmetry directions in the Brillouin zone (BZ). The valence band maximum and conduction band minimum are localized at Γ-point of the BZ for all three examined ZnTe structures, confirming a direct energy gap semiconducting behaviour. The size of their energy gaps was the primary distinction between the analyzed compound topologies. Calculated band gaps for ZnTe in cubic zinc-blende, hexagonal wurtzite, and tetragonal structures are shown in Fig. 2, were 1.8 eV, 1.5 eV, and 1.2 eV, respectively. The computed band gap values were found to be in good agreement (see Table 2) with the reported theoretical23,25 and experimental values.27 Among the structural phases, the band gap of ZnTe in the tetragonal phase is less than that in cubic zinc-blende and hexagonal wurtzite. The valence bands, which are made up of three splitting bands that correspond to the heavy hole band, light hole band, and spin split-off band, respectively, are illustrated in Fig. 2. The conduction bandwidth in ZnTe can change depending on the crystal structure. In general, a material's conduction bandwidth is affected by several variables, including bonding interactions, atomic organization, and crystal symmetry. The conduction band of ZnTe was wider in the face-centered cubic and hexagonal crystal structures of zinc-blende and wurtzite than tetragonal crystal structure. A flatter curvature around the band edge corresponds to a greater effective mass, while a steeper curvature indicates a lower effective mass. The symmetry and degeneracy can help with band broadening by allowing additional states to overlap and they are not the only factors that influence. Bond lengths, such as the Zn–Te bond in the studied zinc telluride structures, may exert a more noticeable effect. Shorter Zn–Te bond lengths promote orbital overlap between the atoms, thus increasing the electron mobility across the lattice and resulting in larger electronic bands. As a result, in these three structures, the differences in Zn–Te bond lengths can have a significantly greater impact on bandwidth than symmetry or degeneracy. It is also true and correct that shorter Zn–Te bonds generate stronger interactions between atoms, broadening the energy bands. The zinc-blende and wurtzite phases are both extremely symmetric structures, although with different specific symmetries. Zinc-blende (cubic) has a highly symmetric cubic structure with the space group F3m, giving it an exceptional level of symmetry in three dimensions. Wurtzite (hexagonal) has somewhat lesser symmetry than zinc-blende, although it nevertheless has a greater degree of symmetry because of its hexagonal lattice. Tetragonal phases frequently have lesser symmetry than zinc-blende and wurtzite. The band structure of the crystal and the type of binding interactions influence the effective mass of the charge carriers. These elements can be used to explain the differences in effective mass among the three phases of ZnTe. The maximum value of the valence band in the wurtzite structure is located at the center of the Brillouin zone, and the valence bands converge strongly around this point. Near the valence band maximum, this curve aids in the formation of charge carriers with higher effective masses. A lower effective mass for charge carriers results from the increased spherical or symmetrical dispersion, which is made possible by higher symmetry. Table 2 summarizes the band gap energy predicted for ZnTe in cubic zinc-blende, hexagonal wurtzite, and tetragonal phases, with comparisons to experimental and theoretical results.
 |
| | Fig. 2 The calculated electronic band profiles of (a) zinc-blende, (b) wurtzite, and (c) tetragonal phases of the ZnTe system. | |
Table 2 The computed band gaps, zero frequency limits for the static dielectric constant, refractive index, and reflectivity for ZnTe, along with other theoretical and experimental data from the literature
| Systems |
Sp. group |
High symmetry point |
Band gaps (eV) |
ε1(0) |
n(0) |
R(0) |
| Our TB-mBJ calculations |
Others theoretical |
Experimental |
| Cubic-ZnTe |
F3m |
Γ–Γ |
2.1 |
2.07 (ref. 25) |
2.13 eV (ref. 23) |
6.21 |
2.51 |
0.18 |
| 6.52 (ref. 25) |
2.52 (ref. 25) |
0.21 (ref. 25) |
| 6.9 (ref. 27) |
2.54 (ref. 27) |
0.19 (ref. 27) |
| Hexa-ZnTe |
P63mc |
Γ–Γ |
1.5 |
1.48 eV (ref. 23) |
1.6 eV (ref. 27) |
7.01 |
2.61 |
0.19 |
| 1.67 eV (ref. 25) |
7.52 (ref. 25) |
2.56 (ref. 25) |
0.20 (ref. 27) |
| |
8.9 (ref. 27) |
|
|
| Tetra-ZnTe |
P4/mmm |
Γ–Γ |
1.3 |
|
|
7.65 |
2.70 |
0.22 |
3.3 Density of states
Fig. 3 depicts the results of the Tran–Blaha modified Beck–Johnson (TB-mBJ) potential calculation of the densities of states for the three phases. Understanding the behavior of state occupancy across a particular energy interval is made easier by the concept of density of states. The states available for occupation are those with a high density of states at a particular energy level. The total density of state and partial density of state for the cubic zinc-blende of ZnTe are shown in Fig. 3(a). The Te contributes most in the valence band region, ranging from 0.0 eV up to −2.0 eV, whereas the Zn contributes minimally. The Te and Zn make up the majority of the contribution from −3.5 up to −4.5 eV. The greater peaks for Zn in the total density of state suggest that the Zn atoms have contributed more to the electronic states at that particular energy level in the cubic structure. This suggests that, as compared to Te atoms, the Zn atoms have a higher density of accessible valence states or a stronger electronic connection in that energy range in the cubic structure. The Zn and Te make the dominating contribution, ranging from 2.3 up to 5.0 eV. The Zn-s, -p states from 0.0 eV to −3.0 eV exhibit a high contribution in the PDOS for cubic zinc-blende ZnTe, while from Te, the only Te-p state has a large contribution from 0.0 eV to −3.0 eV in the cubic structure. This demonstrates that the Zn-s and p-states account for the majority of the valence band of ZnTe in specific energy ranges. Furthermore, the Te-p state of Te makes a significant contribution, implying that Te atoms play a role, although to a lesser extent than Zn. The maximum contributions come from the Zn-s, -p, and -d states, as well as the Te-p and -s at −3.5 eV to −4.5 eV. The Zn-s, -p, states, and Te-p states with energies ranging from 2.3 eV to 5.0 eV account for the majority of contributions in the conduction band region. The electronic states that are associated with the zinc atom, specifically the s-, and p-states, are referred to as Zn-s and p-states. These states cause high peaks in partial density states, significantly increasing the number of electronic states in this energy range. Te-p and Te-s states are the electronic states that are associated with the Te atom. Te-p state plays an especially important role in this energy field. As a result, the higher peaks visible in the partial density states are attributed to the Te-s state, which represents the electronic states associated with the s-orbital of Te atoms in the ZnTe cubic structure. Fig. 3(b) depicts the total density of state and partial density of state for the six-layer ZnTe structure. The Te states contribute the most to the valence area of 0 to −2.5 eV, whereas the Zn levels contribute only slightly.
 |
| | Fig. 3 The calculated density of states of (a) cubic zinc-blende, (b) hexagonal wurtzite, and (c) tetragonal phases of the ZnTe system. | |
The Te and Zn provide the highest peak and donate meaningfully to the wurtzite structure, ranging from −4.0 up to −5.0 eV. The Zn and Te states make a significant and preceding influence in the conduction band, from 2.0 to 5.0 eV. In the partial density of state for wurtzite hexagonal ZnTe in the valence band, the s and d states of Zn have high peaks and their largest influence from 0 to −3.0 eV, whereas Te-s and p-states make significant contributions in the same energy range. The Zn-d states and Te-s states account for the majority of the contributions from −4.0 eV up to −5.0 eV. The higher peak values and significant contributions of Zn-d and Te-s states in the wurtzite structure imply a strong electronic coupling and interactions with these orbitals within the valence band at −4.5 eV. The electronic and bonding characteristics of ZnTe in that energy range are greatly influenced by these electronic states. While Zn-s, -p orbitals and Te-s, -d orbitals from 2.0 up to 5.0 eV cause the majority in the conduction band region. The density of states of the tetragonal structure of ZnTe is revealed in Fig. 3(c). Here, the Zn has a negligible contribution in the range, the Te has greater peaks, and its highest contribution is from 0 eV to −2.0 eV in the tetragonal phase. The higher peaks for Te in the total density of state at energy 1.5 eV imply a greater contribution from Te atom electronic states at that particular energy level. The Te atoms possess a high proportion of available states with a strong electronic linking for the tetragonal phase, as Zn atoms. The intense peaks for the Te in the total density of states are at about 1.5 eV, which indicates that the Te electronic states contribute more at this energy level. This implies that Te atoms have a higher proportion of accessible states or a stronger electronic connection in this energy region in the tetragonal phase than Zn atoms. At 1.5 eV, the electronic states of Te atoms possess high attraction and are more likely to be occupied in the valence band. The major role of the Te and Zn atoms ranges between −3.7 and −4.8 eV. Plotting the total energies for the three phases against volume yields ground-state parameters. The Birch–Murnaghan equations of state are used to reduce the total energy of the three phases to the volume of a unit cell (E–V), which yields the ideal ground state variables.
3.4 Optical properties
The complex dielectric function, ε(ω) = ε1(ω) + iε2(ω) frequently describes the macroscopic optical response functions of materials in the linear response range. The solid's band structure and its spectrum are reflected by the dielectric function, which serves as a link between small physical band transitions and the solid's electronic structure. The real and imaginary parts of the dielectric function can be defined in terms of direct transition probabilities and Kramers–Kronig dispersion relations. Fig. 4(a) depicts the ε1(ω) of ZnTe dielectric function in cubic zinc-blende, hexagonal wurtzite, and tetragonal phases. ZnTe has a static dielectric constant (see Table 2) of 6.21, 7.01, and 7.65 for the zinc-blende, hexagonal wurtzite, and tetragonal phases, respectively. The peaks are seen in the zinc-blende, wurtzite, and tetragonal at 3.51 eV, 3.21 eV, and 4.0 eV, respectively. The tetragonal phase's greatest peaks are likely caused by the crystal's unique electronic band structure and symmetry. When compared to cubic structures, tetragonal structures have less symmetry, which can result in a wider variety of energy transitions between electronic states. Higher peaks in the dielectric function may be the result of the greater diversity of transitions. The possible transitions between electronic states may be constrained by the higher symmetries of cubic zinc-blende and hexagonal wurtzite structures, on the other hand. They have lower dielectric function peaks than the tetragonal phase as a result. Fig. 4(b) depicts the imaginary portion ε2(ω) of the ZnTe dielectric function in the cubic zinc-blende, hexagonal wurtzite, and tetragonal phases. The cubic zinc-blende, hexagonal wurtzite, and tetragonal phases of ZnTe exhibit interband transitions and electronic band structures that govern how the ε2(ω) performs at high energies. The profiles exhibit an equivalent nature for the energy range. The discrepancies in ε2(ω) positions and values can also be seen. The band gaps and the densities of electrons interact in the transitions and undoubtedly contribute to this disparity.
 |
| | Fig. 4 The calculated (a) real component, (b) imaginary component, (c) absorption coefficient, and (d) energy loss function of the studied structures of the ZnTe system. | |
According to the plot, the ZnTe cubic in zinc-blende, hexagonal wurtzite, and tetragonal phases exhibit sharp peaks at energies of 6.10, 4.0, and 5.0 eV, with maximum values for ε2(ω) of 12.2, 12.0, and 10.0, respectively. Based on the density of states plots in Fig. 3, the main characteristics of ε2(ω) can be attributed to changes in the occupied state's hybridized state as it occurs in the vacant orbitals. When the structure is changed, the energy gap enlarges, pushing the total spectrum towards higher energies. In most materials, the absorption coefficient is usually capped by phase space-filling effects. This suggests that fewer electronic states are available for transition as more electronic states are filled. As from Fig. 4(c), the three structures have comparable absorption at energies between 2.50 and 4.0 eV, which is consistent with the extinction coefficient curves. The tetragonal phase absorption is greater at energies between 4.1 eV and 5.0 eV. However, as energy levels rise, the zinc blend absorbs more energy. The greatest peak of the cubic zinc-blende ZnTe curves is 7.0 eV, while the curves of hexagonal wurtzite ZnTe are 6.2 eV and 8.3 eV, respectively. The zinc-blende ZnTe has a considerably higher absorption than wurtzite ZnTe and tetragonal ZnTe. In addition, a peak in zinc-blende curves at 7.0 eV demonstrates that zinc-blende ZnTe may absorb photons more efficiently. Fig. 5(d) shows that the plasma oscillation characteristic is represented by the energy loss function's peak, and the energy at which the peak is found corresponds to the frequency of the plasma oscillation. ZnTe cubic zinc-blende exhibits a peak loss function of 0.49 at 12.58 eV, 0.57 eV at 13.0 eV, and 0.68 at 12.15 eV for the ZnTe tetragonal phase. Compared to hexagonal wurtzite and cubic zinc-blende, the tetragonal phase's peaks are significantly larger. Compared to cubic and hexagonal structures, tetragonal structures frequently exhibit lower levels of symmetry. A greater energy loss function can be produced via lower symmetry, which can also lead to a wider variety of electronic transitions and a wider dispersion of energy loss states. There may be more accessible energy states and transitions in the tetragonal phase's electronic band structure, which helps with energy loss. This might be the result of the particular atom arrangement and the resulting electrical band dispersion.
 |
| | Fig. 5 The calculated (a) refractive index, (d) extinction coefficient, (c) reflectivity, and (b) real optical conductivity for the three structural phases of the ZnTe system. | |
In Fig. 5(a), the ZnTe has a refractive index of 2.51, 2.61, and 2.70 at 0 eV in its cubic zinc-blende, hexagonal wurtzite, and tetragonal phases, respectively (see Table 2). When the energy is between 0 and 3.7 eV, the refractive index of cubic zinc-blende ZnTe increases as the energy increases, and the crystal exhibits characteristics of typical dispersion. Zinc-blende's energy peak is at 3.7 eV. But at energies between 4.0 and 14.0 eV, the refractive index falls as the energy rises, exhibiting anomalous dispersion. The normal dispersion range for hexagonal wurtzite is 0–3.5 eV, while the extreme dispersion range is 3.6–14.0 eV. The highest wurtzite peaks have energies of 3.5 eV. For ZnTe in the tetragonal phase, with energies between 0 and 4.0 eV, the refractive index rises as the energy increases, and the crystal exhibits characteristics of typical dispersion. But at energies between 4.0 and 14.0 eV, the refractive index falls as the energy rises, exhibiting anomalous dispersion. Fig. 5(b) shows that the extinction coefficients of hexagonal wurtzite, cubic zinc-blende, and tetragonal phase only differ a little between 0 and 4.0 eV. The extinction coefficient of cubic zinc-blende, hexagonal wurtzite, and tetragonal phases all rise as energy levels rise. At an energy of 7.0 eV, cubic zinc-blende has a maximum extinction coefficient of 2.52. At an energy of 4.2 eV, hexagonal wurtzite exhibits a maximum extinction coefficient of 2.5. At an energy of 6.2 eV, the greatest extinction coefficient for the tetragonal phase is 2.3. The extinction coefficient for the aforementioned structures decreases as energy increases after 7.0 eV.
The electrical band structure, crystal symmetry, and the way light interacts with the material all have an impact on the reflectivity. The reflectance for ZnTe in cubic zinc-blende, hexagonal wurtzite, and tetragonal phase at 0 to 14.0 eV is shown in Fig. 5(c). The reflectance of the aforementioned structures likewise increases when energy levels rise. At an energy of 13.75, cubic zinc-blende, hexagonal wurtzite, and tetragonal phases, respectively, have maximum reflectivity of 0.65, 0.58, and 0.73. Depending on the particular material and the wavelength range of interest, the behavior of reflectivity might change dramatically. Different electrical band structures and symmetries in crystal formations can lead to variances in reflectance. In the cubic zinc-blende, hexagonal wurtzite, and tetragonal phases of ZnTe, the optical conductivity peaks from 4 to 6.3 eV. The cubic zinc-blende peaks at 6.3 eV, the maximum energy. The largest energy peaks are at 4.0 eV for wurtzite and 4.5 eV for the tetragonal phase. These peaks' energy locations are determined from band structures and the electronic properties of the ZnTe used in the construction of the mentioned structures. Dropping optical conductivity indicates a substance's reduced ability to transmit or absorb light of that specific energy spectrum.
3.5 Thermoelectric properties
By the constant scattering time approximation, the thermoelectric parameters, including the Seebeck coefficient, thermal conductivity, electrical conductivity, and figure of merit, were also estimated as functions of temperature. Fig. 6(a) shows the predicted ZnTe electronic thermal conductivity as a function of temperature for the cubic zinc blende, hexagonal wurtzite, and tetragonal phases. The Wiedemann–Franz law states that the electronic thermal conductivity obeys the proportionality of these two parameters, increasing with temperature like the electrical conductivity: L is the Lorenz number, hence kl = L ∝ T. Up to 300 K, values of around 0.30, 0.10, and 0.20 × (1015 W m−1 K−1 s−1) are obtained for the ZnTe in cubic zinc blende, hexagonal wurtzite, and tetragonal phase, respectively. At 500 K, the maximum thermal conductivity was for the cubic zinc-blende ZnTe. At 500 K, the ZnTe in zinc-blende, wurtzite, and tetragonal phases have, highest electronic thermal conductivity is 0.588, 0.476, and 0.278 × (1015 W m−1 K−1 s−1), respectively. The larger density of states at the Fermi level in cubic zinc-blende than in hexagonal wurtzite and tetragonal phase accounts for its steeper slope over 300 K. Comparing the hexagonal wurtzite and tetragonal phases of zinc telluride (ZnTe), the cubic zinc-blende structure exhibits a higher degree of symmetry. Higher thermal conductivity results from more effective heat transport across the crystal lattice, which is made possible by higher symmetry. The electrical conductivity of ZnTe is shown in Fig. 6(b) for the zinc-blende, wurtzite, and tetragonal phases. When the temperature rises, the electrical conductivity of the phases under study increases almost linearly. In contrast to thermopower, a trait of semiconductors, this performance displays an opposing temperature-dependent fluctuation. The ZnTe tetragonal phase exhibits lower electrical conductivity at a particular temperature. At 300 K, the electrical conductivity of ZnTe in cubic zinc-blende, hexagonal wurtzite, and tetragonal phases is 0.13, 0.15, and 0.078 × (1012 W m−1 s−1 K−2), respectively. At 500 K, we noticed that the electrical conductivity of the wurtzite–ZnTe turned out to be greater than that of the other two phases. At 500 K, we noticed the values of 0.3, 0.35, and 0.2 × (1012 W m−1 s−1 K−2) for the ZnTe in the zinc-blende, wurtzite, and tetragonal phase, respectively. The increase in charge carrier concentration brought on by the thermal excitation that occurs when the temperature rises causes the electrical conductivity to increase. The wurtzite structure of ZnTe has a distinct electronic band structure from the cubic zinc-blende and tetragonal phases. The energy states that electrons can occupy and take part in electrical conduction are determined by the band structure. The wurtzite phase's distinct band structure may permit a higher density of states near the Fermi level, facilitating more effective electron transport and greater electrical conductivity.
 |
| | Fig. 6 The calculated (a) thermal conductivity, (b) electrical conductivity, (c) Seebeck coefficient, and (d) figure of merit for the three structural phases of the ZnTe system. | |
The Seebeck coefficient in zinc-blende, wurtzite, and tetragonal phases is shown in Fig. 6(c). Notably, the Seebeck coefficient exhibits positive values over the whole investigated temperature range of 0 to 500 K, demonstrating that holes act as the prime charge carrier in zinc-blende, wurtzite, and tetragonal phases of ZnTe. When the temperature rises, the Seebeck coefficient of the phases under study increases almost linearly. For temperatures up to 300 K, the values of 60.0 × 10−6 V K−1, 75.0 × 10−6 V K−1, and 80.0 × 10−6 V K−1 are obtained for the ZnTe in zinc-blende, wurtzite, and tetragonal phases, respectively. At 500 K, the ZnTe in zinc-blende, wurtzite, and tetragonal phases have, highest electronic thermal conductivity is 90.0, 120.0, and 115.0 × (10−6 V K−1), respectively. It is feasible for the tetragonal and wurtzite phases of ZnTe to have equal Seebeck coefficients. The material's electronic band structure and carrier transport characteristics play a major role in determining the Seebeck coefficient. Similar electronic band structures between the tetragonal and wurtzite phases of ZnTe can lead to similar energy dispersion relations for electrons and holes. Their Seebeck coefficients may therefore show identical values. A material's thermoelectric efficiency from a vital parameter, which is the figure of merit (ZT). A thermoelectric material with a higher ZT value is more effective. The figure of merit for ZnTe in its zinc-blende, wurtzite, and tetragonal phases is shown as a function of temperature in Fig. 6(d). The thermoelectric performance, as measured by ZT, improves in the sequence of tetragonal phase, wurtzite, and zinc-blende over most of the temperature ranges examined. This shows that ZnTe in monolayer form has a lot of potential as a thermoelectric material. Notably, the tetragonal phase possesses a remarkably high ZnTe figure of merit, particularly at temperatures above 500 K. At 300 K, the ZT values for ZnTe in the zinc-blende, wurtzite, and tetragonal phases are 0.13, 0.18, and 0.21, respectively. Furthermore, the ZT values for ZnTe in these phases increase as temperature rises. ZT values at 500 K are 0.27, 0.36, and 0.387, respectively.
4. Conclusions
In summary, calculations are performed based on the density functional theory, and a comparative investigation of the three diverse ZnTe phases was conducted in terms of their electronic structure, optical, and transport properties. Each of the three phases was expected to demonstrate a semiconducting character with a direct band gap. A decrease in formation energy is associated with a larger degree of ionic character in the bonds, according to a clear and substantial association between the ionicity level of Zn–Te bonds and formation energy. Since the conduction bands of wurtzite ZnTe were smaller than those of zinc-blende and tetragonal ZnTe, the effective electron mass was smaller. The degree of symmetry of the ZnTe wurtzite structure is lower than that of the zinc-blende or tetragonal structures. The Zn peaks in the TDOS imply that Zn atoms have made a stronger contribution to the cubic structure's electronic states at that specific energy level. The strongest peaks of the tetragonal phase in ε1(ω) are probably due to the unique electronic band structure and symmetry of the crystal's structure. The tetragonal configuration has less symmetry than cubic structures, which can lead to a larger range of energy transitions between electronic states. Still, the differences in ε2(ω) positions and values are also apparent. This discrepancy is influenced by band gaps and electron concentrations that are entangled in the transitions. The absorption of the zinc-blende ZnTe is significantly higher than that of the wurtzite and tetragonal ZnTe. Zinc-blende ZnTe can absorb photons more effectively, as seen by a peak in the curves at 7.0 eV. Tetragonal structures usually display lower levels of symmetry than cubic and hexagonal ones. Lower symmetry can yield a larger energy loss function, which can also result in a wider range of electronic transitions and a larger dispersion of energy loss states. When the energy is between 0 and 3.7 eV, the cubic zinc-blende ZnTe crystal exhibits normal dispersion properties, and its refractive index rises as the energy rises. At a specific temperature, the ZnTe tetragonal phase has the lowest electrical conductivity. We observed that, at 500 K, the electrical conductivity of the wurtzite–ZnTe phase was higher than that of the other two phases. Due to the unique band structure of the wurtzite phase, there may be an advanced density of states near the Fermi level, which would aid in more efficient electron transport and improved electrical conductivity. The Seebeck coefficient of the phases under study increases practically linearly as the temperature increases. Also, the figure of merit of ZnTe in zinc-blende, wurtzite, and tetragonal phases increases with temperature.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported and funded by the Deanship of Scientific Research at Imam Mohammad Ibn Saud Islamic University (IMSIU) (grant number IMSIU-DDRSP2503).
References
- J. Liu, H. Liu, J. Wang, H. Sheng, G. Tang, J. Zhang and D. Bai, Phys. B, 2019, 568, 18–24 Search PubMed.
- X.-Y. Tian, C.-X. Du, G. ZhaoRi, M. SheLe, Y. Bao and M. Baiyin, RSC Adv., 2020, 10, 34903–34909 Search PubMed.
- R. Mondal, Y. B. Singh, A. S. Das, S. Kabi, L. S. Singh and D. Biswas, Phys. B, 2021, 612, 412896 Search PubMed.
- A. M. Alanazi, F. Alam, A. Salhi, M. Missous, A. G. Thomas, P. O'Brien and D. J. Lewis, RSC Adv., 2019, 9, 24146–24153 Search PubMed.
- M. T. Khan, I. M. Ashraf, F. Abdel-Wahab, M. F. Sanaa, M. S. A. Al-Juman, A. Almohammedi, M. Shkir and S. AlFaify, Phys. Scr., 2019, 94, 105816 Search PubMed.
- H. A. Alsalmah and S. Mehmood, J. Inorg. Organomet. Polym. Mater., 2024, 34, 2662–2675 Search PubMed.
- K. Mokurala and S. Mallick, RSC Adv., 2017, 7, 15139–15148 Search PubMed.
- C. Nefzi, M. Souli, Y. Cuminal and N. Kamoun-Turki, Mater. Sci. Eng., B, 2020, 254, 114509 Search PubMed.
- Q. Tian, Y. Cui, G. Wang and D. Pan, RSC Adv., 2015, 5, 4184–4190 Search PubMed.
- F. Sarcan, M. Aydin, F. Kuruoğlu, O. Donmez, S. Yildirim and A. Erol, Mater. Sci. Eng., B, 2021, 272, 115322 Search PubMed.
- H. S. Im, Y. Myung, K. Park, C. S. Jung, Y. R. Lim, D. M. Jang and J. Park, RSC Adv., 2014, 4, 15695–15701 Search PubMed.
- H. Yang, Y. Liang, F. Niu, H. Zhang, G. Xu, X. Wei and L. Yang, Appl. Phys. Lett., 2024, 125, 023901 Search PubMed.
- Y. Yu, J. Zhou, H. Han, C. Zhang, T. Cai and C. Song, et al., J. Alloys Compd., 2009, 471, 492–497 Search PubMed.
- Z. Wu and R. E. Cohen, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 235116 Search PubMed.
- F. Ali Sahraoui, S. Zerroug, L. Louail and D. Maouche, Mater. Lett., 2007, 16, 1978–1981 Search PubMed.
- Y. Ding, Y. Wang, J. Ni, L. Shi, S. Shi and W. Tang, Phys. B, 2011, 406, 2254–2260 Search PubMed.
- S. Ramasubramanian, R. Thangavel, M. Rajagopalan and J. Kumar, J. Alloys Compd., 2009, 479, 414–419 Search PubMed.
- H. Li, C. Li, H. Huang, G. Hao and F. Wang, Int. J. Mod. Phys. B, 2023, 38, 2450365 Search PubMed.
- A. S. Hassanien, I. Sharma and P. Sharma, Phys. Scr., 2023, 98, 045911 Search PubMed.
- J. Ji, Q. Gu, R. Khenata, F. Guo, Y. Wang, T. Yang and X. Tan, RSC Adv., 2020, 10, 39731–39738 Search PubMed.
- S. Yousuf and D. C. Gupta, Mater. Sci. Eng., B, 2017, 221, 73–79 Search PubMed.
- N. A. Noor, S. Ali, G. Murtaza, M. Sajjad, S. M. Alay-e-Abbas and A. Shaukat, et al., Comput. Mater. Sci., 2014, 93, 151–159 Search PubMed.
- J. Pellicer-Porres, D. Martinez-Garcia, C. Ferrer-Roca, A. Segura, P. Munsch and J. P. Itie, et al., High Pressure Res., 2003, 23, 339–342 Search PubMed.
- G. Shukla, H. M. Ray, A. Abdullah, S. Tyagi, A. Manchon, S. Sanvito and U. Schwingenschlögl, Phys. Chem. Chem. Phys., 2023, 25, 13533–13541 Search PubMed.
- O. Zakharov, A. Rubio, X. Blase, M. L. Cohen and S. G. Louie, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 10780–10787 Search PubMed.
- A. A. Nafea, A. K. Kareem, M. A. Khalaf and M. M. AL-Ani, Babylon J. Mach. Learn., 2023, 2023, 35–41 Search PubMed.
- S. D. Kshirsagar, M. Ghanashyam Krishna and S. P. Tewari, Mater. Sci. Semicond. Process., 2013, 16, 1002–1007 Search PubMed.
- K. Schwarz, P. Blaha and G. K. H. Madsen, Comput. Phys. Commun., 2002, 147, 71–76 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 Search PubMed.
- F. Tran and P. Blaha, Phys. Rev. Lett., 2006, 102, 226401 Search PubMed.
- B. Barnes, Contemp. Phys., 2011, 52, 612–613 Search PubMed.
- R. Khenata, M. Sahnoun, H. Baltache, M. Rérat, A. H. Reshak and Y. Al-Douri, et al., Phys. Lett. A, 2005, 344, 271–279 Search PubMed.
- O. Boudrifa, A. Bouhemadou, N. Guechi, S. Bin-Omran, Y. Al-Douri and R. Khenata, J. Alloys Compd., 2015, 618, 84–94 Search PubMed.
- A. Parida, S. Senapati, G. K. Pradhan and R. Naik, ChemistrySelect, 2023, 8, e202301933 Search PubMed.
- J. M. Ripalda, J. D. Gale and T. S. Jones, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 245314 Search PubMed.
- M. Kitamura, S. Muramatsu and W. A. Harrison, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 1351–1357 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *cd and
Siti Maisarah Azizd
*cd and
Siti Maisarah Azizd

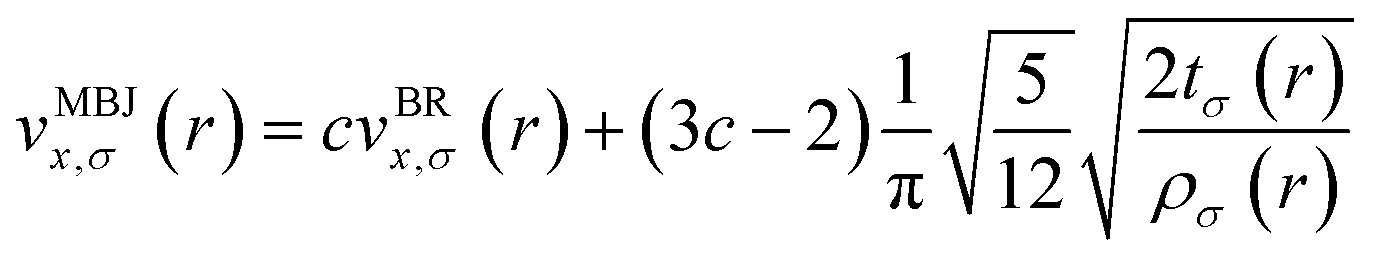
 is electronic density,
is electronic density,  is kinetic energy density, and the vBRx,σ shows Becke–Roussel potential.31 The real and imaginary components are probably presented separately.31
is kinetic energy density, and the vBRx,σ shows Becke–Roussel potential.31 The real and imaginary components are probably presented separately.31





