
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interfacially engineered metal oxide nanocomposites for enhanced photocatalytic degradation of pollutants and energy applications
Mahmoud A. Ahmed
 *a,
Safwat A. Mahmoud
b and
Ashraf A. Mohamed
a
*a,
Safwat A. Mahmoud
b and
Ashraf A. Mohamed
a
aChemistry Department, Faculty of Science, Ain Shams University, Cairo-11566, Egypt. E-mail: mahmoudmahmoud_p@sci.asu.edu.eg
bCenter for Scientific Research and Entrepreneurship, Northern Border University, Arar 73213, Saudi Arabia
First published on 12th May 2025
Abstract
Escalating global energy demands and environmental pollution necessitate innovative solutions for sustainable development. Conventional methods often prove inadequate, driving research towards advanced materials and technologies. This review critically analyzes existing industrial wastewater treatment approaches, highlighting their merits and limitations, before focusing on the recent advancements in metal oxide-based nanocomposite photocatalysis for both pollutant degradation and energy generation. Moreover, the structural, electronic, and optical properties of metal oxides (MOx) are elucidated. The review discusses various MOx synthesis routes and their nanocomposites and elucidates the underlying photocatalytic mechanisms, emphasizing the influence of operational parameters on photocatalytic efficiency. Moreover, it explores how MOx can be utilized for photocatalytic energy generation, in addition to their role in pollutant degradation. Furthermore, it delves into the synergistic effects achieved by combining MOx with complementary nanomaterials (carbon-based structures, polymers, non-metals, semiconductors, and metal sulfides) to create hybrid nanocomposites with enhanced photocatalytic activity for both applications. A cost analysis and SWOT analysis are presented to assess the economic and technological feasibility of this trend. This comprehensive overview provides valuable insights for developing efficient, sustainable, and scalable wastewater treatment solutions using MOx-based nanocomposites, ultimately contributing to improved environmental remediation and water resource management while simultaneously exploring opportunities for energy production.
 Mahmoud A. Ahmed | Mahmoud Adel Ahmed earned his PhD degree in 2024. He has been actively engaged in research for the past eight years and his research focuses on the synthesis, characterization, and environmental applications of nanomaterials and their composites in water treatment and remediation. He has authored several reviews and book chapters on these topics. He also serves as a senior service engineer at Veolia Environmental Services, managing various sectors like reverse osmosis, boilers, cooling towers, and wastewater plants. |
 Safwat A. Mahmoud | Safwat A. Mahmoud is a physicist specializing in experimental solid-state physics. He earned his PhD in experimental solid-state physics from Leipzig University, Germany, in 1992. His research focuses on nanomaterials, nanotechnology, thin-film technology, and optical sensors, with additional interests in materials science and water treatment. He is currently serving as a Professor in the Department of Physics at the College of Science, Northern Border University, Saudi Arabia. Prior to this role, he held a professorship at the Faculty of Science, Minia University, Egypt. |
 Ashraf A. Mohamed | Ashraf A. Mohamed is a professor of environmental analytical chemistry, at the Department of Chemistry, Faculty of Science, Ain Shams University, Cairo, Egypt. He earned his MSc degree in 1991 and his PhD degree in 1995. He has been actively engaged in research for the past 35 years and his current research interests include analytical chemistry, nanomaterials, layered double hydroxides, molecularly imprinted polymers, water treatment and analysis, optical sensors, and paper micro-fluidics. He has authored several reviews and book chapters on these topics. |
1. Introduction
The confluence of escalating global energy demands and the pervasive ramifications of environmental pollution presents a formidable challenge to sustainable development.1–4 The continued reliance on fossil fuels poses a substantial threat to energy security and exacerbates environmental degradation.5 The combustion of fossil fuels releases copious amounts of greenhouse gases, primarily carbon dioxide, contributing to global warming and its associated implications, including extreme weather events, rising sea levels, and disruptions to delicate ecological balances.6,7 Moreover, the extraction, processing, and utilization of fossil fuels generate a plethora of pollutants, including sulfur oxides (SOx), nitrogen oxides (NOx), particulate matter (PM), and volatile organic compounds (VOCs), which contribute to air pollution, acid rain, and respiratory complications.8,9 Simultaneously, the discharge of a diverse array of pollutants from industrial activities, agricultural practices, and urbanization poses a grave threat to environmental integrity and human health. Industrial effluents often contain heavy metals, pharmaceuticals, dyes, and persistent organic pollutants (POPs), contaminating water bodies and disrupting aquatic ecosystems.10–15 Agricultural runoff laden with pesticides, fertilizers, and herbicides contributes to eutrophication, soil degradation, and water pollution. This precarious scenario necessitates a paradigm shift towards clean and renewable energy sources coupled with effective pollution mitigation strategies.Various effluent treatment methods (e.g., biological processes, coagulation–flocculation, sedimentation, disinfection, ion exchange, membrane filtration) are often insufficient for complete removal of recalcitrant pollutants.16–19 These methods suffer from limitations such as fouling, high operating pressures; concentrate stream generation, and inefficient removal of low concentrations of emerging pollutants. Consequently, advanced oxidation processes (AOPs) have emerged as a promising alternative.20 AOPs generate highly reactive hydroxyl radicals (˙OH) that non-selectively mineralize a wide range of organic pollutants to CO2, H2O, and inorganic salts. Various AOPs exist, including photocatalysis, ozonation, sonochemical, Fenton, photoFenton, sonophoto-Fenton, and electrochemical oxidation processes.21,22 Among these, photocatalysis is particularly attractive due to its potential to mitigate both energy scarcity and environmental pollution by utilizing solar or artificial light to activate a semiconductor material, generating ˙OH and other reactive oxygen species (ROS) for targeted catalytic reactions.
Metal oxides are a diverse class of materials with a wide range of optical, structural, and electronic properties, making them crucial for various technologies.23–26 Their diverse functionalities arise from the interplay between metal and oxygen ions, influenced by the metal's oxidation state, coordination geometry, and crystal structure. Metal oxides exhibit diverse structures, from simple rock salt (e.g., MgO) to complex perovskite (e.g., SrTiO3) and layered structures (e.g., MoO3).27–29 This structural diversity significantly impacts their electronic band structure and optical properties. Closely packed structures often lead to wide band gaps and transparent/insulating behavior, while open structures with transition metals can exhibit smaller band gaps, resulting in semiconducting or metallic conductivity. Defects, like oxygen vacancies, further modulate the electronic structure, influencing optical absorption and conductivity.30,31 The band gap, the energy difference between valence and conduction bands, determines the minimum photon energy for electron excitation (absorption edge). Transition metal oxides often absorb visible light and exhibit characteristic colors due to partially filled d-orbitals and phenomena like d–d transitions, charge transfer transitions, and plasmon resonances.32–34 This unique tunability makes metal oxides (MOx) a compelling platform for photocatalysis, using light to drive chemical transformations. MOx are attractive due to their favorable band structures, cost-effectiveness, abundance, and chemical stability. Photocatalysis relies on MOx absorbing photons to generate electron–hole pairs that drive redox reactions. However, limitations like nanoparticles agglomeration, rapid electron–hole recombination, limited visible light absorption, and low charge carrier mobility hinder pristine MOx performance. To overcome these challenges, strategies such as doping, creating heterojunctions, surface functionalization, and morphology control are employed, with the construction of MOx-based composites gaining significant attention (Fig. 1a).35–40 Further, Fig. 1b highlights the key findings from the modification of MOx photocatalysts. Combining MOx with other materials (semiconductors, carbonaceous nanomaterials, noble metals, or polymers) creates synergistic effects that enhance photocatalytic activity. These composites improve charge separation, broaden light absorption (via sensitizers or plasmon resonances), increase surface area and dispersibility, and enhance MO stability. Therefore, designing MO-based composites is crucial for realizing the full potential of metal oxide photocatalysis in applications from environmental remediation to energy conversion.
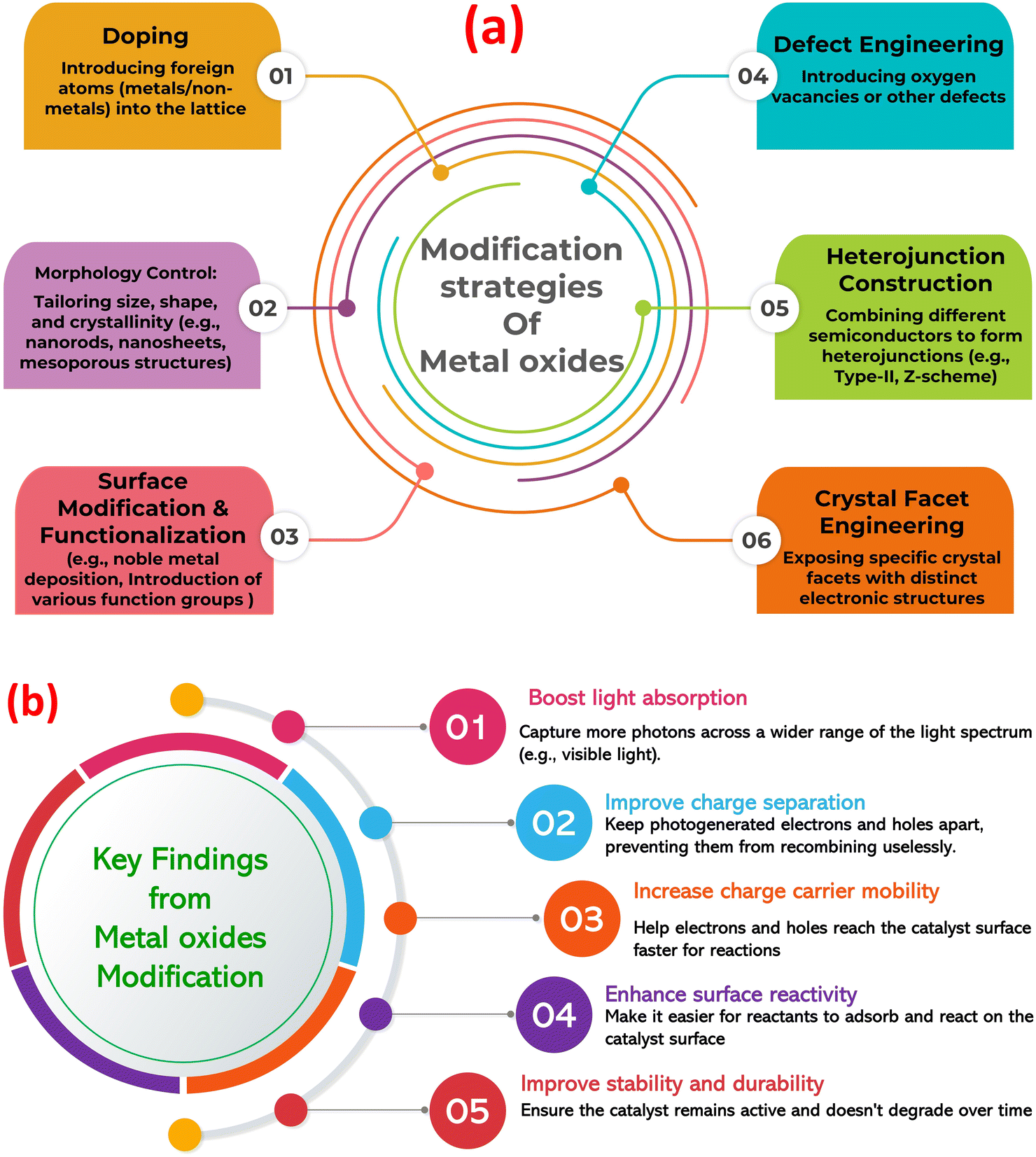 | ||
| Fig. 1 (a) Modification approaches of metal oxides to enhance its photocatalytic role, (b) the key findings from the modification of metal oxide photocatalysts. | ||
Analysis using Scopus data (accessed February 27, 2025) mapped the evolving landscape of metal oxide nanocomposite photocatalysis for wastewater treatment. Employing keywords: “metal oxide”, “pollutants”, “photocatalysis”, “water splitting”, “energy conversion”, “hydrogen production”, “wastewater treatment”, etc., 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 018 documents were retrieved, encompassing 38725 research articles, 4642 reviews, and other publication types, reflecting the exponential growth in publications, particularly over the past decade, underscoring the burgeoning interest in this field, as shown in Fig. 2a and b. A bibliometric analysis of keywords from photocatalysis articles in PubMed reveals core research areas and emerging trends, Fig. 2d. Network visualization clustered the keywords based on co-occurrence frequency. The first cluster centers on fundamental photocatalytic mechanisms. It focuses on photocatalytic degradation of organic pollutants (e.g., dyes, pharmaceuticals) using MOx like ZnO and TiO2 under visible light. The research prioritizes enhancing activity through nano-structural optimization (e.g., particle size, morphology) and addressing inherent limitations like rapid electron–hole recombination. Recent advancements integrate MOx with other materials (e.g., graphene) to improve visible light absorption and charge transfer efficiency. Heterojunctions with transition metal oxides (Fe2O3, CuO) or noble metals (Ag nanoparticles) further narrow bandgaps, enabling dual-functionality for organic pollutant degradation and heavy metal removal (e.g., Cr, Cd) via adsorption-photocatalytic mechanisms. The second cluster explores MOx composites (e.g., MnO2–Fe2O3 hybrids, graphene-supported TiO2) as electro- and photo-catalysts for hydrogen production. Enhanced charge separation and catalytic activity under solar irradiation are achieved through conductive matrices (e.g., carbon nanorods), optimizing electron transport for efficient oxygen/hydrogen evolution reactions. Interdisciplinary approaches combining photocatalysis with electrocatalysis aim to scale solar-driven hydrogen production, aligning with global clean energy goals. Material engineering, via sol–gel methods, pyrolysis, and green synthesis, enables precise control over nanocomposite properties (e.g., crystallinity, porosity) to maximize active sites. Characterization tools guide structure–activity relationships, while eco-friendly synthesis and recycling address sustainability. Challenges include scalability, long-term stability, and mitigating secondary pollution from metal leaching. Future research may leverage bio-inspired designs, machine learning, and pilot-scale trials to bridge lab innovations with real-world applications. This analysis underscores the transformative role of metal oxide composites in advancing sustainable water treatment and renewable energy systems.
018 documents were retrieved, encompassing 38725 research articles, 4642 reviews, and other publication types, reflecting the exponential growth in publications, particularly over the past decade, underscoring the burgeoning interest in this field, as shown in Fig. 2a and b. A bibliometric analysis of keywords from photocatalysis articles in PubMed reveals core research areas and emerging trends, Fig. 2d. Network visualization clustered the keywords based on co-occurrence frequency. The first cluster centers on fundamental photocatalytic mechanisms. It focuses on photocatalytic degradation of organic pollutants (e.g., dyes, pharmaceuticals) using MOx like ZnO and TiO2 under visible light. The research prioritizes enhancing activity through nano-structural optimization (e.g., particle size, morphology) and addressing inherent limitations like rapid electron–hole recombination. Recent advancements integrate MOx with other materials (e.g., graphene) to improve visible light absorption and charge transfer efficiency. Heterojunctions with transition metal oxides (Fe2O3, CuO) or noble metals (Ag nanoparticles) further narrow bandgaps, enabling dual-functionality for organic pollutant degradation and heavy metal removal (e.g., Cr, Cd) via adsorption-photocatalytic mechanisms. The second cluster explores MOx composites (e.g., MnO2–Fe2O3 hybrids, graphene-supported TiO2) as electro- and photo-catalysts for hydrogen production. Enhanced charge separation and catalytic activity under solar irradiation are achieved through conductive matrices (e.g., carbon nanorods), optimizing electron transport for efficient oxygen/hydrogen evolution reactions. Interdisciplinary approaches combining photocatalysis with electrocatalysis aim to scale solar-driven hydrogen production, aligning with global clean energy goals. Material engineering, via sol–gel methods, pyrolysis, and green synthesis, enables precise control over nanocomposite properties (e.g., crystallinity, porosity) to maximize active sites. Characterization tools guide structure–activity relationships, while eco-friendly synthesis and recycling address sustainability. Challenges include scalability, long-term stability, and mitigating secondary pollution from metal leaching. Future research may leverage bio-inspired designs, machine learning, and pilot-scale trials to bridge lab innovations with real-world applications. This analysis underscores the transformative role of metal oxide composites in advancing sustainable water treatment and renewable energy systems.
 | ||
| Fig. 2 Trends in publications over the last four decades that analyze SCOUPS data; keywords are “metal oxide”, “pollutants”, “photocatalysis”, “water splitting”, “energy conversion”, “hydrogen production”, and “wastewater treatment”. The figure includes: (a) yearly publication trends, (b) document type, (c) contributions from various countries to the field, and (d) bibliometric network analysis map of keywords. | ||
Therefore, the present review provides a critical appraisal of current and emerging technologies for wastewater remediation, focusing on the burgeoning field of MOx-based nanocomposite photocatalysis. Beyond simply summarizing recent advancements, this work dissects the fundamental photocatalytic mechanisms governing enhanced performance in these materials, elucidating the intricate interplay between material design, synthetic strategies, and operational parameters. It explores the synergistic impacts arising from the integration of MOx with diverse nanomaterials, highlighting the resulting improvements in photocatalytic activity for both pollutant degradation and energy generation. A key objective is to address the critical research gap related to the long-term stability and recyclability of MOx nanocomposite photocatalysts in wastewater treatment, evaluating current strategies and proposing innovative approaches for enhancing their practical applicability. Furthermore, it explores emerging strategies to overcome the associated limitations and propose future research directions to unlock the full potential of MOx nanocomposites for sustainable wastewater treatment and resource recovery, paving the way for a circular economy approach to water management.
2. Optical features and electronic structure of some metal oxides
MOx display intriguing optical features and possesses a distinctive electronic structure, rendering it a subject of great interest in numerous scientific disciplines.41 For instance, NiO is optically renowned for its characteristic deep green color, which arises from its electronic transitions within the visible range.42,43 The absorption spectrum of NiO exhibits pronounced absorption in the ultraviolet (UV) region, extending into the visible range.44–46 This absorption is attributed to charge transfer transitions between the valence band originating mainly from oxygen 2p orbitals to the conduction band originating mainly from nickel 3d orbitals. The bandgap energy of NiO typically resides around 3.4–4.0 eV, classifying it as a wide-bandgap material suitable for applications in the UV region.47–49 The electronic structure of NiO is intricately linked to its cubic crystal structure, which typically adopts a rock salt-like configuration (NaCl structure). In this arrangement, each nickel ion is encompassed by six oxygen ions in an octahedral coordination. The electronic configuration of nickel in NiO is [18Ar]3d8, with two unpaired electrons occupying the 3d orbitals.50 NiO is also recognized as a p-type semiconductor.51 This means that it exhibits a predominance of positive charge carriers, known as “holes”, in its electronic structure.52 The p-type behavior of NiO arises from the presence of oxygen vacancies and nickel interstitials, which create acceptor states within the bandgap. The presence of oxygen vacancies or other defects can perturb the band structure, thereby influencing the optical and electronic properties of NiO. Many studies have examined the effect of oxygen vacancies on the reactivity of NiO in applications like catalysis, photocatalysis, sensing, and electronic devices.53–56 These studies used computational and experimental approaches to investigate the impact of oxygen vacancies on the performance of NiO. The primary limitation of NiO semiconductors in photocatalytic processes is its quick charge carriers recombination and wide band gap, which restrict its performance in the visible region. This poses a challenge for the degradation of complex organic molecules, as it necessitates efficient photon generation and charge carrier separation to initiate effective radical-driven redox reactions.Titanium dioxide (TiO2) stands as a prominent metal oxide, extensively studied and utilized due to its diverse and tunable properties.57 TiO2 exists primarily in three crystalline polymorphs: anatase, rutile, and brookite.58 Anatase, characterized by a tetragonal structure with edge-sharing TiO6 octahedra, is generally considered as the most photocatalytically active phase due to its higher electron mobility and longer charge carrier lifetime compared to rutile.59,60 Rutile, also tetragonal but with a more compact structure featuring both edge- and corner-sharing octahedra, exhibits a higher refractive index and greater UV absorption, making it suitable for pigments and UV-blocking applications.61–63 Brookite, with its orthorhombic structure, has shown promising photocatalytic activity in specific reactions but is often challenging to synthesize in pure form. The structural differences between these polymorphs directly influence their electronic band structures and consequently their optical properties. TiO2's optical properties are dominated by its semiconducting nature. The band gap energies of anatase (∼3.2 eV) and rutile (∼3.0 eV) correspond to absorption in the near-UV region, resulting in their white appearance and excellent UV-blocking capabilities.64 The electronic transitions responsible for this absorption involve the excitation of electrons from the O 2p valence band to the Ti 3d conduction band. Defects within the TiO2 lattice, such as oxygen vacancies and Ti3+ interstitials, can introduce localized states within the band gap, influencing the optical absorption and photocatalytic activity.64,65 For example, oxygen vacancies can create shallow donor levels below the conduction band, enhancing visible light absorption and potentially increasing photocatalytic efficiency.66 Furthermore, the refractive index of TiO2, particularly in the rutile phase, is relatively high (∼2.4–2.9), making it a valuable material for optical coatings and anti-reflection layers.67
Zinc oxide (ZnO), a versatile II–VI semiconductor, presents a compelling platform for photocatalytic applications.68 Crystalline ZnO predominantly adopts the wurtzite structure, characterized by a hexagonal unit cell with tetrahedrally coordinated Zn2+ and O2− ions.69 This non-centrosymmetric arrangement gives rise to intrinsic piezoelectric and pyroelectric properties, potentially influencing charge separation and photocatalytic activity. ZnO's direct band gap of ∼3.37 eV at room temperature dictates its optical absorption in the near-UV region, rendering it transparent in the visible spectrum.70 This character, while advantageous for certain applications like UV filters, limits its utilization of the full solar spectrum for photocatalysis. However, the high exciton binding energy (∼60 meV) in ZnO results in robust excitonic absorption features even at room temperature, suggesting efficient exciton formation and potential for enhanced photocatalytic activity through exciton-mediated processes.71,72 Furthermore, defects inherent to ZnO, such as oxygen vacancies and zinc interstitials, play a pivotal role in defining its electronic properties, often contributing to n-type conductivity.73,74 These defects can also introduce localized states within the band gap, influencing charge carrier dynamics and potentially impacting photocatalytic performance. The electronic structure of ZnO, characterized by a filled O 2p valence band and an empty Zn 4s conduction band, governs its photocatalytic behavior. Upon UV irradiation, electrons are excited across the band gap, creating electron–hole pairs that can participate in redox reactions at the ZnO surface. The efficiency of this process, however, is often limited by the rapid recombination of these charge carriers. Furthermore, the relatively high reduction potential of photogenerated electrons in ZnO restricts its ability to reduce certain species, limiting its applicability in some photocatalytic reactions. Despite these challenges, ZnO's high electron mobility, large surface area-to-volume ratio in nanostructured forms, and relatively low toxicity make it an attractive candidate for photocatalysis. Moreover, its inherent photostability compared to some other metal oxides further strengthens its potential for sustained photocatalytic activity. Understanding the interplay between ZnO's intrinsic properties, including its crystal structure, electronic band structure, defect chemistry, and optical absorption characteristics, is crucial for developing strategies to enhance its photocatalytic performance.
CuO crystallizes in a monoclinic structure, characterized by Cu2+ ions in a distorted square planar coordination with oxygen.75 This structural arrangement contributes to its distinct electronic properties and influences its interaction with light. CuO is a p-type semiconductor with a relatively narrow band gap, typically ranging from 1.2 to 1.9 eV, depending on the synthesis method and particle size.76–78 Furthermore, CuO exhibits strong absorption in the visible and near-infrared regions due to its narrow band gap and d–d electronic transitions within the Cu2+ ions. The absorption characteristics can be further influenced by factors such as particle size, morphology, and crystal defects. Nanostructured CuO, for instance, can exhibit enhanced optical absorption due to increased surface area and quantum confinement effects. The optical properties of CuO are also relevant for applications beyond photocatalysis, including solar cells, gas sensors, and electrochromic devices. Moreover, CuO is characterized by its p-type semiconductivity, arising from the presence of copper vacancies, which act as acceptor levels. The relatively high hole mobility in CuO facilitates charge transport, but the rapid recombination of photogenerated electron–hole pairs limits its photocatalytic efficiency.
WO3 exhibits polymorphism, adopting different crystal structures (monoclinic, orthorhombic, tetragonal, and cubic) depending on temperature and synthesis conditions.79,80 The monoclinic phase is stable at room temperature, and is most commonly investigated for photocatalysis.81,82 Crucially, the WO3 lattice readily accommodates oxygen vacancies, creating WO3−x, where the degree of oxygen deficiency (x) significantly influences its electronic structure and, consequently, its optical and electronic properties. These oxygen vacancies introduce defect states within the band gap, impacting charge carrier behavior and overall photocatalytic activity.83 Moreover, WO3 is an n-type semiconductor with a band gap typically ranging from 2.6 to 3.0 eV for the stoichiometric composition, corresponding to absorption in the visible to near-UV region.84,85 The presence of oxygen vacancies, however, plays a pivotal role in modulating the electronic band structure. These vacancies introduce localized states within the band gap, effectively narrowing the band gap energy and extending the absorption spectrum further into the visible light region. This shift towards visible light absorption is highly desirable for solar-driven photocatalysis, as it enables more efficient utilization of the solar spectrum. Furthermore, the presence of these defect states can influence charge carrier dynamics, affecting both charge separation and recombination rates, which are critical parameters governing photocatalytic efficiency. Further, the most striking feature of WO3 is its electrochromic behavior, stemming from the reversible insertion and extraction of ions, often accompanied by changes in oxygen vacancy concentration. This process modulates the optical absorption properties, leading to a dramatic and reversible color change, ranging from transparent or pale yellow in the oxidized state to deep blue or black in the reduced state. This dynamic optical tunability, while exploited in electrochromic devices, also has implications for photocatalysis. The precise control over oxygen vacancies, and therefore the optical absorption, allows for tailoring the light absorption properties to match the desired spectral range for specific photocatalytic reactions. Moreover, the high refractive index of WO3 can be advantageous in certain photocatalytic configurations, enhancing light trapping within the material and potentially increasing the interaction of light with the photoactive sites.
Cerium dioxide (CeO2) stands as a compelling metal oxide with unique redox properties stemming from the facile switching between Ce4+ and Ce3+ oxidation states.86,87 CeO2 adopts a fluorite crystal structure, characterized by a face-centered cubic arrangement of Ce4+ cations and O2− anions.88,89 This structure facilitates the formation of oxygen vacancies, which are crucial for the material's redox activity and catalytic performance. The presence of oxygen vacancies and the associated Ce3+ ions introduce localized states within the band gap, influencing the optical absorption and electronic conductivity. Furthermore, the ability of CeO2 to readily store and release oxygen makes it an effective oxygen buffer, a property that is exploited in various catalytic applications, including three-way catalysts for automotive exhaust gas treatment. Typically, CeO2 is pale yellow to off-white in color due to its absorption edge in the near-UV region.90,91 The band gap of CeO2 is generally reported to be around 3.2 eV, although the precise value can vary depending on the synthesis method and the presence of defects.91 The absorption edge arises from charge transfer transitions between the O 2p valence band and the Ce 4f conduction band. The presence of oxygen vacancies and Ce3+ ions introduces defect states within the band gap, leading to increased absorption in the visible region. This enhanced visible light absorption can be advantageous for photocatalytic applications, as it allows for utilization of a broader portion of the solar spectrum. Furthermore, the refractive index of CeO2 is relatively high, making it suitable for optical coatings. Electronically, CeO2 exhibits n-type semiconducting behavior, with the conductivity largely influenced by the concentration of oxygen vacancies and Ce3+ ions. The presence of these defects introduces donor levels within the band gap, increasing the electron carrier concentration. The electronic conductivity of CeO2 can be further tuned by doping with other elements or by controlling the oxygen vacancy concentration through annealing under different atmospheres. The facile switching between the Ce4+/Ce3+ oxidation states allows CeO2 to participate in redox reactions, acting as an oxygen buffer and promoting the activation of reactants. This redox activity is particularly important in photocatalysis, where CeO2 can promote charge separation and enhance the efficiency of redox reactions at the surface.
Co3O4 crystallizes in the normal spinel structure, where Co2+ ions occupy tetrahedral sites and Co3+ ions occupy octahedral sites within a cubic close-packed oxygen lattice.92,93 This specific cation distribution and the interplay between the two cobalt oxidation states significantly influence the electronic, magnetic, and catalytic properties of Co3O4. Its electronic structure is characterized by a complex interplay of electron correlations and spin–orbit coupling, leading to interesting phenomena such as antiferromagnetic ordering at low temperatures. Co3O4 exhibits two prominent absorption bands in the visible region, at around 400 and 700 nm, respectively.93,94 These absorption features arise from ligand-to-metal charge transfer transitions involving O2− and Co2+/Co3+ ions. The first absorption band is attributed to O2− → Co3+ transitions, while the second band is associated with O2− → Co2+ transitions. The precise position and intensity of these absorption bands can be influenced by factors such as particle size, morphology, and the presence of defects. The optical properties of Co3O4 make it a potential candidate for applications in solar energy conversion and photocatalysis.95 Furthermore, the refractive index and extinction coefficient of Co3O4, crucial parameters for optical applications, can be tuned by controlling the synthesis conditions and morphology. Moreover, Co3O4 exhibits p-type semiconducting behavior with a band gap typically ranging from 1.4 to 2.2 eV, depending on the synthesis method and morphology.96,97 The relatively small band gap allows for absorption of a significant portion of the visible light spectrum, making it suitable for photocatalytic applications. The electrical conductivity of Co3O4 is influenced by the concentration of oxygen vacancies and other defects, which can act as charge carriers. The unique electronic structure of Co3O4, with the coexistence of Co2+ and Co3+ ions, facilitates redox reactions, making it a promising catalyst for various applications, including CO oxidation, oxygen evolution reaction (OER), and the degradation of organic pollutants. Furthermore, Co3O4 has shown potential for application in energy storage devices, such as supercapacitors and lithium-ion batteries, due to its good electrochemical performance and relatively low cost.
Fig. 3 illustrates the bandgap energies along with the CB and valence VB edge positions of some metal oxides.98 The data highlight variations in electronic structure, which are critical for understanding their photocatalytic and optoelectronic properties. Precise alignment of CB and VB levels relative to redox potentials (e.g., water oxidation/reduction levels) is emphasized, as this directly influences charge transfer efficiency in applications such as solar energy conversion and pollutant degradation.
 | ||
| Fig. 3 Energy levels of the CB and VB of some metal oxides, compared to the redox potentials of water splitting and selected free radicals (versus NHE) at pH = 0, reprinted with the permission of ref. 98, copyright 2025, Elsevier. | ||
3. Interfacial engineering of metal oxides and their composite photocatalysts
Interfacial engineering plays a pivotal role in enhancing the performance of metal oxides and their composite photocatalysts. The interface between the composite components is crucial for charges creation and separation, and surface reactions. We can significantly improve photocatalytic efficiency for various applications, including environmental remediation, water splitting, and organic synthesis, by carefully modifying the interface structure, composition, and properties. This can be achieved via, for example well-tailored synthesis methods, heterojunction formation, doping with metallic and nonmetallic species, enhancing the surface area and surface functional groups, creating surface defects and oxygen vacancies and their densities, improving light harvesting, especially in the visible region, lowering bandgap energies, and improving photocatalyst stability and reusability.3.1 Fabrication methods for metal oxide-based composites
The synthesis of metal oxides-based composites for environmental applications leverages a range of preparative techniques, broadly categorized as in situ or ex situ crystallization strategies. In situ methods entail the simultaneous formation of both the metal oxides and the composite matrix, promoting a homogeneous distribution of metal oxides and strong interfacial interactions with the supporting material. This approach often allows for finer control over crystallite size and morphology, potentially leading to enhanced performance. Conversely, ex situ methods involve the incorporation of pre-synthesized metal oxide nanoparticles into a separate matrix material. This offers flexibility in tailoring the properties of both components independently before composite formation, although achieving uniform dispersion and preventing particle agglomeration can be challenging. Several well-established techniques are employed within both strategies, e.g., sol–gel, hydrothermal and solvothermal methods, co-precipitation microwave-assisted, ultrasound-assisted methods. After a successful synthesis of MOx-based composites, multifaceted analysis and detailed characterization techniques are important for understanding the properties of compounds and the structure–property relationships. Fig. 4 summarizes common characterization techniques used to analyze the structural, optical, and electronic properties of fabricated composite photocatalysts.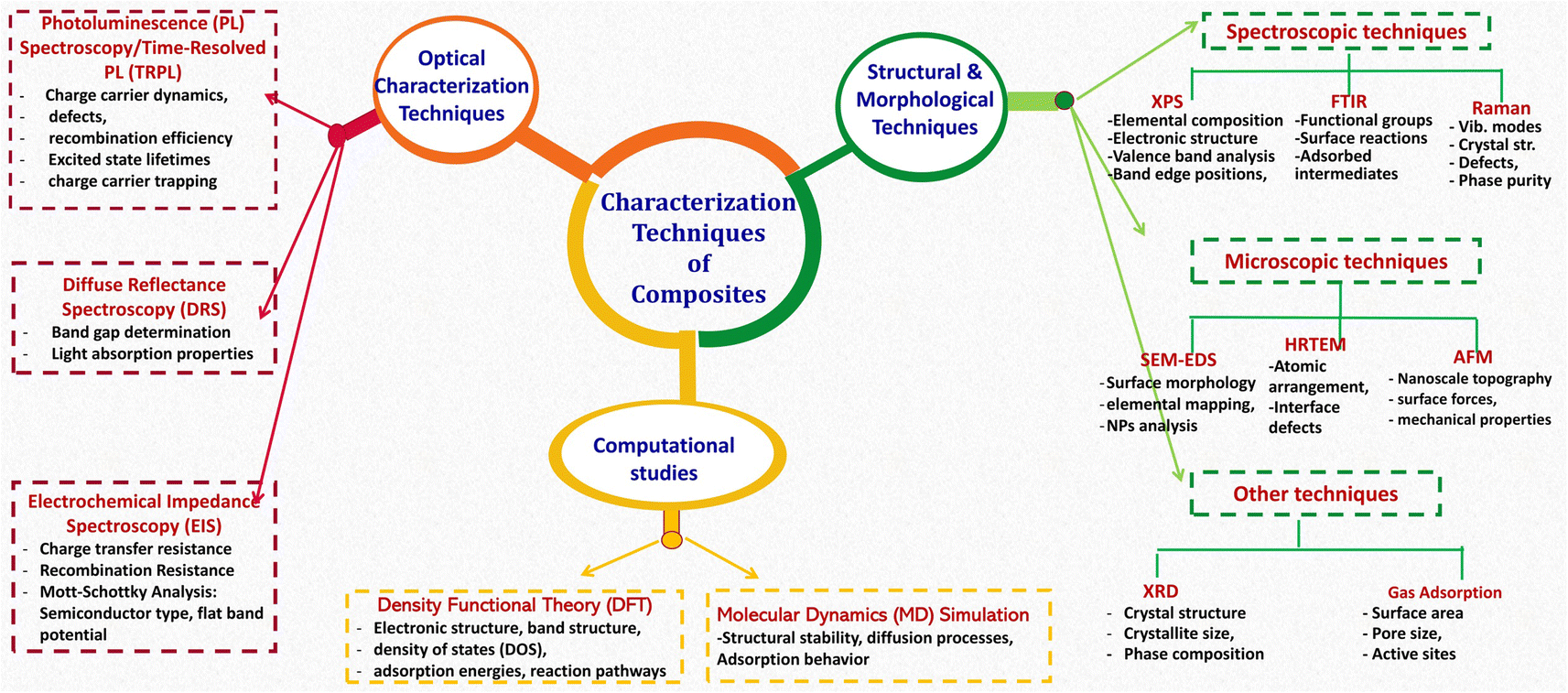 | ||
| Fig. 4 Summary of characterization techniques of MOx-based materials. | ||
Co-precipitation is a foundational wet chemical method for synthesizing metal oxide-based composites, enabling precise control over physicochemical properties by tuning reaction parameters.99 This process involves simultaneous precipitation of multiple metal cations from a solution using agents like hydroxide (OH−), carbonate (CO32−), or oxalate (C2O42−) ions.100,101 Precise pH control ensures homogeneous cation co-precipitation by balancing hydrolysis equilibria, while the choice of agent dictates precursor crystallinity; e.g., hydroxides yield amorphous phases, while carbonates form ordered frameworks. Precipitation kinetics, governed by concentration, mixing, and temperature, modulate particle morphology: slow nucleation favors monodisperse crystallites (Ostwald ripening), while rapid quenching produces metastable nanoparticles. Calcination thermally transforms precursors; temperature regulates crystallinity and sintering, while atmosphere (e.g., O2, H2/N2) tunes oxidation states and oxygen vacancies, enhancing functionality for catalysis, sensing, or energy storage via defect-engineered charge transport and surface reactivity. For example, a CeO2/ZnO nanocomposite, synthesized via co-precipitation, exhibited roughly double the activity of ZnO alone and ten times the activity of pure CeO2.102 The synthesized catalyst morphology was analyzed via HRTEM, as shown in Fig. 5a, showing that spherical-shaped and crystalline ZnO nanoparticles (5–200 nm) were decorated with smaller CeO2 NPs (8–17 nm). The 40CeO2/ZnO hybrid comprised aggregated ZnO and CeO2 phases forming a porous structure. Separately, a ternary NiO/ZnO/g-C3N4 composite was developed and tested for its azo dye degradation capabilities.103 The two-dimensional structure of g-C3N4 provided ample nucleation sites, facilitating the growth of nickel oxide and zinc oxide NPs. The resulting NZC nanocomposite displayed a clustered, curled morphology.103 XPS analysis, shown in Fig. 5b–g, confirmed that the NiO/ZnO/g-C3N4 composite contains C (C–N bonds in g-C3N4), N (C–N–C/N–(C)3 groups), Ni2+ (NiO), Zn2+ (ZnO), and O (Ni–O/Zn–O bonds), with oxygen-deficient defects, validating its chemical structure. Further, iron-doped ZnO was fabricated via chemical co-precipitation.104 XRD characterization revealed a single-phase crystalline structure for both the undoped and iron-doped ZnO NPs. The SEM imaging showed agglomerates of NPs with varying sizes. A minimization in the band gap energy was noticed with increasing iron content, attributed to modifications in the lattice parameters.104 Further, the synthesis of nitrogen-doped ZnO was reported, noting the development of a nanorod morphology upon nitrogen incorporation.106 Photoluminescence (PL) analysis revealed nitrogen-doped ZnO nanoparticles exhibit band gap narrowing, reduced electron–hole recombination at 1% doping (via non-radiative centers), and enhanced UV-blue emission (CIE chromaticity shift), making them suitable for UV-light devices, Fig. 5h and i. Additionally, the growth of CuO/CdO nanosheets using a co-precipitation route was described.105 The introduction of the non-ionic surfactant Triton X-100 led to the formation of hexagonal, nanoporous grains. While pristine CuO and CdO exhibited monoclinic and cubic crystal structures, respectively, XRD analysis of the CuO/CdO composite revealed a mixed-phase composition.105
 | ||
| Fig. 5 (a) HRTEM images of representative ZnO, Ce2O, and Ce2O/ZnO composite, reprinted with the permission of ref. 103 copyright 2025, Elsevier; (b–g) XPS analysis of the NiO/ZnO/g-C3N4 nanocomposite material: (b) full survey spectrum, (c) C 1s, (d) N 1s, (e) Ni 2p, (f) Zn 2p, and (g) O 1s core-level spectra, reprinted with the permission of ref. 104, copyright 2025, Elsevier; (h) PL spectra (300–700 nm), (i) detailed emission profiles (500–620 nm) for varying nitrogen concentrations of ZnO and nitrogen-doped ZnO (ZnNxO1−x NPs), and (j) CIE chromaticity coordinates comparing color emission characteristics of ZnO and ZnN0.01O0.99 NPs, reprinted with the permission of ref. 105, copyright 2025, Elsevier. | ||
Microwave-assisted synthesis has emerged as a powerful technique leveraging electromagnetic radiation in the frequency range of 300 MHz–300 GHz to drive chemical reactions.46 This method offers distinct advantages, including cost-effectiveness, time efficiency, energy savings, and the ability to produce controlled-size products.46 Unlike conventional heating methods, microwave irradiation facilitates rapid and homogeneous heating of materials, leading to optimal nucleation conditions, short crystallization times, and enhanced control over macroscopic morphology and size distribution during fabrication.46 This precise control over the synthetic environment allows for the tailoring of metal oxides composite properties, making microwave-assisted methods particularly attractive for optimizing performance in environmental applications. Thus, a CoFe2O4@TiO2@rGO nanocomposite (CoTG) was synthesized using a combination of microwave and sol–gel methods. TEM analysis confirmed the successful impregnation of spherical TiO2 and CoFe2O4 nanoparticles onto rGO sheets.107 DRS spectra revealed that the CoTG nanocomposite exhibited an effective bandgap for visible light activity.107 Furthermore, solution pH significantly influenced the crystallinity and structure of ZnO synthesized using microwave irradiation, as demonstrated by the SEM micrographs in Fig. 6a–c.108 Similarly, the photocatalyst morphology exhibited a strong pH-dependency.112 Furthermore, a cost-effective microwave-assisted method as employed to create N-doped TiO2/rGO hybrid composites (N/TiO2/rGO) with varying rGO content.109 Anatase was the only crystalline phase detected in the synthesized materials. While rGO loading did not affect morphology, it improved photocatalytic activity, especially at lower concentrations.109 The FTIR, Raman, XRD, and DRS spectra, shown in Fig. 6d–g, confirmed the anatase TiO2 structure in N/TiO2/rGO composites, with Raman and XRD indicating rGO integration (via D/G bands and disorder peaks) and FTIR showing reduced O–H intensity due to rGO's hydrophobicity. Tauc plot revealed rGO reduces the bandgap (optimal at ∼5 wt%), enhancing charge separation and narrowing the bandgap for improved photocatalytic activity (Fig. 6g). Additionally, the successful microwave synthesis of TiO2–CuO materials with well-defined crystalline structures was reported.110 Nitrogen adsorption/desorption isotherms analysis revealed that TiO2–CuO composites exhibit decreasing BET surface areas (119 to 19 m2 g−1) with higher CuO content, while pore diameters increase (8.6–19.7 nm), confirming mesoporosity (Fig. 6h and i). The optimal TiO2:CuO ratios (7:3, 5:5, 3:7) showed higher surface areas than pure CuO, suggesting enhanced photocatalytic potential due to well-crystalline anatase and CuO phases.
 | ||
| Fig. 6 SEM images for ZnO samples obtained at pH: (a) 8; (b) 10; and (c) 12, reprinted with the permission of ref. 108, copyright 2025, Elsevier; (d) FTIR and (e) Raman spectra, (f) XRD patterns, and (g) reprinted with the permission of ref. 109, copyright 2025, Elsevier; Tauc plots for bandgap analysis of N/TiO2 and N/TiO2/rGO photocatalysts with varying rGO loadings (0.25–10 wt%), (h and i) N2 adsorption/desorption isotherms and pore diameter distributions of fabricated TiO2, CuO, and TiO2/CuO composites, reprinted with the permission of ref. 110, copyright 2025, Elsevier; (j) AFM images and AFM roughness profile of CeO2@ZnO CNS, reprinted with the permission of ref. 111, copyright 2025, Elsevier. | ||
Solvothermal synthesis has emerged as a powerful and versatile technique for fabricating MOx-based composites with precisely tailored microstructures and enhanced functionalities. This method leverages the unique properties of solvents at elevated temperatures and pressures to facilitate controlled chemical reactions and crystal growth. Solvothermal synthesis offers distinct advantages in achieving intricate morphologies, high crystallinity, and homogenous elemental distributions. The solvothermal process typically involves dissolving metal precursors and desired dopants or supporting materials in a suitable solvent, which is then sealed within an autoclave. The autoclave is subsequently heated to a specific temperature, typically ranging from 100 °C to 300 °C, generating autogenous pressure within the sealed vessel. This elevated temperature and pressure environment promotes the dissolution and recrystallization of the precursors, leading to the formation of well-defined MOx nanostructures and their integration with the composite matrix. The choice of solvent plays a crucial role in determining the final product characteristics. Different solvents exhibit varying physicochemical properties, including polarity, viscosity, and coordinating ability, which can influence the reaction kinetics, nucleation, and growth mechanisms. For example, polar solvents like water and alcohols can facilitate the hydrolysis and condensation of metal precursors, while non-polar solvents like toluene and hexane are often employed for the synthesis of organic–inorganic hybrid composites. The versatility of solvothermal synthesis extends to the ability to tailor the morphology and composition of metal oxides composites through careful manipulation of reaction parameters. Adjusting the precursor concentration, reaction temperature, and dwell time can influence particle size, shape, and crystallinity. Furthermore, the introduction of surfactants or capping agents can further modulate the growth process, leading to the formation of hierarchical structures, core–shell morphologies, or other complex architectures. The controlled environment within the autoclave minimizes the introduction of impurities and allows for the incorporation of dopants or supporting materials with high precision. This precise control over composition enables the design of metal oxides composites with tailored electronic properties and enhanced catalytic or adsorptive performance. For instance, incorporating transition metals or other dopants into the metal oxides lattice can modify its electronic structure and improve its catalytic activity towards specific reactions. Similarly, incorporating carbonaceous materials or other high-surface-area supports can enhance the composites' adsorption capacity and facilitate mass transport. For instance, CeO2@ZnO core–shell nanostars with a crystalline structure were fabricated via hydrothermal and precipitation techniques.111 Their crystallinity and nanoscale dimensions were verified by XRD analysis. AFM analysis revealed CeO2@ZnO core–shell nanostructures exhibit a nanosized, star-like morphology with high surface roughness (91.64 nm over 2 × 2 μm), enhancing organic molecule adsorption to promote photocatalytic activity, Fig. 6j.111 In a separate study, TiO2 nanosheets were directly grown on CaTiO3 surfaces using a hydrothermal process.113 Furthermore, a hydrothermal approach in an ethanol/water solution was employed to load TiO2-functionalized graphene oxide onto TiO2 nanoparticles.114 In addition, a hydrothermal method was used to synthesize a mesoporous CeO2/rGO nanocomposites with a surface area of 100.129 m2 g−1.115 In a separate study, a surfactant-assisted hydrothermal approach was used to synthesize multiple layers of a coral-like shaped MgO/g-C3N4.116
Sonochemical synthesis, utilizing the unique effects of acoustic cavitation, has emerged as a powerful technique for fabricating MOx-based composites with tailored properties for advanced environmental applications.21 The rapid formation and collapse of microbubbles in a liquid medium under ultrasonic irradiation generate localized hotspots characterized by extreme temperatures and pressures.21 These transient, localized conditions promote rapid nucleation and growth, leading to the formation of highly crystalline nanostructures with controlled morphology and enhanced surface area. Furthermore, the intense microstreaming and shockwaves generated by cavitation enhance mass transfer and facilitate uniform dispersion of the metal oxides component within the composite matrix. Specifically, sonochemical methods offer several distinct advantages such as precise control over particle size and morphology, enhanced surface area and porosity, uniform dispersion and intimate interfacial contact, and activation of catalysts and enhanced catalytic activity.117 For instance, A CoO–ZnO nanocomposite was synthesized using sonochemical co-precipitation.118 SEM micrographs revealed the clumping of spherical particles, which was attributed to the differing magnetic properties of the composite materials.
Sol–gel processing offers a versatile and powerful route for synthesizing MOx-based composites with tailored microstructures and enhanced functionalities for environmental applications. This method leverages the controlled hydrolysis and condensation of metal alkoxides or metal salts in a solution, ultimately leading to the formation of a gel network. This gel, upon subsequent drying and calcination, yields the desired MOx composite. The inherent advantages of the sol–gel method lie in its ability to achieve high purity, homogeneity, and precise control over composition at relatively low temperatures compared to solid-state methods. Moreover, the sol–gel process allows for facile incorporation of dopants and the creation of multi-component composites with intricate architectures. The structural and textural properties of the final metal oxides composite are significantly influenced by several key parameters within the sol–gel process. These include the choice of precursors, solvent, catalyst, water-to-alkoxide ratio, aging time, drying conditions, and calcination temperature. Manipulating these parameters allows for fine-tuning of pore size distribution, particle size, crystallinity, and surface area, which are critical factors in determining the material's performance in applications such as catalysis and adsorption.
In summary, the fabrication of metal oxide (MOx)-based composites for environmental applications demands a strategic balance between synthesis scalability, structural precision, and functional performance. Ex situ synthesis approaches, while enabling modular design of pre-optimized components, often struggle with interfacial incompatibility and uneven nanoparticle distribution. In situ synthesis approaches, e.g., sol–gel, co-precipitation, and others, excel in achieving homogeneous dispersion and strong interfacial interactions, which are crucial for catalytic and electronic properties, but face challenges in controlling agglomeration during scale-up. Co-precipitation remains a versatile, low-cost route for tailoring crystallinity and defect chemistry, yet its reliance on precise pH and temperature control limits reproducibility. Microwave-assisted synthesis offers rapid and energy-efficient crystallization with fine morphological control but requires optimization of radiation parameters to prevent uneven heating in complex composites. Solvothermal methods provide unparalleled microstructural precision and crystallinity but are constrained by high-pressure/temperature conditions and solvent selection trade-offs. Sonochemical routes promote uniform dispersion via cavitational effects but lack scalability, while sol–gel processing enables atomic-level homogeneity at the expense of prolonged processing times. A critical challenge across all methods lies in reconciling the trade-off between achieving nanoscale precision, e.g., defect engineering, core–shell architecture, and maintaining cost-effective and eco-friendly scalability. Additionally, the integration of characterization techniques (e.g., HRTEM, XPS, BET) is indispensable for validating structure–property relationships. Future efforts should prioritize hybrid fabrication strategies, such as microwave-solvothermal or sonochemical co-precipitation, to synergize the advantages of individual methods while mitigating their limitations. Additionally, advancing in situ characterization during synthesis and adopting machine learning for parameter optimization could accelerate the development of MOx composites tailored for real-world environmental remediation.
3.2 Strategies to enhance photocatalytic performance of metal oxide composites
Extensive research has been conducted to enhance the photocatalytic performance of pristine metal oxides. Strategies such as constructing heterojunctions, doping with other elements, as well as hybridizing with carbon-based nanomaterials, have been explored.47,119 | ||
| Fig. 7 Various photocatalyst heterojunction interfaces: (a) Type-II heterojunction, (b) P–N heterojunction, (c) direct Z-scheme heterojunction, (d) all-solid-state Z-scheme heterojunction, and (e) S-scheme heterojunction, reprinted with the permission of ref. 121, copyright 2025, Elsevier. | ||
The Type-II heterojunction is a foundational photocatalytic architecture designed to enhance charge separation by leveraging staggered band alignment between two semiconductors. This design is widely used in applications where rapid charge transport outweighs the need for extreme reduction or oxidation capabilities. In a Type-II heterojunction, two semiconductors with offset band structures are combined. Semiconductor A (SC1) has a higher conduction band (CB) and valence band (VB) than Semiconductor B (SC2). When these materials form an intimate interface, their Fermi levels equilibrate, inducing band bending and creating a built-in electric field at the junction. This electric field drives directional charge transfer electrons in SC1's CB migrate to SC2's CB (lower energy), and goes in SC2's VB transfer to SC1's VB (higher energy). For instance, thin TiO2/WO3·H2O layers were created using supercritical CO2. This combo splits light-made electrons and holes faster, making reactions quicker. Sunlight excites both materials, creating electrons and holes. Electrons move from TiO2 to WO3·H2O, while holes go to TiO2, keeping them apart. Electrochemical impedance spectroscopic tests (EIS) showed the combo moves charges easier than pure parts, lowering resistance.128 Further, a ternary CdS QDs, ZnO and g-C3N4 nanocomposite was synthesized.129 The boosted performance of this nanocomposite likely stems from the optimal alignment of electronic energy bands among the three components, which forms combined Z-Scheme and Type-II heterojunctions. These heterojunctions improve charge transfer efficiency, explaining why the CdS@ZnO/g-C3N4 nanocomposite outperforms single or dual-component systems in photocatalytic activity, as shown in Fig. 8a.129 Further, the photocatalytic mineralization of rhodamine B dye (RB) by the α-Fe2O3@NiO primarily relies on ˙OH and photogenerated h+, as confirmed by scavenger tests.132 Without scavengers, efficiency was 94%, but adding tert-butyl alcohol (˙OH scavenger) and EDTA (h+ scavenger) reduced efficiency to 77% and 43%, respectively, highlighting their critical roles. Under UV light, the nanocomposite adsorbs RB dye, initiating charge separation: electrons (e−) move to NiO's CB, while holes remain in α-Fe2O3's VB. The band alignment, α-Fe2O3's VB is more negative (higher energy) than NiO's VB, and NiO's CB is more positive (lower energy) than α-Fe2O3's CB, thereby enabling efficient charge transfer. Electrons from α-Fe2O3 migrate to NiO, while holes shift to α-Fe2O3, prolonging carrier lifetimes. Reactive radicals (˙OH, ˙O2−) form via reactions between charge carriers, water, and oxygen, attacking adsorbed dye molecules and driving photodegradation. The band alignment between α-Fe2O3 and NiO indicates a Type II heterojunction. The staggered band structure allows spatial separation of electrons (accumulating in NiO's CB) and holes (accumulating in α-Fe2O3's VB), minimizing recombination and enhancing photocatalytic efficiency.
 | ||
| Fig. 8 (a) Proposed mechanism of the photocatalytic degradation of RhB using the ternary nanocomposite CdS@ZnO/g-C3N4, reprinted with the permission of ref. 129, copyright 2025, Elsevier; (b) the bands alignment of ZnFe2O4, TiO2, and ZT-10 nanocomposite, reprinted with the permission of ref. 130, copyright 2025, Elsevier; (c) PL spectra of GN0, GN5, GN10 and GN15 nanocomposite samples, (d) effect of scavengers of degradation of MB on GN10, reprinted with the permission of ref. 122, copyright 2025, Elsevier; and (e) Z-scheme mechanism for photocatalysis of TC by CF/ZnO/Ag2O photocatalysts; reprinted with the permission of ref. 131, copyright 2025, Elsevier. | ||
p–n heterojunctions, formed by the interface between p-type (hole-conducting) and n-type (electron-conducting) metal oxides, are pivotal in designing functional materials with enhanced electronic and optoelectronic properties. The junction arises from the contact of two semiconductors with opposing doping characteristics, such as p-type NiO or CuO and n-type TiO2 or ZnO. At the interface, Fermi level equilibration drives electron diffusion from the n-type to the p-type material and hole diffusion in the reverse direction, creating a depletion region and a built-in electric field. This field acts as a driving force for charge separation, enabling efficient extraction of photogenerated carriers without external bias. Turns out, some semiconductors aren't strictly n-type or p-type.133 For example, the researchers can be messed with a material's structure to tweak it into p-type or n-type, e.g., if ZnO had oxygen missing (oxygen vacancies), it acted as n-type, but if metal atoms were missing (metal vacancies), it flipped to p-type.134 In another instance, TiO2 with messed-up titanium (Ti vacancies) acted as a p-type, while regular TiO2 stayed n-type semiconductor.135 The difference in Fermi levels between the p-type and n-type materials drives the diffusion of majority carriers across the interface. Electrons diffuse from the n-type to the p-type material, leaving behind positively charged ionized donors, while holes diffuse from the p-type to the n-type material, leaving behind negatively charged ionized acceptors. This diffusion creates a space charge region, also known as the depletion region, at the interface. Crucially, the resulting separation of positive and negative charges establishes a built-in electric field directed from the n-type to the p-type material. This field opposes further diffusion of majority carriers, ultimately reaching an equilibrium state. The magnitude and spatial extent of this built-in electric field are determined by the doping concentrations, the permittivities of the materials, and the difference in their work functions. This internal electric field plays a critical role in separating photogenerated electron–hole pairs, driving electrons towards the n-type material and holes towards the p-type material, thereby hindering recombination and enhancing the efficiency of charge collection. The synthesized ZnFe2O4/TiO2 (ZT-10) p–n heterojunction enhanced NH4+–N removal through optimized band alignment and an internal electric field (IEF).130 Mott–Schottky analysis confirmed ZnFe2O4 (p-type, Efb = 0.34 eV vs. NHE) and TiO2 (n-type, Efb = −0.17 eV vs. NHE) exhibit staggered Type II band edges (ZnFe2O4: ECB = −1.34 eV, EVB = 0.54 eV; TiO2: ECB = −0.37 eV, EVB = 2.81 eV). Pre-contact, TiO2's Fermi level (EF) lay near its conduction band (electron-rich), while ZnFe2O4's EF resided near its valence band (hole-rich). Upon contact, EF equilibration triggered band bending, forming the heterojunction, as shown in Fig. 8b. The IEF spatially separates charges, driving electrons to TiO2 and holes to ZnFe2O4, minimizing recombination and sustaining redox reactions for efficient pollutant degradation.130 A nanoflower-structured p–n heterojunction photocatalyst, BiOBr/TiO2 (BT-x), was synthesized via a facile coprecipitation method.136 Under visible light, BT-x demonstrated superior efficiency in degrading methyl Orange (MO) and gaseous formaldehyde. The p-type BiOBr, with its narrow bandgap, enhances visible-light absorption, while the p–n heterojunction with TiO2 promotes enhanced charge carrier separation and transfer, reducing electron–hole recombination. The 3D nanoflower morphology amplifies light utilization through internal scattering, boosting absorption, while also providing abundant exposed active sites for catalytic reactions. The system forms a Type II heterojunction due to the staggered band alignment between p-type BiOBr and n-type TiO2.136 Furthermore, CuO/ZnO p–n heterojunction nanofibers, were fabricated by integrating p-type CuO with n-type ZnO nanofibers and demonstrated robust efficiency in degrading pyridine from fuel oil.137 The enhanced performance stems from boosted-light harvesting and robust separation phenomena. Mechanistic studies revealed that h+ primarily drive the formation of reactive intermediates, e.g., ˙O2−, facilitating pyridine's complete mineralization. The nanofiber structure further optimizes light utilization and charge transfer pathways.137 A mesoporous rod-shaped ZnO/CuO/CeO2 n–p–n heterojunction was fabricated using a two-step co-precipitation method for photocatalytic applications.138 Characterization via XRD, FTIR, UV-vis, and SEM confirmed its structure, with interfaces between ZnO (n-type), CuO (p-type), and CeO2 (n-type), extending light absorption to 800 nm. Enhanced performance stemmed from broad light absorption, optimized band alignment, and efficient charge carrier separation/transfer. The mesoporous rod morphology improved light harvesting and active site exposure, while the n–p–n configuration facilitated directional charge flow.138
Z-scheme heterojunctions represent a promising architecture for enhancing the performance of metal oxide-based photocatalysts by mimicking the natural photosynthesis process. These systems can be categorized into two main types: mediated and direct Z-scheme photoreaction systems, each exhibiting unique charge transfer mechanisms and electric field dynamics. In mediated Z-schemes, a redox couple or a solid-state electron shuttle facilitates the recombination of photogenerated electrons from the photoreaction system II, PSII, analogue with holes from the photoreaction system I, PSI, analogue. This indirect recombination, driven by favorable energy level alignments, preserves the high redox potential of the system by leaving behind highly reductive electrons in the PSI analogue and highly oxidative holes in the PSII analogue.139 The internal electric field within each semiconductor component directs charge carriers towards the mediator interface, enhancing the recombination process. Mediator selection plays a critical role, with noble metals offering high conductivity but facing cost and stability issues, while redox couples provide a cost-effective alternative but may exhibit slower kinetics. Solid-state mediators, like reduced graphene oxide (rGO), aim to combine the advantages of both. Thus, adding 10% NiO to g-C3N4 (forming the GN10 nanocomposite) minimized PL peak by 66%, reflecting suppressed electron–hole recombination, as shown in Fig. 8c.122 This modification significantly boosted MB dye degradation efficiency from 33% (using pristine g-C3N4) to 91.6% (using the GN10 nanocomposite). Scavenger experiments revealed a Z-scheme photocatalytic mechanism, Fig. 8d and e, where visible light-induced charge carriers (electrons and holes) were effectively separated. Superoxide radicals (˙O2−), generated via electron transfer to oxygen, were identified as the primary active species driving MB degradation.122 Furthermore, the N–ZnO/g-C3N4 hybrid, fabricated via high-calcination, demonstrated superior performance owing to a Z-scheme charge-transfer mechanism.140 The band alignment of N–ZnO enables e− in its CB to migrate to the VB of g-C3N4, minimizing recombination phenomena. This process retains e− in g-C3N4's CB and holes in N–ZnO's VB, boosting redox activity. PL studies employing terephthalic acid (TA) emphasized ˙OH radical generation, with the N–ZnO/g-C3N4 hybrid showing the robust PL peak (∼460 nm), attributed to 2-hydroxyterephthalic acid (a ˙OH adduct). Pristine ZnO failed to generate ˙OH under visible light, while g-C3N4 alone exhibited poor ˙OH signals owing to secondary reactions of superoxide radicals (˙O2−) with water. The composite's boosted ˙OH production stems from efficient hole accumulation in N–ZnO's VB, which oxidizes water.140 Further, a Z-scheme Ag2O/ZnO heterostructure grown directly on a large-area carbon fiber (CF) substrate (CF/ZnO/Ag2O) was developed to boost photocatalytic degradation of TC in water.141 The carbon fiber cloth enables easy catalyst recovery and scalability for industrial use, while the Z-scheme design boosts separation and catalytic efficiency. Under light, electrons from ZnO's CB migrate via the CF to Ag2O, where they recombine with holes (h+) in Ag2O's VB. This creates a solid-state Z-scheme heterojunction on the CF surface, preserving the strong redox potentials of both ZnO (for oxidation) and Ag2O (for reduction), thereby improving TC degradation, as shown in Fig. 8e. Furthermore, solid-state Z-scheme PW12/Ag/ZnO, was fabricated by integrating ZnO with Keggin-type tungstophosphate (PW12) and incorporating silver (Ag) as a conductive mediator.131 The ternary hybrid leverages Ag to enable efficient spatial charge separation and directional carrier transfer, differing markedly from binary counterparts (PW12/Ag and PW12/ZnO). Experimental results reveal that the ternary system alters the charge migration pathway, enhancing photocatalytic performance. Silver bridges ZnO and PW12, facilitating electron transfer between the semiconductors while minimizing recombination losses. This design preserves the redox capabilities of both components, a hallmark of Z-scheme systems, and improves visible-light-driven activity.131
The S-scheme (Step-scheme) heterojunction is an advanced photocatalytic architecture designed to optimize charge separation while retaining the strong redox potentials of two coupled semiconductors. Unlike traditional Type-II heterojunctions, which sacrifice redox power for charge separation, the S-scheme selectively recombines low-energy charges (electrons from the reduction photocatalyst and holes from the oxidation photocatalyst) while preserving high-energy charges for redox reactions. This mechanism mimics natural photosynthesis but with enhanced efficiency, driven by interfacial electric fields and tailored band alignment. The S-scheme relies on a staggered band structure between two semiconductors: a reduction photocatalyst (RP) with a higher Fermi level (e.g., g-C3N4, CdS) and an oxidation photocatalyst (OP) with a lower Fermi level (e.g., TiO2, WO3). When these semiconductors form a heterojunction, their Fermi levels equilibrate, inducing band bending at the interface. This bending generates a built-in electric field that directs charge flow. Electrons from the OP's conduction band (CB) migrate to the RP's CB, and Holes from the RP's valence band (VB) transfer to the OP's VB. The electric field acts as a “charge filter”, promoting recombination of low-energy electrons (OP's CB) and low-energy holes (RP's VB) at the interface. This leaves behind high-energy electrons in the RP's CB (for reduction reactions like H2 evolution) and high-energy holes in the OP's VB (for oxidation reactions like O2 generation or pollutant degradation). The electric field also spatially separates charges, reducing bulk and surface recombination. For instance, the SnO2/SnS2 heterojunction exemplifies the S-scheme charge-transfer mechanism, where staggered band alignment preserves the strong oxidative VB of SnO2 (∼3.1 eV) and the reductive CB of SnS2 (∼−0.2 eV), enabling robust carrier separation while retaining high redox potentials.142 Under illumination, interfacial recombination of low-energy carriers (SnO2 CB electrons and SnS2 VB holes) is driven by the built-in electric field, leaving high-energy holes in SnO2's VB and electrons in SnS2's CB for redox reactions. This selective charge dynamics enhances oxidative ˙OH radical formation (via direct H2O/OH− oxidation) and reductive  generation (via O2 reduction). Synergy between the components further promotes secondary ROS, from
generation (via O2 reduction). Synergy between the components further promotes secondary ROS, from  recombination.142 Further, the TiO2/BaTiO3 heterojunction demonstrates an S-scheme charge-transfer pathway, where a work function disparity (TiO2: 6.57 eV, BaTiO3: 3.83 eV) drives electron transfer from BaTiO3 to TiO2, forming an interfacial electric field (IEF), Fig. 9c.143 This IEF steers recombination of low-energy carriers (TiO2's CB electrons and BaTiO3's VB holes), preserving high-energy holes in TiO2's VB (1.88 eV) and electrons in BaTiO3's CB for redox reactions. ESR spectra confirm enhanced ˙OH and ˙O2− generation, surpassing thermodynamic thresholds for H2O/OH− oxidation and O2 reduction, Fig. 9a–c. Density Functional Theory (DFT) calculations revealed stronger adsorption of H2O (−1.14 eV) and O2 (−1.10 eV) on TiO2/BaTiO3 versus TiO2 (−0.64/−0.06 eV), with elongated O–O/O–H bonds facilitating ROS formation, Fig. 9d–g. Spatial charge localization directs H2O adsorption on TiO2 (hole-mediated ˙OH) and O2 adsorption on BaTiO3 (electron-mediated ˙O2−), bypassing Type-II heterojunction limitations. This synergy ensured efficient toluene mineralization via ROS pathways.143 A ZnO/WO3 S-scheme, fabricated via hydrothermal and calcination, exhibited boosted photocatalytic H2O2 generation via a direct two-electron O2 reduction pathway.144 The interfacial internal electric field in the heterojunction promotes charge migration while preserving electrons with robust reduction capability. PL spectra showed ZW30's lower 390 nm emission versus ZnO (Fig. 9h), while WO3's minimal PL intensity reflects its low photoexcitation efficiency. The time-resolved PL (TRPL) spectra revealed ZW30's shorter carrier lifetime (4.4 ns vs. ZnO's 5.88 ns), Fig. 9i, indicating reduced recombination. Transient photocurrent (PC) and EIS measurements demonstrated ZW30's rapid photo response, higher current density (15.6 vs. 9.4 μA cm−2), and lower charge-transfer resistance (1296 Ω), confirming efficient electron transfer, Figure 9k.144 A separate investigation demonstrated that black nickel oxide nanoparticles (NiO NPs) were firmly attached to nitrogen-rich graphitic carbon nitride (g-C3N5) nanosheets (CNNS), forming an S-scheme NOCN heterojunction. This structure enhanced charge carrier separation and redox potential, as shown in Figure 9l.145 The study revealed that the NiO NPs boosted light absorption and heat generation capabilities in the NOCN composites compared to their unmodified counterparts.
recombination.142 Further, the TiO2/BaTiO3 heterojunction demonstrates an S-scheme charge-transfer pathway, where a work function disparity (TiO2: 6.57 eV, BaTiO3: 3.83 eV) drives electron transfer from BaTiO3 to TiO2, forming an interfacial electric field (IEF), Fig. 9c.143 This IEF steers recombination of low-energy carriers (TiO2's CB electrons and BaTiO3's VB holes), preserving high-energy holes in TiO2's VB (1.88 eV) and electrons in BaTiO3's CB for redox reactions. ESR spectra confirm enhanced ˙OH and ˙O2− generation, surpassing thermodynamic thresholds for H2O/OH− oxidation and O2 reduction, Fig. 9a–c. Density Functional Theory (DFT) calculations revealed stronger adsorption of H2O (−1.14 eV) and O2 (−1.10 eV) on TiO2/BaTiO3 versus TiO2 (−0.64/−0.06 eV), with elongated O–O/O–H bonds facilitating ROS formation, Fig. 9d–g. Spatial charge localization directs H2O adsorption on TiO2 (hole-mediated ˙OH) and O2 adsorption on BaTiO3 (electron-mediated ˙O2−), bypassing Type-II heterojunction limitations. This synergy ensured efficient toluene mineralization via ROS pathways.143 A ZnO/WO3 S-scheme, fabricated via hydrothermal and calcination, exhibited boosted photocatalytic H2O2 generation via a direct two-electron O2 reduction pathway.144 The interfacial internal electric field in the heterojunction promotes charge migration while preserving electrons with robust reduction capability. PL spectra showed ZW30's lower 390 nm emission versus ZnO (Fig. 9h), while WO3's minimal PL intensity reflects its low photoexcitation efficiency. The time-resolved PL (TRPL) spectra revealed ZW30's shorter carrier lifetime (4.4 ns vs. ZnO's 5.88 ns), Fig. 9i, indicating reduced recombination. Transient photocurrent (PC) and EIS measurements demonstrated ZW30's rapid photo response, higher current density (15.6 vs. 9.4 μA cm−2), and lower charge-transfer resistance (1296 Ω), confirming efficient electron transfer, Figure 9k.144 A separate investigation demonstrated that black nickel oxide nanoparticles (NiO NPs) were firmly attached to nitrogen-rich graphitic carbon nitride (g-C3N5) nanosheets (CNNS), forming an S-scheme NOCN heterojunction. This structure enhanced charge carrier separation and redox potential, as shown in Figure 9l.145 The study revealed that the NiO NPs boosted light absorption and heat generation capabilities in the NOCN composites compared to their unmodified counterparts.
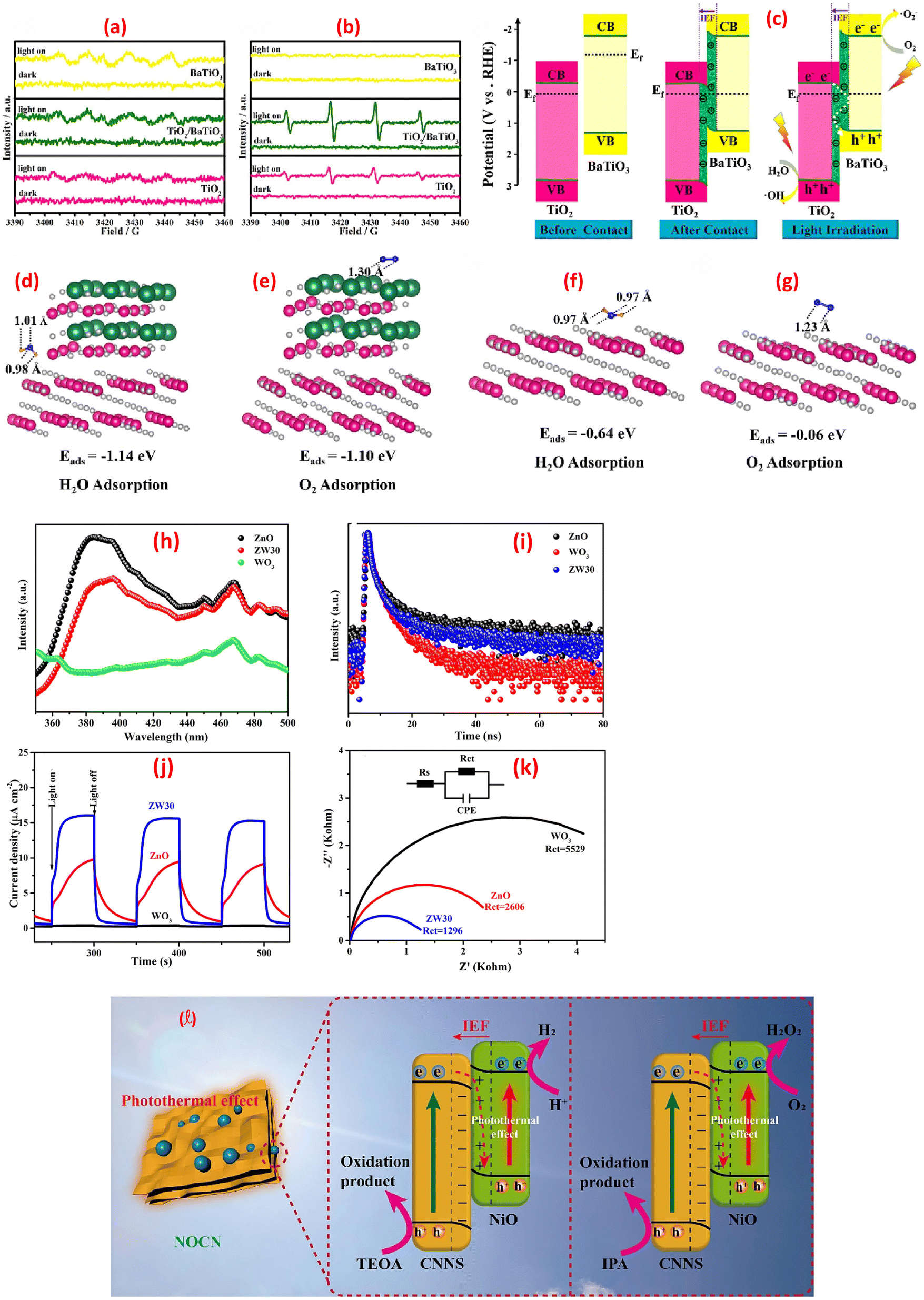 | ||
| Fig. 9 ESR spectra (under dark and light irradiation): (a) DMPO–˙OH (aqueous dispersion), (b) DMPO–˙O2− (methanol dispersion). (c) Schematic illustration of the S-scheme charges transfer process on the TiO2/BaTiO3 heterojunction. (d) H2O and (e) O2 adsorption on TiO2/BaTiO3, (f) H2O and (g) O2 adsorption on TiO2, reprinted with the permission of ref. 143, copyright 2025, Elsevier; (h–k) spectra of ZnO, WO3, and ZW30, (h) PL spectra, (i) TRPL spectra, (j) transient photocurrent response, and (k) EIS spectra, reprinted with the permission of ref. 144, copyright 2025, Elsevier; (l) photothermal-assisted photocatalytic H2 and H2O2 production via NOCN S-scheme heterojunction, reprinted with the permission of ref. 145, copyright 2025, Elsevier. | ||
However, noble metals also introduce challenges, including high costs and nanoparticle aggregation during prolonged reactions. Transition metals (e.g., Cu, Ni, Co, Fe) offer a cost-effective and versatile route to enhance metal oxide photocatalysts, primarily through bandgap engineering and heterojunction formation. Doping transition metals into oxides like ZnO or WO3 introduces mid-gap states or modifies d-orbital hybridization, narrowing the bandgap to enhance visible-light absorption.
Combining noble- and transition-metals on metal oxides can yield synergistic effects that surpass the performance of single-metal systems. For example, Ni/Pt co-deposition on TiO2 achieved higher CO2 yields than either single deposition on TiO2.157 Moreover, the synergistic deposition of Ni and Pt nanoparticles on TiO2 enhanced photocatalytic hydrogen production compared to pure TiO2 by reducing the bandgap (via dopant-induced quasi-static energy levels), forming a Pt-mediated Schottky junction to accelerate electron transfer, and suppressing charge recombination through Ni–Pt interfacial interactions, while the preferential reduction of Pt4+ (over Ni2+) and methanol's role as a sacrificial agent further optimized charge separation and proton reduction kinetics.157 Similarly, the bimetallic deposition of Au and Cu on Al2O3 significantly enhanced the catalytic CO oxidation compared to pure Al2O3 by stabilizing Au nanoparticles against sintering via Cu incorporation, optimizing charge transfer between metallic Au and Cu+/Cu2+ species, and mitigating carbonate-induced deactivation through reactive intermediate formation that preserved active sites under operational conditions.158 Further, The co-deposition of Ag and Pt nanoparticles on Ag3PO4–WO3 heterostructures enhanced photocatalytic hydrogen production by synergistically leveraging Ag's plasmonic resonance for broad visible-light absorption, where Pt acted as an electron sink to suppress charge recombination, and the formation of a Schottky junction at the metal-semiconductor interface to facilitate efficient electron transfer, while the Ag3PO4/WO3 heterojunction promoted spatial separation of photogenerated carriers and the green-synthesized Pt–Ag nanoparticles improved dispersion and stability, collectively optimizing redox kinetics for bioethanol reforming.159 The co-deposition of Pt and Al on WO3 significantly enhanced photocatalytic performance compared to pure WO3 by synergistically reducing the bandgap (from 2.36 eV to 1.95 eV) via Al-induced lattice distortion and Pt-mediated conduction band modulation, while Pt nanoparticles acted as electron sinks to suppress recombination and oxygen vacancies/Al3+ sites improved charge transfer kinetics, enabling 43.61% optical modulation and 85% transmittance recovery via efficient Li+ intercalation in the porous heterostructure.160 The co-deposition of Ag and Cu on TiO2 significantly enhanced photocatalytic performance compared to pure TiO2 by combining Ag's strong visible plasmonic resonance and Cu's extended light absorption, while optimized photo-deposition time (30 min) ensured controlled nanoparticle growth and uniform dispersion, creating a ternary Ag–Cu–TiO2 interface that suppressed charge recombination and accelerated electron transfer, thereby boosting solar-driven degradation efficiency for organic dyes.161 Similarly, the co-deposition of Ag and Cu onto ZnO enhanced photocatalytic performance compared to pure ZnO by leveraging Ag's UV plasmonic resonance and Cu's visible-light absorption, while optimized Ag/Cu bimetallic deposition under UV formed combined interfaces that reduced charge recombination and synergized plasmonic effects, achieving 95% degradation; however, under sunlight, monometallic Cu/ZnO (99%) and Ag/ZnO (98%) outperformed due to their direct plasmonic alignment with solar spectra and simpler charge transfer pathways, avoiding the electron loss observed in bimetallic systems.162 These hybrid systems also mitigate individual drawbacks: transition metals reduce reliance on costly noble metals, while noble metals compensate for the slower kinetics of transition metal-based catalysts. However, optimizing dual-metal systems require precise control over metal ratios, spatial distribution, and interfacial charge transfer pathways.
| Mechanical methods | Advantages | Disadvantages |
|---|---|---|
| Sedimentation | (1) No energy requirement | (1) Ineffective for fine particles (<0.01 mm) and colloids due to low settling rates |
| (2) Excellent reproducibility | (2) Space requirements: requires significant area for large-scale operations | |
| (3) Low operational and maintenance costs due to gravity dependence | (3) Continuous removal of sludge is necessary to prevent quality issues | |
| (4) Capable of treating large volumes of wastewater efficiently | ||
| Dissolved air flotation (DAF) | (1) Effluent quality: provides superior removal of oils, fats, and suspended solids, yielding high-quality effluent | (1) Significant energy and maintenance costs associated with air generation and system upkeep |
| (2) Rapid process: efficient treatment for varying influent qualities and flow rates | (2) Pretreatment requirement: often necessitates pretreatment for optimal performance due to the presence of specific contaminants | |
| (3) Space efficiency: smaller footprint compared to conventional technologies like sedimentation alone | (3) Requires careful engineering to ensure effective separation | |
| Filtration | (1) Effective removal: capable of removing a wide range of particulates and some pathogens | (1) Clogging: filters can quickly become clogged, necessitating regular maintenance and backwashing |
| (2) Flexibility: modular design allows scalability and integration into various systems | (2) Energy use: depending on pressure-driven systems, filtration can be more energy-intensive than passive methods | |
| (3) Can enhance downstream biological and chemical treatments by eliminating solids | ||
| Screening | (1) Protects downstream equipment from large solids and debris which can cause failures | (1) Only removes larger solids (typically > 1 mm); finer particles may require additional processes |
| (2) Ease of operation: generally easy to operate and manage, minimal technical skills required | (2) Regular cleaning: screens must be regularly cleaned to ensure continuous operation (mechanical or manual) | |
| (3) Cost-effective: significantly reduces the load on subsequent treatment stages | ||
| Centrifugation | (1) Very effective at separating various contaminants, including fine solids; can achieve low solid concentrations in effluent | (1) Requires considerable energy and maintenance, making it expensive to operate |
| (2) Capable of rapid processing of sludge and other suspended materials | (2) More complex systems may require skilled operators and regular maintenance to prevent breakdowns | |
| (3) Smaller equipment footprint compared to traditional settling tanks | (3) Can generate significant noise and vibration during operation | |
|
||
| Physicochemical methods | ||
| Membrane filtration | (1) Can remove pathogens, bacteria, and solids down to the nanometer range, producing high-quality effluent | (1) High capital and operating costs |
| (2) Generates less sludge compared to conventional methods | (2) Membranes are prone to fouling | |
| (3) Consume small area | ||
| Coagulation and flocculation | (1) High efficiency for fine particles | (1) Chemical use |
| (2) Removes metals, colour and turbidity | (2) Needs careful management of chemical dosing to avoid residual chemicals in treated water | |
| (3) Increases the overall effectiveness of sedimentation and filtration processes | (3) Sludge production | |
| (4) Multiple process step | ||
| Adsorption | (1) Low cost | (1) Low selectivity of adsorbent |
| (2) Relatively high performance | (2) Disposal problems | |
| (3) Design simplicity | (3) Not suitable for high concentration pollutants | |
| (4) Non-toxic | ||
| (5) Regeneration of adsorbents is often possible | ||
| Ozonation | (1) Highly effective in disinfection and mineralization of organic pollutants | (1) Ozone is unstable and must be generated on-site |
| (2) Produces no harmful residues | (2) Requires careful handling and safety considerations | |
| (3) No need to alter pH and temperature | (3) In ozone generation, toxicity issues and fire hazards may occur | |
| Advanced oxidation processes (AOPs) | (1) Effective for a wide range of organic contaminants, including those that are resistant to conventional treatment methods | (1) Higher operational costs due to the need for chemicals and specialized equipment |
| (2) Can achieve high degradation rates | (2) Potential for toxicity of intermediates | |
| Ion exchange | (1) Possible to regenerate resin | (1) Fouling issue |
| (2) Small area requirement | ||
|
||
| Biological methods | ||
| Aerobic treatment | (1) Simplicity of activity | (1) Foaming problems may arise |
| (2) Enhances nitrogen and phosphorus removal | (2) Cost expensive | |
| Anaerobic treatment | (1) Producing biogas | (1) Longer retention times required |
| (2) Less environmental pollution | (2) Potential for odor emission | |
| (3) Reduces sludge volume | ||
| (4) Can be applied for large capacities | ||
| Catalyst | Pollutant | Light source | Time minutes | Removal (%) | Heterojunction type | Ref. |
|---|---|---|---|---|---|---|
| a Pollutants: MB, methylene blue; TC, tetracycline; OTC, oxytetracycline; RhB, rhodamine B; MO, methyl orange; Flu, fluorescence. | ||||||
| Ce/ZnO | MB | Sun light | 120 | 92.62% | Redox-mediated charge separation | 169 |
| TiO2-GO | Crystal violet dye | Sun light | 150 | 63 | 170 | |
| NiO/-gC3N4 | MB dye | Sun simulator | 90 | 91 | Z-scheme | 122 |
| ZnO–C/MnO2 | Tetracycline hydrochloride | Halogen lamp (340–800 nm) | 92 | 60 | Z-scheme | 171 and 172 |
| 10% NiO/TiO2 | Brilliant green | Sunlight | 180 | 87 | p–n type | 173 |
| TiO2/MnO2 | RhB | 75 W metal halide lamp for visible light and 24 W UV lamp for UV light | 180 | 90.50 | Z-scheme | 174 |
| Bi2WO6/NiO/Ag | Naphthenic acids | Visible light | 180 | 90 | Z-scheme | 175 |
| CuO–CdS | TC | 300 W Xe lamp | 30 | 86.0 | S-scheme | 176 |
| CoFe2O4/g-C3N4 | Enrofloxacin | 250 W xenon lamp with a 420 nm cut-off filter | 60 | 89 | p–n type | 177 |
| ZnCo2O4/MnO2/FeS2 | MO | 500 W halogen lamp | 200 | 96 | Z-scheme | 178 |
| NiO/Bi2WO6 | Ciprofloxacin | Visible light | 90 | 93 | S-scheme | 179 |
| Carbon nanosheet/MnO2/BiOCl | RhB | 300 W Hg lamp | 25 | 97 | Z-scheme | 180 |
| MB | 40 | 98 | ||||
| TC | 30 | 80 | ||||
| BiOI/T-ZnOw | RhB | Visible light | 97.1 | p–n type | 181 | |
| OTC | 88.0 | |||||
| ZnO/CeO2 | RhB | 300 W Xe lamp | 80 | 96 | Z-scheme | 182 |
| Bi2O2CO3/ZnO | RhB | Sunlight of 1000 W power | 180 | 97 | S-scheme | 179 |
| Fe2O3@e-HNbWO6 | RhB | 300 W Xe lamp | 80 | 99.5 | Z-scheme | 183 |
| 0.1% Ba/ZnO | RhB | Vis-light | 60 | 98.8 | Metal doping | 184 |
| Ag/ZnO/AgO/TiO2 | RhB | Xenon lamp (UV-vis) with power of 350 W | 100 | 99.32 | Z-scheme | 185 |
| MoO3/Bi2O4 | RhB | 100 W LED lamps | 40 | 99.5 | Z-scheme | 186 |
| NiO/TiO2 | MO | UV light (254 nm, 15 W) | 30 | 96.5 | p–n type | 187 |
| NiFe2O4/TiO2 | Congo red | Sunlight irradiation | 180 | 97.0 | p–n type | 188 |
| AgO/TiO2 | MB amoxicillin | Sunlight | 360 | 90 | p–n type | 189 |
| 120 | 50 | |||||
| AgI/SnO2 | Flu | Sunlight of 1000 W power | 120 | 95 | Z-scheme | 179 |
| TiO2/Bi2O3 | Levofloxacin | UV-vis | 120 | 92.7 | p–n type | 190 |
| AgI/ZnO/WO3 | MB | 100 W tungsten lamp | 120 | 91.5 | Double Z-scheme (WO3–ZnO and ZnO–AgI) | 191 |
On the other hand, non-metals (e.g., N, S, C) replace oxygen anions, altering the VB through orbital hybridization. Nitrogen doping in TiO2, for example, raises the VB edge via N 2p–O 2p hybridization, narrowing the bandgap and enabling visible-light harvesting. Sulfur's larger ionic radius induces lattice strain, generating oxygen vacancies that serve as electron traps and adsorption sites. These vacancies also promote charge separation by localizing electrons, enhancing surface reactions. Non-metal dopants further influence surface acidity and hydrophilicity, improving reactant adsorption (e.g., H2O or O2). For instance, N-doping in the ZnO@BiVO4 composite induced oxygen vacancy defects in the ZnO component, enhancing charge separation and photocatalytic efficiency, with optimal performance (99.34% tetracycline degradation) achieved at a 20% BiVO4 mass ratio under simulated solar light via a Type-I heterojunction mechanism.192 N-doping in ZnO significantly reduced the bandgap (from 3.12 eV to 2.91 eV), enhancing visible-light absorption and charge separation, which enabled 99% degradation of methylene blue and methyl orange under visible light. Additionally, N-doping introduced oxygen vacancies and mesoporous structures, synergistically boosting supercapacitor performance with a specific capacitance of 762 F g−1 in redox-additive electrolytes, driven by improved conductivity and redox-active sites.193 N-doping in ZnO reduced its bandgap to 2.99 eV, enhancing visible-light absorption and creating Zn–N bonds that facilitated charge separation via a heterojunction with g-C3N4. This doping introduced oxygen vacancies and increased surface area (147.9 m2 g−1 vs. 66.5 m2 g−1 for pure g-C3N4), promoting reactive oxygen species (˙OH and ˙O2−) generation. The 2D–2D interface between N-doped ZnO and g-C3N4 suppressed electron–hole recombination (evidenced by reduced PL intensity), achieving 96.2% crystal violet and 99.3% brilliant green degradation under visible light.194 Nitrogen-doped ZnO supported on biochar photocatalyst derived from Lantana camara leaves (N–ZnO@LBC) exhibited a bandgap reduction from 2.83 eV to 2.78 eV (UV-DRS), enhancing visible-light absorption by introducing mid-gap states, while XRD confirmed lattice expansion due to N substitution at O sites, improving charge separation.195 EDX and FTIR spectra validated the N incorporation and hydroxyl group retention, enabling efficient persulfate activation to generate dominant O2−˙ radicals (scavenger tests).195 Furthermore, the 2D/2D heterojunction with biochar suppressed electron–hole recombination, achieving 95.7% MB degradation under visible light and retaining >90% efficiency over 5 cycles, with a treatment cost of US$9.79 per m3.195 Additionally, sulfur doping in ZnO nanoparticles via a low-temperature solvothermal process introduces S 2p-derived mid-gap states, as confirmed by UV-vis and XPS, reduced the band gap (3.24 eV to 3.09 eV) and enabled visible light absorption. This modification enhanced photocatalytic NOx degradation under visible light by 386% due to improved charge separation, while increased surface area (40.7 m2 g−1 vs. 29.5 m2 g; TEM and BET analysis), and boosted the UV performance by 42%, demonstrating dual structural and electronic optimization.196 Moreover, sulfur doping in carbon xerogel/TiO2 composites introduced Ti–O–S bonds (as evidenced by XPS data) and mid-gap states, reducing the band gap from 3.0 eV (rutile TiO2) to 1.9 eV (as evidenced by DRS data), enabling visible light absorption. This, alongside suppressed TiO2 crystallite growth (smaller anatase/rutile phases via XRD) and enhanced hydroxyl radical formation under humidity, boosting ethylene photo-oxidation by 25% under visible light and 8% under UV, driven by improved charge separation and sulfur migration from carbon to TiO2 during carbonization.197 Additionally, sulfur doping in TiO2/BiVO4 introduced mid-gap states via Ti–O–S bonds (confirmed by XPS and Raman), reducing the band gap from 3.22 eV (pure TiO2) to 2.10 eV (confirmed by DRS data) and enabled visible-light absorption. Coupled with the TiO2/BiVO4 heterojunction, sulfur enhanced charge separation (as calculated by DFT simulations) and oxygen vacancy formation, achieving 89.3% dibenzothiophene removal under visible light through improved ˙OH radical generation and interfacial electron transfer.198 Table 3 shows a comparison of metal deposition and doping approaches.
| Feature | Metal deposition | Doping |
|---|---|---|
| Mechanism of enhancement | Schottky barrier formation, SPR effect (for specific metals), enhanced light absorption, co-catalytic activity, increased surface area | Modified band structure (band gap narrowing, intra-band gap states), enhanced charge separation (trapping sites), increased charge carrier mobility, modified surface properties |
| Location of modification | Primarily on the surface of metal oxide | Within the metal oxide lattice |
| Nature of modification | Formation of a metal-semiconductor interface; physical presence of metal nanoparticles | Introduction of foreign atoms into the metal oxide crystal structure |
| Light absorption enhancement | Primarily through plasmon resonance (for specific metals) and light scattering; can also indirectly enhance absorption in metal oxide through improved charge separation | Directly modifies the band gap or introduces intra-band gap states, leading to increased absorption in the visible region |
| Charge separation enhancement | Schottky barrier promotes electron transfer from metal oxide to metal | Dopants act as electron or hole traps, preventing recombination |
| Charge carrier mobility impact | Can indirectly improve mobility in metal oxide by reducing recombination | Can directly enhance mobility by altering the electronic structure and conductivity of metal oxide |
| Material stability | Potential for metal leaching or sintering over time, especially under harsh conditions | Generally more stable as dopants are incorporated into the lattice |
| Examples | Au, Pt, Ag, Pd, Fe deposition on NiO | N, C, Cu, Fe, Al, Li doping of NiO |
| Disadvantages | Potential for metal leaching, light shielding at high loadings, cost of noble metals | Can be challenging to control dopant concentration and distribution, optimization can be complex |
| Catalyst | Synthesis method | Experimental conditions | Heterojunction type | Performance metrics | Advantages | Ref. |
|---|---|---|---|---|---|---|
| NiO–TiO2 | Hydrothermal calcination | UV-vis light, methanol/water electrolyte | p–n type | 23.5 ± 1.2 mmol h−1 g−1 | Enhanced charge separation, stability. However, higher NiO loading but limited by aggregation | 201 |
| g-C3N4/ZnO/Au | Au solution mixed with g-C3N4/ZnO | Light source: 300 W Xe lamp (UV-vis) | Z-scheme (Au-mediated) | 46.46 μmol g−1 h−1 | The Au enhances the absorption of visible light by the LSPR effect, and act as an electron mediator to accelerate the electrons transfer | 202 |
| Electrolyte: 20 mL TEOA + 80 mL H2O | ||||||
| Cu@TiO2–Cu2O | Solvothermal synthesis | Light source: 300 W Xe lamp (UV-vis) | p–n/Schottky | 12.6 mmol g−1 h−1 | Multiple charge transfer channels, enhanced light absorption, highest stability, higher Cu2O loading but suffers from aggregation | 203 |
| TiO2 | Electrolyte: 20 vol% methanol in water | Schottky p–n | 2.7 mmol g−1 h−1 | |||
| Cu@TiO2 | 5.5 mmol g−1 h−1 | |||||
| TiO2–Cu2O | 6.0 mmol g−1 h−1 | |||||
| polyaniline/ZnO | Combined sol–gel and oxidative polymerization of aniline | Aqueous solution of methanol (20%). A 300 W Xe lamp | Z-scheme | 9.4 mmol h−1 g−1 | Polyaniline is promotes the light absorption and offering an additional electrons that combine with H+ to generate H2-gas | 204 |
| ZnO/g-C3N4 | Solution mixing | Light: 400 W Xe lamp | Z-scheme | 1358 μmol g−1 h−1 | Green synthesis (rajma seeds) | 205 |
| Catalyst: 10 mg with Eosin Y dye and 20% TEOA. | Enhanced charge separation (Z-scheme) | |||||
| High surface area (48 m2 g−1) | ||||||
| Visible-light absorption (Eg = 3.2 eV) | ||||||
| Stable for 24 h (HER) | ||||||
| CeO2/CdSe-DETA (CS-2) | Hydrothermal mixing with CdCl2, Se, DETA, and N2H4 | Light source: 300 W Xe lamp (λ ≥ 420 nm) | S-scheme | 3.71 mmol g−1 h−1 | S-scheme pormotes charge separation | 206 |
| Electrolyte: Na2S/Na2SO3 solution | Broad visible-light absorption (up to 746 nm) | |||||
| High stability (6 cycles) | ||||||
| Large surface area (∼48 m2 g−1) | ||||||
| N-doped CeO2−δ@ZnIn2S4 | Hydrolysis | 300 W xenon lamp, 0.25 M Na2SO3 + 0.35 M Na2S (for H2 evolution) | S-scheme | 798 μmol g−1 h−1 | EPR confirm that S-scheme is built between ZnIn2S4 and N-doped CeO2−δ which stimulates the separation of the e−–h+ pairs, and demonstrates exceptional full range operation for photocatalytic HER activity and durability | 207 |
| CeO2/ZnIn2S4 | Solvothermal (CeO2) + oil bath (composite) | 3 W UV LEDs (λ > 420 nm, 80.0 mW cm−2) | S-scheme | 69 μmol h−1 | S-scheme mechanism enhances charge separation | 208 |
| Electrolyte 0.5 M Na2SO3/Na2S | Hollow structure improves light absorption and SSA | |||||
| Internal electric field drives redox reactions |
| Composite material | Light source | Reactor type | Co-reactants/sacrificial agents | Product and yield | Main findings | Ref. |
|---|---|---|---|---|---|---|
| Ag QDs/hierarchically porous defective TiO2 (Ag/TiO2) | 300 W xenon lamp (UV-vis, unfiltered) | Catalyst weight: 50 mg | H2O (electron donor; holes oxidize H2O to ˙OH) | CO: 2.3 μmol g−1 (4 h) | Hierarchical pores + defects (Ti3+, O vacancies) enhance CO2 adsorption and charge separation | 209 |
| Reactor: 80 mL gas-closed quartz reactor | Selectivity: 100% CO (no H2 byproduct) | Ag QDs enable SPR for visible light absorption | ||||
| CO2 pressure: ambient (purged with CO2 for 30 min) | ||||||
| WO3/THFB-COF-Zn (3:7 mass ratio) |
Simulated visible light (λ = 420–800 nm) | Gas–solid phase (quartz reactor) | None/(sacrificial-agent-free) | CO production rate: 54.1 μmol g−1 h−1 (7× higher than pristine COF) | S-scheme heterojunction: Internal electric field drives e− from WO3 CB to THFB-COF-Zn VB. Retains high-potential e− (THFB-COF-Zn CB) for CO2 reduction | 210 |
| Catalyst loading: 10 mg dispersed on quartz plate | Selectivity: 100% CO (no H2, CH4, or HCOOH detected) | |||||
| CO2 pressure: pure CO2 atmosphere (1 atm) | ||||||
| H2O source: water vapor (no liquid phase) | ||||||
| TiO2/AC-Ag | 300 W Xe lamp (UV-vis) | Catalyst weight: 100 mg | H2O (electron donor) | CO: 6× higher than pristine TiO2 (relative yield) | AC enhances CO2 adsorption (12× higher than pristine TiO2) and electron transfer | 211 |
| Reactor: 100 mL quartz reactor | Separate reaction sites: H2O oxidation on TiO2, CO2 reduction on AC. | |||||
| CO2 pressure: ambient (continuous CO2 bubbling) | Ag SPR extends visible light absorption | |||||
| g-C3N4/WO3 | 300 W Xe lamp (simulated sunlight) | Liquid-phase (gas-closed reactor) | Triethanolamine (TEOA, 2 mL) | CO production rate: 23.0 μmol h−1 (2300 μmol h−1 g−1) | S-scheme charge transfer: e− from WO3 CB recombines with h+ from g-C3N4 VB. Retains high-potential e− (g-C3N4 CB) for CO2 reduction | 212 |
| Catalyst loading: 10 mg in 12 mL solvent (H2O/acetonitrile/TEOA = 2:8:2) |
CO selectivity: 90.6% (vs. H2 byproduct) | |||||
| CO2 pressure: 1 atm (high-purity CO2) | ||||||
| Key intermediates: CO2*− (1595 cm−1), COOH (1380/1628 cm−1), CO (2130 cm−1) via in situ DRIFTS. | ||||||
| Ag/TiO2-x nanoparticles-assembly | 300 W Xe lamp (simulated sunlight) | Catalyst weight: 50 mg | H2O (proton source) | CH4: 8.61 μmol g−1 h−1 | Synergy of oxygen vacancies (enhanced light absorption, charge separation) and Ag Schottky junctions (electron trapping) | 213 |
| Reactor: glass dish in sealed reactor | CO: 2.27 μmol g−1 h−1 (CH4 yield 18× higher than TiO2) | |||||
| CO2 pressure: atmospheric (1 atm) | ||||||
| α-Fe2O3/Cu2O | 300 W xenon arc lamp (λ > 400 nm) | 0.10 g of photocatalyst; 10 mL of deionized water; CO2 pressure, 0.3 MPa; stainless steel cylindrical reactor | N.A | CO: 1.67 μmol gcat−1 h−1 | The tailored heterojunction improved photocatalytic efficiency by optimizing charge pathways, demonstrating how strategic band alignment in oxide composites can amplify redox capabilities for solar fuel synthesis | 214 |
| ZnO–Cu2O | 300 W Xe lamp (UV-vis) | Catalyst weight: 19 mg | H2O (0.2 M Na2CO3, pH 7.4) | CH4: 1080 μmol g−1 h−1 | Z-scheme charge separation via ZnO–Cu2O band alignment | 215 |
| Reactor: 41 mL quartz flask | CO: 1.4 μmol (3 h) | Defect-free surfaces reduce recombination | ||||
| CO2 pressure: 2.6 bar (saturation), ambient (reaction) | Selectivity: >99% CH4 | High surface area (colloidal morphology) | ||||
| QE: 1.5% | ||||||
| PO43−–TiO2–Agx | 300 W Xe lamp (UV-vis) | Catalyst weight: 100 mg | H2O (Na2CO3 solution) | CH4: 3.36 μmol g−1 h−1 | PO43− enhances surface hydroxyls, Ti3+ sites, and CO2 adsorption | 216 |
| Reactor: liquid-phase (aqueous suspension) | CO: 0.69 μmol g−1 h−1 (CH4 yield 24× higher than TiO2) | Ag nanoparticles form Schottky junctions for charge separation and LSPR. | ||||
| CO2 pressure: ambient | Synergy of Ag and PO43− boosts charge transfer and surface reactivity | |||||
| Ag/CoOx-NTO (A/B) | 300 W Xe lamp (UV-vis) | Catalyst weight: ∼100 mg (inferred) | H2O (Na2CO3 solution) | CH4: 1.37 μmol g−1 h−1 (NTO-B) | Dual cocatalysts: Ag (electron traps via Schottky junctions) and CoOx (hole traps for H2O oxidation) | 217 |
| Reactor: liquid-phase (aqueous suspension) | CH4: 1.34 μmol g−1 h−1 (NTO-A) | CoOx supplies H+ for CO2 reduction, reducing competition with H2O | ||||
| CO2 pressure: Ambient (Na2CO3 solution) | (9–12× higher than pristine NTO) | Enhanced charge separation (4.68 s electron lifetime) | ||||
| Material: quartz | Moderate stability (activity decline due to catalyst loss) | |||||
| pg-C3N4/Ag–TiO2 | 300 W Xe lamp (UV-vis) | Catalyst weight: 50 mg | H2O (sacrificial agent) | CH4: 35.4 μmol g−1 h−1 | S-scheme heterojunction between pg-C3N4 (porous defective g-C3N4) and Ag–TiO2 enhances charge separation | 218 |
| Reactor: gas-phase (quartz reactor) | CO: 17.3 μmol g−1 h−1 | pg-C3N4 provides high surface area (59.67 m2 g−1) and nitrogen defects for CO2 adsorption | ||||
| CO2 pressure: ambient (purged CO2) | Total CO2 conversion: 52.7 μmol g−1 h−1 | Ag adjusts TiO2's work function, enabling SPR for visible light absorption and electron transfer | ||||
| AQE: 2.364% (at 420 nm) | Stable for 5 cycles with retained crystallinity | |||||
| Ag-cluster/TiO2 | Xenon lamp (PLS-SXE300, Beijing Perfectlight) with AM 1.5 G filter | Gas-phase reactor: PLR MFPR-I multifunctional Photochemical reactor (150 mL chamber) | No sacrificial agents or photosensitizers used | CH4 production rate: 25.25 μmol g−1 h−1 (vs. 0.72 μmol g−1 h−1 for pristine TiO2) | Ag–O hybridization enhances charge transfer and stabilizes intermediates (*COOH, *CHO, *CH3O) | 219 |
| Catalyst weight: 5 mg dispersed on quartz dish | Water vapor: 200 μL deionized water added to maintain humidity | CO production rate: 15.92 μmol g−1 h−1 (vs. 3.71 μmol g−1 h−1 for pristine TiO2) | In situ DRIFTS confirmed key intermediates (*CO, *CHO) and stronger *CHO adsorption on Ag-cluster/TiO2 | |||
| CO2 pressure: 105 kPa (99.999% purity) | Electron selectivity for CH4: 86% (vs. 44% for pristine TiO2) | DFT calculations: Ag clusters lower Gibbs free energy for *COOH formation (0.84 eV vs. 2.16 eV on TiO2) and favor *CO → *CHO over CO desorption | ||||
| Reactor material: quartz (culture dish), sealed chamber with ZnSe windows for in situ DRIFTS | ||||||
| BWO/NiMU2 | 300 W Xe lamp (PLS-SXE300D, full spectrum) | Gas–solid phase (stainless steel chamber with quartz window) | None (sacrificial agent-free) | • CO: 4493 μmol g−1 h−1 | S-scheme charge transfer: e− from BWO CB recombines with h+ from NiMU2 VB. Ni sites act as CO2 adsorption/activation centers | 220 |
| Catalyst loading: 10 mg on sample table + 1 mL H2O | • H2: 9191 μmol g−1 h−1 | Photothermal synergy: Light drives H2O splitting to H+. Heat (250 °C) enhances H+ spillover to Ni sites | ||||
| CO2 pressure: continuous flow (99.999% purity) | CO:H2 ratio: 1:1.6 to 1:2.2 (tunable) |
|||||
| Temperature: 250 °C (photothermal conditions) | Selectivity: >95% for syngas |
GO and rGO play pivotal roles in boosting the performance of MOx, with their distinct structural and electronic features enabling tailored functionalities for specific applications. GO is distinguished by its oxygen-rich functional groups, including hydroxyl, carboxyl, and epoxy moieties, which are anchored to its basal planes and edges.221–225 These groups widen its bandgap and reduce electrical conductivity compared to rGO, but they significantly enhance adsorption capacity by facilitating π–π interactions and provide reactive sites for redox reactions critical in pollutant degradation.226,227 Additionally, the oxygenated structure of GO broadens its light absorption spectrum, albeit with limited efficiency in the visible range. In contrast, rGO is derived from the chemical reduction of GO, a process that reduces oxygen-containing groups and restores a sp2-hybridized carbon network closer to pristine graphene. This structural modification drastically improves electrical conductivity and narrows the bandgap, endowing rGO with superior visible-light absorption and charge-carrier mobility.228 The mechanisms by which GO and rGO enhance activity are multifaceted. GO's functional groups not only stabilize MOx nanoparticles, preventing agglomeration, but also create diverse reaction pathways through surface-bound radicals and enhanced reactant adsorption. For instance, in MgO@GO composites, GO reduces the bandgap from 2.36 eV (MgO alone) to 1.71 eV, enabling visible-light harvesting and achieving 98% Rhodamine 6G degradation within 15 minutes.226 SEM-EDS and UV-vis analyses confirmed GO's role in improving photocurrent response and hydroxyl radical generation, attributed to its high surface area and electron–hole separation efficiency.226 Similarly, in CuO/Fe3O4/GO Z-scheme systems, GO prevented nanoparticle agglomeration, as evidenced by uniform dispersion in SEM/EDS, while its oxygenated sites promote ROS generation. This synergy improved tetracycline degradation to 97.3%, compared to 78.1% for CuO alone, with ˙O2− and h+ identified as dominant species via scavenger tests and EPR analysis.227 Conversely, rGO excels in applications requiring rapid charge transfer and minimized recombination. In BiOBr/rGO composites, rGO acted as an electron acceptor, reducing PL intensity by 49% and increasing photocurrent by 2.02× compared to pristine BiOBr.228 Its conductive network enhanced adsorption capacity, concentrating pollutants like RhB and TC near active sites, leading to 96% and 73% degradation efficiencies, respectively, under visible light. The dominant pathways, mediated by ˙O2− and h+, highlight rGO's ability to sustain redox cycles while mitigating charge recombination.228
Chitosan serves as a multifunctional scaffold for metal oxide photocatalysts, leveraging its physicochemical properties to enhance catalytic efficiency through synergistic mechanisms. Its amino (–NH2) and hydroxyl (–OH) groups play a central role in chelating metal ions (e.g., Ti4+, Zn2+) during synthesis, facilitating controlled nucleation and growth of nanoparticles (e.g., TiO2, ZnO) while preventing agglomeration.229,230 This ensures uniform dispersion and maximizes active surface area for redox reactions.231–235 During photocatalysis, chitosan mitigates electron–hole recombination by acting as a charge mediator: amino groups trap holes (h+), while hydroxyl groups transfer e− to adsorbed oxygen, generating superoxide radicals (O2˙−).231–233 Concurrently, its porous matrix enhances pollutant adsorption via electrostatic interactions (e.g., –NH3+ with anionic dyes) or hydrogen bonding, concentrating pollutants near catalytic sites to promote interfacial electron transfer and reactive oxygen species (ROS) generation (˙OH, O2˙−).231,234,235
The chitosan polymer's hydrophilic nature stabilizes hydroxyl ion (OH−) adsorption, favoring ˙OH radical formation through h+-mediated oxidation, while its structural integrity reduces metal oxide leaching and enhances durability via cross-linking (e.g., with glutaraldehyde).231–234,236 Spectroscopic analyses reveal that chitosan induces bandgap narrowing in metal oxides through surface complexation (e.g., Ti–O–C bonds in TiO2–chitosan composites) or defect-state formation, enabling visible-light absorption.231–235 For instance, in TiO2–chitosan hybrids, chitosan immobilized TiO2 nanoparticles on substrates, prevented aggregation, and concentrated methyl orange (MO) dye via electrostatic adsorption, achieving enhanced interfacial degradation.231 FTIR and SEM-EDX analyses confirmed the retention of functional groups and uniform TiO2 dispersion, while XRD revealed optimal crystallite sizes (4–18 nm) that balanced light absorption and surface reactivity.231 Similarly, in CuO/CS hybrids, chitosan reduced the recombination rate (as confirmed by photoluminescence (PL) data), narrowed the bandgap (as confirmed by DRS data), and enhanced RhB dye mineralization.
Chitosan's multifunctionality extends to complex composites. For instance, in AgZnFe2O4@CS nanocomposites, CS stabilized MOx nanoparticles, prevented aggregation, and facilitated pollutant adsorption via –NH2/–OH groups, achieving 81.5% and 82.3% degradation of metronidazole and penicillin G, respectively, under UV light.234 FTIR and SEM analyses corroborate chitosan's role in maintaining structural stability and promoting persulfate activation, with SO4˙− identified as the dominant radical.234 Further, in TiO2/CS-biochar composites, chitosan-derived biochar introduced Ti3+ and oxygen vacancies during calcination, narrowing TiO2's bandgap and acting as an electron transporter to reduce recombination, resulting in a 30-fold higher RhB degradation rate compared to pristine TiO2.234 XPS and PL analyses confirmed the enhanced charge separation and defect states.234
Further, chitosan enhanced hierarchical architectures such as TiO2/CNT/rectorite aerogels, forming a porous lamellar matrix that prevented TiO2 aggregation and adsorbed Rhodamine B (RhB) via functional groups. The optimized aerogel achieved 95% RhB degradation in 100 minutes, retaining 75% efficiency over three cycles due to structural stability (6). BET and XPS analyses highlighted its high surface area (84.59 m2 g−1) and TiO2 dispersion.237 Additionally, in Pt@CS/ZnTiO3 systems, chitosan's –NH2/–OH groups adsorbed pollutants (e.g., imidacloprid) via electrostatic interactions, while its porous matrix synergized with Pt nanoparticles to suppress charge recombination, reducing the bandgap to 2.71 eV (vs. ZnTiO3's 2.93 eV).235 PL and BET data revealed extended carrier lifetimes and a mesoporous structure (3.07 m2 g−1), enabling 75% methylene blue (MB) degradation retention over five cycles.235
Thus, chitosan transcended the role of passive support, actively participating in adsorption, charge separation, and ROS generation. Its tunable porosity, mechanical stability, and surface functionality enabled tailored integration with metal oxides, establishing it as a critical component in designing robust, solar-driven photocatalytic systems for environmental remediation.
3.3 Mechanistic insights into heterostructure performance
As discussed in the previous section, the photocatalytic performance of MOx-based composites is significantly enhanced through heterostructure engineering. This improvement arises from three key mechanisms: optimized charge carrier dynamics, enhanced light absorption efficiency, and amplified surface reactivity. By tailoring the interface between distinct materials, heterostructures mitigate electron–hole recombination, extend the spectral range of photon utilization, and increase active sites for redox reactions. Fig. 10 summarizes the profound influence of heterostructure formation on the photocatalytic activity of metal oxides, demonstrating how strategic material design can elevate their functional efficacy in environmental applications such as pollutant degradation and solar energy conversion. | ||
| Fig. 10 Impact of heterostructure formation on the photocatalytic performance of metal oxides. | ||
4. Photocatalytic application of metal oxide-based composites
4.1 Photocatalytic mineralization of pollutants
The rapid expansion of industrial activities has resulted in a substantial rise in wastewater laden with a wide array of persistent organic pollutants that pose serious risks to human health and environmental sustainability. Heterogeneous photocatalysis, employing metal oxide (MOx) nanomaterials, offers efficient degradation and mineralization pathways of these pollutants. For example, nickel oxide with its favorable band gap and electronic properties, exhibits potential for enhanced photocatalytic activity under UV and visible light irradiation. The generated ROS are the primary agents responsible for the oxidative degradation of organic pollutants. The degradation process involves a complex sequence of reactions, such as hydroxylation, dehydrogenation, and bond cleavage, ultimately leading to the transformation of complex organic molecules into simpler intermediates, resulting eventually in complete mineralization to CO2, H2O, and inorganic ions. For instance, a ternary NiO/ZnO/g-C3N4 composite exhibited substantially promoted azo dye degradation compared to its binary combinations and individual constituents.103 The enhanced activity, achieving near-complete mineralization of Congo red and methylene blue, is attributed to synergistic impacts including powerful interfacial charge transfer and the role of g-C3N4 in promoting light harvesting, ROS generation, and charge carrier separation. Similarly, ZnO/NiO/g-C3N4 photocatalysts, optimized for TC mineralization, exhibited 91.49% removal efficiency within 60 minutes. The mineralization process followed PFO kinetics, with an observed rate constant of 0.05356 min−1.238 Radical scavenging tests reflected that ˙OH and ˙O2− were the dominant reactive oxygen species involved in mineralization of TC. Additionally, an alginate–ZnO–polypyrrole (PPy) hybrid demonstrated effective removal of acid blue 92 (AB92) from aqueous solutions under both UV and visible light irradiation.239 The noticed robust removal performance is attributed to the enhanced PPy dye-adsorption and the hindered electron–hole recombination within the ZnO–PPy system, as mechanistically shown in Fig. 11a and b. Furthermore, the Pd@NSC-WO3 composite leveraged a dual-function mechanism to efficiently degrade phenolic pollutants (PPs) (1). Under UV light, the engineered heterojunction between Pd and N,S-doped WO3 facilitated exceptional charge separation, where Pd acted as an electron reservoir, prolonging the lifetime of photoexcited electrons, while the doped WO3 matrix optimized hole mobility (Fig. 11c–j). This synergy amplified radical generation (˙OH and ˙O2−), which selectively attack aromatic rings via various pathways, depending on the pollutant's substituents (e.g., C4/C5 cleavage in catechol). Crucially, the composite's nanoscale architecture (5.1 nm particles) and surface defects enhanced adsorption of PPs and intermediate aliphatic acids, ensuring rapid mineralization to CO2/H2O. Moreover, Higher catalyst doses enhanced active sites availability, accelerating ROS generation, though excessive loading may hinder light penetration (Fig. 11c). Lower initial PP concentrations favored faster degradation due to reduced competition for reactive species, while acidic pH optimized WO3's surface charge, promoting adsorption of anionic intermediates and stabilizing radicals (Fig. 11d). However, neutral to mild alkaline conditions may enhance hydroxyl radical production via OH− oxidation. Furthermore, light intensity directly dictates charge carrier generation, where stronger UV irradiation intensifies electron–hole pair formation, amplifying radical yields and degradation rates.240 Further, the bifunctional Rh/WO3 catalyst integrated hydrogenation and photocatalytic oxidation to overcome inherent limitations of standalone processes.240 Thus, a bifunctional Rh/WO3 catalyst was synthesized via a liquid-phase reduction method.241 TEM and HRTEM analysis (Fig. 11k–m) confirmed the uniform dispersion of Rh nanoparticles (5–10 nm) on WO3 nanosheets and revealed distinct lattice fringes (Rh(111): 0.21 nm; WO3(002): 0.38 nm), directly visualizing a Schottky heterojunction interface. HAADF-STEM and elemental mapping further validated the atomic-scale integration of Rh with WO3, where Rh acted as an electron sink, creating built-in electric fields at the interface to drive charge separation, which is crucial for synergistic hydrogenation and photocatalytic oxidation activity. Additionally, a CeO2/BiYO3 hybrid was developed for the photocatalytic mineralization of tetracycline (TC).242 This material exhibited substantially improved photocatalytic role compared to its individual precursors, BiYO3 and CeO2, highlighting its promise as an effective photocatalyst. Analysis of band potentials revealed a Type II heterojunction, facilitating efficient charge migration. Moreover, the Ce3+/Ce4+ redox couple served as an electron trap, further boosting the heterojunction role by minimizing electron–hole recombination. While peak catalytic activity was reported only during the initial cycle, a simple annealing treatment fully restored the catalyst's effectiveness, suggesting excellent reusability of the CeO2/BiYO3 hybrid. | ||
| Fig. 11 (a and b) Possible photocatalytic degradation and adsorption mechanisms of acid blue 92 (AB92) dye removal by polypyrrole–zinc oxide–sodium alginate (PPy–ZnO–SA) nanocomposite, reprinted with the permission of ref. 239, copyright 2025, Elsevier; (c and d) effects of Pd2@NSC-WO3 catalyst dose and hydroquinone (HQ) initial concentration on the photocatalytic degradation efficiency of HQ; (e and f) effects of pH and light intensity on the rate constant of HQ degradation; (g) proposed photocatalytic process for efficient photo-oxidation of phenolic pollutants (PPs) using the Pd@NSC-WO3 under UV light irradiation and (h–j) the plausible photodegradation pathways for PPs, reprinted with the permission of ref. 240, copyright 2025, Elsevier; (k–m) TEM and HRTEM of 1 wt% Rh/WO3 heterostructure photocatalyst, reprinted with the permission of ref. 241, copyright 2025, Elsevier. | ||
4.2 Hydrogen production
Photocatalytic water splitting offers a direct route to converting solar energy into chemical energy in the form of hydrogen fuel. This process mimics natural photosynthesis but uses a semiconducting photocatalyst material instead of chlorophyll. The process begins when the photocatalyst absorbs photons of light with sufficient energy to excite electrons from the VB to the CB, creating electron–hole pairs. These charge carriers then migrate to the surface of the photocatalyst, where they participate in redox reactions that split water molecules. Crucially, the photocatalyst's band gap and electronic structure must be carefully tuned to straddle the water redox potentials, ensuring that the excited electrons have enough energy to reduce water to hydrogen and the holes can oxidize water to oxygen species. Efficient charge separation is also essential, as recombination of electron–hole pairs diminishes the overall efficiency of the process. At the photocatalyst surface, two distinct half-reactions occur simultaneously. The photogenerated electrons in the conduction band react with water molecules (or protons in acidic conditions) to produce hydrogen gas. This reduction reaction involves the transfer of two electrons to two protons, forming a hydrogen molecule (2H+ + 2e− → H2). Concurrently, the holes in the valence band drive the oxidation of water molecules, generating oxygen gas and protons. This four-electron oxidation reaction (2H2O + 4h+ → O2 + 4H+) is more complex and often kinetically slower than the hydrogen evolution reaction, representing a bottleneck in the overall water splitting process. The efficient extraction of these protons and electrons to the respective reaction sites on the photocatalyst surface is crucial for maximizing the hydrogen production rate. MOx-based composites have emerged as a significant research focus in the field of hydrogen production, owing to their tunable electronic structure and diverse morphologies that can be achieved through strategic synthetic design. For instance, a TiO2/NiS core–shell structure was fabricated employing hydrothermal and electrospinning strategies, yielding 655 μmol (g h)−1 of hydrogen.243 The vertically aligned NiS nanoplates promoted charge migration performance. Furthermore, a hierarchical ZnO/CdS Z-scheme with a microsphere morphology was created.244 The optimal CdS loading (30.9%) achieved a hydrogen yield rate of 4134 μmol (g h)−1, attributed to improved charge separation and visible light harvesting.244 Similarly, a 15 wt% W18O49/CeO2 hybrid exhibited a 1.93-fold increase in hydrogen generation over pristine CeO2, resulting from Z-scheme creation.245 Furthermore, a WO3/TiO2/rGO S-scheme heterostructure was developed and achieved a substantially robust hydrogen yield of 245.8 μmol (g h)−1, 3.5 times that of pristine TiO2.246 This improvement stemmed from robust TiO2–WO3 interactions and a Schottky junction between rGO and TiO2, which facilitated charge migration and separation. Moreover, a hydrothermally fabricated TiO2 nanosheets on graphene demonstrated a robust hydrogen evolution rate of 736 μmol (h g)−1 and a 3.1% quantum efficiency under UV irradiation, with graphene acting as an electron acceptor to enhance charge separation.247 Additionally, a GO/CdS/ZnO ternary hybrid achieved a hydrogen evolution rate of 6.511 mmol (g h)−1, exceeding the performance of its binary counterparts owing to a narrower bandgap, high surface area, and suppressed electron–hole recombination.248 Similarly, an ultrasonically synthesized porous WO3/rGO hybrid249 exhibited promoted hydrogen evolution (640.5 μmol (g h)−1) compared to pristine WO3, owing to improved charge diffusion.4.3 Carbon dioxide reduction
The photocatalytic reduction of carbon dioxide (CO2) using metal oxide-based composites represents a transformative approach to mitigating climate change while producing value-added chemicals. This process hinges on the interplay of light-driven charge carriers, surface chemistry, and reaction engineering to overcome the thermodynamic stability of CO2 (ΔG = −394 kJ mol−1) and reconfigure its molecular structure.250 At its core, CO2 reduction proceeds through a cascade of proton-coupled electron transfers. Initial steps involve photogenerated electrons (from the composite's conduction band) and protons (derived from water oxidation) destabilizing the linear CO2 molecule. The first critical step is a one-electron reduction of CO2 to form a bent, activated CO2˙− radical intermediate (*CO2−), followed by C–O bond cleavage and subsequent C–H bond formation.214,251 However, the single-electron reduction pathway is thermodynamically prohibitive due to the highly negative redox potential of CO2/CO2˙− (−1.90 V vs. NHE). To circumvent this barrier, proton-coupled multielectron reduction pathways are favored, as they operate at lower redox potentials, enabling feasible activation under ambient or mild conditions (25–100 °C, 1 atm) with UV/visible light irradiation in aqueous or gas-phase reactors (Fig. 12a).252 The practical significance of this process lies in its dual role in sustainability and energy innovation. The reduction pathways are highly dependent on the number of electrons transferred. For example, a 2-electron process yields carbon monoxide (CO) or formic acid (HCOOH), while 6–8 electrons are required to generate methane (CH4) or methanol (CH3OH). These products are dictated by the catalyst's ability to stabilize critical intermediates such as *CO2−, *COOH, or *CH3O. By converting CO2, a major greenhouse gas, into fuels (e.g., CH4, CH3OH) or industrial feedstocks (e.g., CO for syngas), it closes the carbon cycle and reduces reliance on fossil fuels. For instance, the researchers electrochemically deposited Ag nanoparticles within TiO2 nanotubes, enhancing SPR-driven CO2 reduction via amplified light scattering and “hot electron” injection into TiO2 (as evidenced by synchronous illumination X-ray photoelectron spectroscopy, SIXPS).253 Plasmonic near-field effects reduced charge recombination, yielding 2.3× higher CH4 production than surface-loaded Ag. This highlights spatial control of plasmonic metals in oxide scaffolds to synergize light harvesting and charge dynamics for efficient solar fuel synthesis. Further, the researchers developed a novel metal-covalent organic framework heterojunction (WO3/THFB-COF-Zn) for efficient CO2 photoreduction.210 The THFB-COF-Zn framework was synthesized via a solvothermal method, by condensing 1,3,5-triazine-2,4,6-tris(4′-hydroxy-5′-formylphenyl)benzene (THFB), ethylene diamine and Zn(ClO4)2·6H2O, whereby the product exhibited enhanced porosity (∼20 Å pore size), and stability in harsh conditions. By integrating WO3 nanoparticles, an S-scheme heterojunction was formed and exhibited enhanced charge separation, where electrons move from WO3 to THFB-COF-Zn, while holes remain in WO3. This design boosted photocatalytic activity, achieving a CO production rate of 54.1 μmol g−1 h−1 (7× higher than pure THFB-COF-Zn) with nearly 100% selectivity for CO. Key intermediates like *COOH were detected via in situ diffuse reflectance Infrared Fourier Transform spectroscopy (DRIFTS), confirming the reaction pathway. The study demonstrates how S-scheme heterojunctions improve CO2 conversion efficiency by optimizing charge transfer and light absorption.210 In another study, TiO2 nanosheets anchored on activated carbon (AC) with Ag nanoparticles (TiO2/AC-Ag) enhanced photocatalytic CO2 reduction.211 Structural analysis confirmed anatase-phase stability, while Ag's SPR effect broadened visible-light absorption and reduced charge recombination (as confirmed by quenched PL intensity). AC's high surface area (427 m2 g−1) boosted CO2 adsorption (12× higher than pristine TiO2), and its conductivity facilitated electron–hole separation. Thus, the ternary composite achieved 6× higher CO production than TiO2 alone, underscoring synergies between SPR, conductive supports, and adsorption capacity for efficient CO2 conversion. Further, engineered α-Fe2O3/Cu2O composites enhanced CO2 photoreduction by leveraging interfacial charge dynamics.214 An internal electric field at the α-Fe2O3/Cu2O junction directed photogenerated electrons from Fe2O3's conduction band to Cu2O's valence band, effectively separating charges. This mechanism retained high-energy electrons in Fe2O3 for CO2 reduction while holes in Cu2O drove water oxidation to ˙OH and O2. The tailored heterojunction improved photocatalytic efficiency by optimizing charge production and separation pathways, demonstrating how strategic band alignment in oxide composites can amplify redox capabilities for solar fuel synthesis. The g-C3N4/WO3 heterostructure, synthesized via a two-step process involving calcination, demonstrated enhanced photocatalytic CO2 reduction using a H2O/acetonitrile/TEOA system under a 300 W xenon lamp. Characterization (XRD, FT-IR, XPS, BET) revealed high crystallinity, meso-porosity (179 m2 per g surface area), and effective CO2 adsorption (23 cm3 per g at 273 K) (16). The composite achieved a CO production rate of 23.0 μmol h−1 (90.6% selectivity) with stability over four cycles, outperforming individual g-C3N4 and WO3 due to an S-scheme charge transfer mechanism that improved electron–hole separation. Isotope (13CO2) and in situ DRIFTS analyses confirmed CO2 as the carbon source and identified key intermediates (COOH*, CO2*−), validating the catalytic pathway. This work underscores the potential of crystalline carbon nitride-based heterostructures for efficient solar-driven CO2 conversion (16). Furthermore, hierarchically porous Ag/TiO2 with Ti3+ defects and oxygen vacancies demonstrated enhanced CO2 photoreduction to CO under UV-vis light.209 Ag quantum dots (≤3 nm) improved visible absorption via SPR and facilitated charge separation, while the obtained mesoporous structure with high SSA (68 m2 g−1) and defects boosted CO2 adsorption. The composite achieved 2.3 μmol per g CO yield in 4 h, which is 1.5× higher than pristine TiO2, attributed to synergistic electron transfer (Ag → TiO2) and defect-mediated activation. Stability tests confirmed minimal Ag leaching, underscoring the role of structural and electronic optimization in sustainable solar fuel synthesis. Further, the synergistic integration of oxygen vacancies and Ag into potholed TiO2 nanoparticles via solvothermal treatment significantly enhanced photocatalytic CO2 reduction by addressing critical bottlenecks in charge dynamics and surface reactivity (Fig. 12b).213 Oxygen vacancies narrowed the bandgap (2.7 eV vs. 3.1 eV for pristine TiO2), extending visible-light absorption and acting as electron traps to suppress recombination (as evidenced by 3× higher CH4 yield for TiO2-x over pure TiO2). Ag nanoparticles further amplified charge separation through Schottky junctions, funneling electrons to Ag's SPR-induced “hot spots” and creating electron-rich active sites. The optimized Ag/TiO2-x-10% composite achieved CH4 and CO yields of 8.61 and 2.27 μmol g−1 h−1, which are 18.3× and 32× higher than TiO2, respectively, due to dual pathways: (1) oxygen vacancies promoting CO2 adsorption/H2O dissociation for proton supply and (2) Ag-driven SPR and Schottky effects enhancing electron density.213 Additionally, PO43−-modified TiO2 nanorods loaded with Ag NPs (PO43−–TiO2–Agx) was synthesized via a hydrothermal method, where Ag enhanced visible-light absorption via plasmonic effects, while PO43− introduced surface hydroxyl groups and Ti3+ sites to improve CO2 adsorption and charge separation.216 The optimized PO43−–TiO2–Ag4 catalyst achieved a CH4 production rate of 3.36 μmol g−1 h−1, which is 24 times higher than unmodified TiO2, due to synergistic effects, where Ag acted as an electron sink, reducing recombination, and PO43− provided Lewis basic sites for CO2 activation. In situ FTIR revealed *HCOO− and *OCH3 as key intermediates in the CO2 reduction to CH4 pathway, while stability tests confirmed robustness under vacuum reactivation. The approach was further extended to other TiO2 phases (e.g., brookite), demonstrating broad applicability for photocatalytic CO2 reduction (Fig. 12b).216 Another study developed Ag/CoOx dual-cocatalyst-modified TiO2@Na2Ti3O7 nanoassemblies (NTO).217 The Ag/CoOx synergy enhanced charge separation, where Ag formed Schottky junctions to trap electrons, while CoOx acted as hole sinks for H2O oxidation, providing H+ for CO2 reduction. UV-DRS spectra revealed reduced bandgaps (3.05 → 2.8 eV), and broadened light absorption. Photoelectrochemical tests (EIS, transient photocurrent) confirmed prolonged charge lifetimes (4.68 vs. 2.37 s) and lower charge-transfer resistance. Further, an S-scheme heterojunction of Ag-doped TiO2 NPs anchored on porous, N-deficient g-C3N4 (pg-C3N4) was developed.218 The freeze-drying synthesized pg-C3N4 introduced strategically engineered nitrogen vacancies that dramatically improved CO2 adsorption while maintaining structural stability, overcoming a key limitation in defect-engineered photocatalysts. DFT calculations revealed that Ag-doping critically modulated the work function difference between the composite components, creating an interfacial electric field that directed charge separation while preserving strong redox potentials. Another work demonstrated a highly efficient Ag-cluster-modified TiO2 photocatalyst (Ag-cluster/TiO2) for selective CO2 reduction to CH4, achieving a remarkable yield of 25.25 μmol g−1 h−1 with 86% electron selectivity.219 The 1 nm-sized Ag clusters, uniformly dispersed onto TiO2 microspheres (as confirmed by AC-HAADF-STEM, aberration corrected high-angle annular dark-field scanning transmission electron microscopy), created strong metal–support interactions that significantly enhanced charge separation (25.01 ns carrier lifetime vs. 20.48 ns for pure TiO2) and CO2 adsorption capacity (317 °C desorption peak in CO2-temperature-programmed desorption). In situ DRIFTS and DFT calculations revealed that interfacial Ag–O hybridization promoted key intermediate (*CHO) formation while suppressing CO desorption, steering the reaction pathway toward CH4 production. Furthermore, a ternary Ag–CeO2–ZnO hybrid demonstrated enhanced CO2 reduction via a Z-scheme charge transfer mechanism.153 Ag absorbed visible light, generating hot electrons transferred to ZnO/CeO2, with suppressed PL emission and higher photocurrent (Fig. 12c) that indicated reduced charge recombination and improved carrier separation. EPR spectra confirmed strong ˙OH and ˙O2− radical generation (Fig. 12d and e), which are crucial for redox reactions. The composite achieved CO and CH4 production yields of 75.4 and 4 μmol g−1 h−1, respectively, with 4.47% quantum efficiency at 420 nm, outperforming individual components due to synergistic interfacial electron dynamics and efficient light utilization. Stability tests showed consistent performance over multiple cycles.153 Furthermore, a Z-scheme Cu2O–Pt/SiC/IrOx heterojunction was synthesized via sequential photodeposition of Pt NPs (2–4 nm) on SiC, followed by Cu2O and IrOx, as shown in TEM and HTEM images of Fig. 12f and g.254 The band alignment between IrOx, SiC, and Cu2O enabled efficient interfacial charge separation, yielding higher photocurrent density compared to Pt/SiC or Cu2O–Pt/SiC composites. This enhanced charge dynamics drove CO2 photoreduction to HCOOH at 61.5 μmol g−1 h−1, with Pt acting as a cocatalyst to facilitate electron transfer. The Z-scheme design minimized recombination, optimizing redox capability for solar fuel production (Fig. 12h). Additionally, an S-scheme BWO/NiMU2 heterojunction, integrating Bi2WO6 (BWO) with ultrathin Ni-anchored polymeric carbon nitride (Ni-PCN), demonstrated excellent photothermal CO2 reduction to syngas (CO + H2) at 250 °C under full-spectrum light.220 The heterojunction combined ultrathin PCN nanosheets (2.15 nm, 137.8 m2 per g surface area) with atomically dispersed Ni sites, enhancing CO2 adsorption (87.45 μmol g−1) and charge separation via an S-scheme mechanism. This design achieved CO and H2 production rates of 4493 and 9191 μmol g−1 h−1, respectively, with >95% syngas selectivity and stability over 12 hours. Key innovations included Ni single sites (as confirmed by HAADF-STEM data) acting as CO2 activation centers, improving electron density (as supported by XPS/DFT data) and reducing charge-transfer resistance (as confirmed by Nyquist plots). EPR/UV-vis/Tauc plots supported the S-scheme charge transfer suppressed recombination, retaining high redox potentials for CO2 reduction (*COOH intermediates via in situ FT-IR) and H2O oxidation. Photothermal synergy (light-driven H+ generation and thermal H+ diffusion) boosted reaction kinetics, enabling 21× higher H2 yield than thermal catalysis alone.220 | ||
| Fig. 12 (a) Proposed mechanism of CO2 conversion over a MOx-based photocatalyst, reprinted with the permission of ref. 252, copyright 2025, Elsevier; (b) Schematic diagram of photocatalytic CO2 reduction on PO43−–TiO2–Ag4, reprinted with the permission of ref. 214, copyright 2025, Elsevier; (c) photocurrent measurements over samples of ZnO, CeO2, and 3AgCZ heterostructure; (d and e) EPR spectra of CeO2, ZnO, and sample 3AgCZ, (d) DMPO–˙OH and (e) DMPO–˙O2−, reprinted with the permission of ref. 153 copyright 2025, Elsevier; (f and g) TEM and HRTEM images of Cu2O–Pt/SiC/IrOx; and (h) the electron transfer processes in Cu2O–Pt/SiC/IrOx under light illumination, reprinted with the permission of ref. 254, copyright 2025, Elsevier. | ||
In summary, metal oxide composites are critical for photocatalytic CO2 reduction, overcoming challenges like rapid charge recombination and limited light absorption through heterostructures, doping, oxygen vacancy engineering, and integration with conductive layered materials (e.g., graphene, MOFs). Future efforts should focus on combinatorial synthesis guided by DFT and machine learning to optimize band structures, defect densities, and interfacial charge dynamics. Integrating photothermal effects could further enhance kinetics. Moreover, operando characterization (e.g., in situ XAS, transient absorption spectroscopy) will deepen mechanistic understanding of intermediate stabilization and charge transfer pathways. Selectivity may be controlled through surface modifications, co-catalyst tuning, and adjusting reaction parameters (pH, light intensity and range).
4.4 Hydrogen peroxide production
Hydrogen peroxide (H2O2) is a critical chemical with multifaceted applications, ranging from disinfection and environmental remediation to energy storage and green synthesis. Its appeal lies in its potent oxidative capability, eco-friendly decomposition into water and oxygen, and versatility across various sectors.255 Several strategies are applied to the production of hydrogen peroxide, such as the anthraquinone (Riedl–Pfleiderer) process, electrolytic processes, byproduct recovery, methane oxidation, etc. However, conventional production methods, such as the anthraquinone process, are plagued by high energy demands, toxic intermediates, and complex infrastructure, driving the need for sustainable alternatives.256 Photocatalytic H2O2 generation using metal oxide composites has emerged as a promising solution, leveraging solar energy, water, and oxygen to enable decentralized and low-cost synthesis. In photocatalytic systems, H2O2 is primarily generated through two competing pathways: the oxygen reduction reaction (ORR) and the water oxidation reaction (WOR) (Fig. 13a).256 The efficiency of these processes is further dictated by operational conditions.255 Neutral or mildly acidic pH environments are optimal to prevent H2O2 decomposition, while dissolved oxygen concentration directly impacts ORR kinetics. Light intensity and wavelength also play dual roles: while greater irradiance enhances charge carrier generation, it may accelerate H2O2 photodegradation. A significant hurdle in scaling photocatalytic H2O2 production is the reliance on sacrificial agents, such as ethanol or EDTA, to scavenge holes and mitigate recombination. While these agents improve yields, they introduce economic and environmental costs, undermining the sustainability of the process. Recent efforts focus on developing self-sufficient metal oxide systems (e.g., BiVO4 or α-Fe2O3) that balance WOR and ORR kinetics without additives. For instance, the strategic interplay of electronic structure and surface hydrophobicity was validated through multi-modal characterization.257 As depicted in Fig. 13b and c, the staggered band alignment between poly(9,9-dioctylfluorene-alt-benzothiadiazole) (PFBT, CB: −3.55 eV, VB: −5.9 eV) and TiO2 (CB: −4.2 eV, VB: +2.7 eV) creates a directional charge cascade: electrons migrate from PFBT's LUMO to TiO2's CB for O2 reduction (2e− pathway), while holes shift to PFBT's HOMO, enabling water oxidation without sacrificial agents. This mechanism is amplified by the polymer's high dipole moments, which enhance charge separation efficiency by 68% compared to bare TiO2. The heterojunction's performance is quantified in Fig. 13d, where PFBT/TiO2 achieves 67 μM H2O2 under visible light (>420 nm), clearly contrasting with negligible yields from pristine components. Critically, in the difluorinated derivative, PF2FBT (poly(9,9-dioctylfluorine-alt-difluorobenzothiadiazole)), the two fluorine atoms induced hydrophobicity (contact angle: 100.6°, Fig. 13e) and reduced H2O2 adsorption on TiO2 by 82%, slashing decomposition rates from 0.14 min−1 (bare TiO2) to 0.014 min−1.257 Another study demonstrated how precise oxygen vacancy (OV) modulation in WO3−x amplifies the built-in electric field (BEF) in an S-scheme heterojunction, enabling simultaneous CO and H2O2 production from CO2 and water (Fig. 1f). The controlled OV introduction narrowed bandgaps (2.75 → 2.48 eV) and shifted Fermi levels, with n-WO3−x achieving optimal alignment for BEF maximization. DFT calculations revealed that OVs introduced W-5d defect states, enhancing visible-light absorption and charge separation. The NH2-MIL-125(Ti)(NM)/n-WO3−x heterojunction forms an intimate interface where BEF-driven S-scheme charge transfer (Fig. 13f) directs electrons from WO3−x's CB to NM's LUMO for CO2 reduction (12.57 μmol per g per h CO) while holes oxidize water to H2O2 (8.41 μmol g−1 h−1). In situ XPS confirms light-induced electron redistribution, suppressing recombination by 80% and lowering overpotential.258 Further study unveils how Gd3+ doping in WO3 (Gd-WO3) strategically enhances both photocatalytic oxidation and H2O2 production by engineering electronic and surface properties.259 The Gd3+ substitutes W sites, introducing mid-gap states that extend light absorption to the near-infrared (1100 nm) and narrow the bandgap (2.30 eV vs. 2.42 eV for WO3). This doping elevates photocurrent sixfold by leveraging Gd's 4f half-filled orbitals, which act as electron reservoirs to accelerate charge separation. Crucially, DFT calculations (Fig. 13g–j) reveal Gd weakens *OOH adsorption (ΔG*OOH = 4.49 eV vs. 3.73 eV for WO3), favoring a direct 2e− O2-reduction pathway for H2O2 (0.58 mmol L−1 g−1 h−1) over the 1e− route. Furthermore, in constructing a 2D/1D heterostructure, ultrathin BiOBr nanosheets intimately interface with WO3 nanorods, forming coherent junctions that minimize charge recombination.260 Advanced characterization (XPS, ESR) reveals that interfacial oxygen vacancies (OVs) act as electron reservoirs, facilitating O2 adsorption and activation while inducing localized band bending to optimize charge dynamics. Crucially, the Z-scheme charge transfer mechanism preserves the strong redox potentials of both components: electrons in BiOBr's conduction band drive O2 reduction via a two-electron pathway, while holes in WO3's valence band oxidize H2O to ˙OH radicals, enabling dual-channel H2O2 synthesis. This structural and electronic synergy, combined with extended UV-vis DRS light absorption, yields an exceptional H2O2 production rate (8.24 mmol L−1 in 2 h), outperforming individual components and physical mixtures (6). Another work integrates CuO/Cu(OH)2 nanosheets with g-C3N4 to harness dual redox pathways: reductive (O2 → O2˙− → H2O2 via g-C3N4 electrons) and oxidative (H2O → ˙OH → H2O2 via Cu(OH)2 holes). Validated by ESR and PL quenching, a Z-scheme charge transfer minimizes recombination, while Cu(OH)2's hydroxyl-rich surface amplifies oxidative radical yields. Crucially, the hierarchical hollow structure boosts light absorption and charge mobility (EIS/photocurrent), achieving a record 1354 μmol per g per h H2O2, which is 13.6× higher than CuO alone. Moreover, it exhibited remarkable stability (8 h operation, 0396 mg per L Cu leaching that is still within the specified emission standard for metal ions (1 mg L−1, GB 25467-2010)), this approach bypasses noble metals and single-path limitations.261 Furthermore, S-scheme NiO/C3N5 heterojunctions (NOCN) were developed through interfacial anchoring of black NiO onto nitrogen-rich g-C3N5 nanosheets (CNNS), achieving dual photocatalytic H2 (112.2 μmol g−1 h−1) and H2O2 (91.2 μmol L−1 h−1) production under visible light.145 TEM/HRTEM analysis revealed NiO NPs (∼10–50 nm) uniformly dispersed on ultrathin CNNS, with lattice fringes of 0.24 nm (NiO (111)) and amorphous CNNS regions, confirming coherent interfacial contact critical for charge transfer, as shown in Fig. 13k–p.145 The S-scheme mechanism directs electrons from CNNS to recombine with NiO holes, preserving high-potential electrons (NiO CB: −0.88 V) and holes (CNNS VB: +1.44 V) for robust redox reactions. NiO's broad light absorption induces a photothermal effect (76 °C under irradiation), accelerating charge. | ||
| Fig. 13 (a) Schematic illustration of the three fundamental processes in photocatalytic H2O2 production, reprinted with the permission of ref. 256, copyright 2025, Elsevier; (b–e) possible transfer over the organic–inorganic heterojunction following along the band alignment charge transfer pathway I (b), and interfacial charge transfer pathway II (c); (d) photocatalytic generation of H2O2 of bare TiO2, pristine PFBT, and PFBT/TiO2 heterostructure without the inclusion of holes scavengers under visible light of 420 nm, 100 mW cm−2; (e) the H2O2 decomposition over bare TiO2 and PFBT/TiO2 heterostructure under UV-B light of 350 nm, 100 mW cm−2, reprinted with the permission of ref. 257, copyright 2025, Elsevier; (f) photocatalytic formation of CO and H2O2 over NM/n-WO3−x S-scheme heterojunction, reprinted with the permission of ref. 258, copyright 2025, Elsevier; (g) ΔG*OOH comparison for oxygen reduction on WO3 vs. Gd–WO3; (h) 2e− pathway free energy diagram for H2O2 generation; (i) differential charge densities after *OOH adsorption (yellow/blue: e− – gain/loss); (j) schematic of the 2e− pathway for H2O2 generation on Gd–WO3, reprinted with the permission of ref. 259, copyright 2025, Elsevier; (k–p) TEM and HRTEM images of (k and l) CNNS, (m and n) black NiO NPs and (o and p) 12-NOCN heterostructure, reprinted with the permission of ref. 145, copyright 2025, Elsevier. | ||
5. Cost analysis
The economic viability of modified MOx composite photocatalysts demands a rigorous and quantitative cost analysis, extending beyond rudimentary material expenses to encompass the entire life cycle. This analysis must be intricately interwoven with quantifiable performance metrics to justify the economic benefits of enhanced photocatalytic activity. While heterojunction construction, exemplified by TiO2 coupled with WO3 or CdS, demonstrably improves charge separation and thus photocatalytic efficiency, the associated cost implications necessitate careful and quantitative scrutiny. Utilizing more economical metal oxides like WO3 (approximately ∼20–30 $ per kg), Fe2O3 (∼5–10 $ per kg), or CuO (∼10–20 $ per kg) presents a less significant cost increase compared to sulfides like CdS (approximately ∼50–100 $ per kg) or more complex oxides like BiVO4 (∼50–150 $ per kg). However, the true economic impact is best evaluated by normalizing the cost to the active surface area ($ per m2), considering the material density and specific surface area, and correlating it with quantifiable performance enhancements. These enhancements, including improved charge carrier lifetimes (quantified in picoseconds to nanoseconds via transient absorption spectroscopy or time-resolved photoluminescence), increased photocatalytic reaction rates (expressed as mol s−1 m−2 or a defined kinetic rate constant), and enhanced quantum efficiencies (apparent quantum yield or formal quantum efficiency, ideally measured across a relevant wavelength range), should be directly compared against the incremental cost per unit area. Such a data-driven approach enables a robust cost-benefit assessment and facilitates direct comparison between different material combinations. Moreover, MOx incorporating non-metal dopants offers a potential avenue for cost-effective performance enhancement. Nitrogen doping, achievable through a simple annealing process in ammonia or nitrogen gas, incurs minimal cost increases (<1%) and can significantly modify the electronic band structure, leading to enhanced visible light absorption. Similarly, carbon doping, utilizing readily available precursors like carbohydrates, can introduce defect sites and improve charge separation. Quantifying the impact of these non-metal dopants on band gap energy (via DRS), charge carrier dynamics, and ultimately, photocatalytic activity, is crucial for evaluating their cost-effectiveness. Comparing the cost per unit enhancement (e.g., $ per % increase in quantum yield) for non-metal doping against the cost associated with metal doping or heterojunction formation provides a valuable metric for material selection. Moreover, incorporating co-catalysts, such as earth-abundant metal sulfides like MoS2 or NiS, presents an alternative to noble metals like platinum (Pt, approximately ∼30000 $ per kg) or gold (Au, significantly higher cost). While these earth-abundant co-catalysts might require optimized loading and careful synthesis to achieve comparable performance enhancements, their significantly lower cost makes them attractive candidates for large-scale applications. Quantifying their impact on hydrogen evolution rates (in photocatalytic water splitting), pollutant degradation kinetics, or other relevant performance metrics, alongside a detailed cost breakdown of their synthesis and integration, is essential for evaluating their economic viability. The integration of carbon materials further complicates the cost landscape. High-quality graphene and carbon nanotubes (CNTs), priced between ∼100 and ∼1000 $ per g, significantly impact overall material cost, while activated carbon, at ∼1 $ per kg, provides a more economically viable alternative. Quantifying the performance benefits conferred by these carbon materials, such as increased surface area, enhanced charge transfer efficiency (measured through EIS), and improved photocatalytic activity, against their respective costs and loadings is crucial. Optimizing the loading of expensive materials like graphene and rigorously assessing their long-term stability, including potential leaching under real operational conditions, is paramount for ensuring economic viability.
Synthesis technique critically dictates cost and performance of modified metal oxide photocatalysts. Precise, but expensive, techniques like atomic layer deposition (ALD) and chemical vapor deposition (CVD), >0.10 $ per cm2 offer superior control over film properties and heterostructures but face scalability challenges. Conversely, solution-based methods such as coprecipitation, or sol gel approach (<$0.01 per cm2) offer cost advantages but compromise precise morphological control. Quantifying energy consumption and comparing structural properties (surface area, size distribution, crystallinity) across methods is crucial for optimizing cost-performance trade-offs. Emerging methods like microwave and sonochemical syntheses warrant investigation for enhanced efficiency and control. Finally, a comprehensive techno-economic analysis must incorporate a detailed life cycle assessment (LCA) and extend beyond simple material costs. This should include precise quantification of raw material costs, synthesis costs (including energy consumption, labor, equipment depreciation, and facility overhead), catalyst lifetime and replacement costs, and end-of-life management, including disposal or recycling costs. Quantifying the cost per unit of treated effluent ($ per m3) or product generated ($ per kg or $ per mole), coupled with LCA data on environmental impacts such as CO2 emissions, water usage, and potential eco-toxicity, provides a holistic framework for evaluating the true cost and sustainability of these photocatalytic materials. This quantitative, data-driven approach facilitates informed decision-making, optimizing material selection, synthesis methods, and operational parameters for economically viable and environmentally responsible photocatalytic technologies.
6. SWOT analysis
This review explores the promising field of metal oxide nanocomposite photocatalysis for environmental remediation and energy generation. It begins by examining the fundamental mechanisms governing photocatalysis and delves into the synergistic effects achieved by combining MOx with other nanomaterials. This comprehensive scope encompasses a range of topics from conventional wastewater treatment approaches to the cutting-edge advancements in MOx nanocomposite photocatalysis, including material synthesis, mechanistic insights, influencing factors, and future research directions. Furthermore, it addresses critical aspects such as material stability and recyclability, key considerations for practical implementation. The inherent sustainability of this technology, aligning with global environmental goals, is also emphasized. Despite the significant potential, several key areas require further investigation to fully realize the promise of MOx nanocomposite photocatalysis. A crucial gap lies in the need for a rigorous cost–benefit analysis. While the potential for cost-effectiveness is acknowledged, a detailed assessment comparing production costs, material expenses, operational costs (including energy consumption and maintenance), and the economic benefits of resource recovery (e.g., energy generation, reclaimed water) against established technologies is lacking. This comparative analysis is essential for evaluating economic viability and promoting practical implementation. Furthermore, the potential of these materials extends beyond pollutant degradation. Integrating photocatalysis with electrochemistry, for example, offers exciting possibilities for simultaneous pollutant removal and energy harvesting. However, this aspect of photoelectrochemical applications remains relatively unexplored and warrants further investigation. Leveraging advanced characterization techniques, such as TRPL and in situ microscopy, can provide deeper insights into the structure–activity relationships governing photocatalytic performance. This knowledge is crucial for optimizing nanocomposite design and maximizing efficiency. Furthermore, the literature lacks actual examples of successful applications that include thorough performance statistics. Showcasing specific case studies, including removal efficiency of target pollutants (heavy metals, or specific organic species) under realistic settings and in varied water matrices, will considerably increase the practical relevance. Quantitative comparisons against existing technologies for these specific cases are particularly valuable. Beyond cost–benefit analysis and practical examples, a comprehensive comparative analysis with other AOPs is warranted. While this review primarily focuses on MOx nanocomposites, a more detailed comparison of their advantages and disadvantages against other AOPs in terms of efficiency, cost, environmental impact, and applicability to different pollutants would provide a more balanced perspective. Similarly, while the potential for energy generation using MOx photocatalysis is reviewed, a deeper exploration of specific strategies, realistic energy yields, and associated technical challenges is needed to fully assess this application. Finally, the review acknowledges the importance of recyclability and stability but needs to expand on other practical implementation challenges. These include reactor design and optimization (light penetration, mass transfer limitations), long-term performance and fouling, safe handling of nanomaterials, potential environmental risks of nanoparticles, and regulatory hurdles for real-world applications. Several opportunities emerge from these identified gaps. Further research should prioritize exploring synergistic effects of specific MOx combinations and optimizing their synthesis methods for enhanced photocatalytic activity. Developing scalable and cost-effective production methods is crucial for practical implementation. Investigating novel applications beyond wastewater treatment and energy generation, such as air purification or CO2 reduction, could broaden the technology's impact. Addressing the recyclability and stability challenges, as well as conducting life cycle assessments, are also crucial. Finally, it's important to acknowledge potential threats to the widespread adoption of this technology. Competition from other emerging wastewater treatment and energy generation technologies, challenges in scaling up production while maintaining cost-effectiveness, navigating regulatory requirements, ensuring public acceptance of nanomaterials, and the lack of standardized testing protocols are all potential obstacles that must be addressed. Fig. 14 shows a summary of SWOT analysis of the synthesis, characterization and applications of MOx in photocatalytic processes. | ||
| Fig. 14 Summary of SWOT analysis of the synthesis, characterization and applications of MOx in photocatalytic processes. | ||
7. Conclusion and prospectives
The transformative potential of metal oxide-based composites in photocatalysis lies at the intersection of material innovation, environmental urgency, and energy sustainability. By engineering heterojunctions (e.g., S-scheme, Z-scheme), tailoring defects (oxygen vacancies, dopants), and integrating hybrid matrices (graphene, biochar, polymers), these composites surmount the inherent limitations of pristine MOx, achieving unprecedented charge separation, broad-spectrum light absorption, and robust redox activity. Advanced synthesis methods, from solvothermal crystallization to bio-inspired green chemistry, enable atomic-level control over morphology and interfacial dynamics, directly translating to enhanced photocatalytic efficiency. Yet, critical challenges remain, e.g., long-term stability under harsh operational environments, scalability of nanoscale precision to industrial volumes, and the economic-ecological balance of material production. To propel MOx composites from laboratory curiosities to societal solutions, the following prospective directions demand urgent attention:(1) Scalable synthesis & green manufacturing: develop scalable and cost-effective methods for synthesizing high-efficacy MOx nanomaterials and fabricating functional systems, while prioritizing earth-abundant elements (e.g., Fe, Zn) and waste-derived precursors (e.g., biomass, slag) for sustainable and environmentally friendly large-scale production.
(2) Durability enhancement: develop more effective and stable MOx-based nanocomposites that can withstand harsh environmental conditions (extreme pH, UV exposure) while retaining their favorable adsorptive and photocatalytic properties over extended life cycles.
(3) Advanced hybrid design: develop ternary/quaternary MOx hybrids with green carbon-based materials to enhance charge dynamics and enhance photocatalytic efficiency. Explore bio-inspired architectures (enzyme-mimetic co-catalysts, cyanobacteria-MOx composites) for selective CO2 conversion.
(4) Reactor innovation & process integration: design modular, solar-optimized reactors with light-trapping features (plasmonic waveguides) and integrate MOx systems into hybrid platforms (e.g., photocatalytic membrane reactors) for simultaneous pollutant degradation and resource recovery.
(5) Real-world validation & standards: conduct pilot-scale studies in wastewater/air purification systems. Establish global benchmarks for efficiency (quantum yield) and durability, supported by academia-industry consortia.
(6) Risk mitigation & circular economy: screen for ecotoxicity and design recyclable composites (magnetic cores, pH-responsive materials). Adopt lifecycle assessments (LCAs) to align with zero-waste goals. Future studies should focus on developing mitigation strategies and conducting comprehensive risk assessments to ensure responsible development.
(7) Computer-aided material optimization: leverage machine learning to predict dopant combinations, heterojunctions, and defect densities. Use generative prediction and simulation for novel architecture (e.g., MOx-MOF hybrids).
(8) Operando mechanistic insights: employ synchrotron techniques (XANES) and ultrafast spectroscopy (fs-TAS) to map defect evolution and carrier dynamics, guiding real-time catalyst design. Further, employing advanced techniques, such as TRPL and in situ microscopy, can provide deeper insights into structure–activity relationships, guiding optimized MOx nanocomposites design and maximizing photocatalytic efficiency.
(9) Policy & interdisciplinary collaboration: advocate for standardized toxicity protocols and funding models to accelerate industrial adoption of MOx technologies.
(10) Economic viability frameworks: balance efficiency with costs (material, energy) and end-of-life recyclability, ensuring scalability for global environmental applications.
By addressing these advanced research directions, we can push the boundaries of MOx-based nanomaterials and pave the way for their widespread implementation in addressing critical environmental and energy challenges. A key emphasis should be placed on bridging the gap between fundamental research and practical applications. Fig. 15 shows a summary of perspectives of the synthesis, characterization and applications of MOx in photocatalytic processes.
 | ||
| Fig. 15 Summary of prospectives of the synthesis, characterization and applications of MOx in photocatalytic processes. | ||
Data availability
The data analyzed in this review article are from previously published studies. The specific datasets and sources are cited throughout the manuscript and listed in the reference section. Readers can access the underlying data from the original published sources as cited. The authors confirm that they did not have any special access privileges to these datasets.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors extend their appreciation to Northern Border University, Saudi Arabia, for supporting this work through project number (NBU-CRP-2025-2292).References
- S. Yang, K. Wang, Q. Chen and Y. Wu, J. Mater. Sci. Technol., 2024, 175, 104–114 CrossRef CAS.
- K. Peng, L. Zhang, Y. Xie, Y. Ma, J. Ye, Y. Dai, Y. Chen, L. Wang and W. Zhang, Fuel, 2024, 374, 132460 CrossRef CAS.
- D. Wang, H. Tian, J. Zhu, Z. Lu, Z. He and S. Song, Appl. Surf. Sci., 2024, 673, 160862 CrossRef CAS.
- M. A. Ahmed and A. A. Mohamed, Inorg. Chem. Commun., 2023, 148, 110325 CrossRef CAS.
- A. Wawrzyńczak and A. Feliczak-Guzik, Coatings, 2024, 14, 366 CrossRef.
- Z. Wang, J. Hong, S.-F. Ng, W. Liu, J. Huang, P. Chen and W.-J. Ong, Acta Phys.-Chim. Sin., 2021, 37, 2011033 Search PubMed.
- A. Pugazhendhi, S. Kamarudin, M. A. Alshehri, G. Ramya and K. Brindhadevi, Environ. Res., 2024, 119427 CrossRef CAS PubMed.
- M. Ali, I. Hussain, I. Mehmud, M. Umair, S. Hu and H. M. A. Sharif, Nanomaterials, 2021, 11, 3301 CrossRef CAS PubMed.
- A. Chebbi, A. Sinopoli, A. Abotaleb and Y. Bicer, Catal. Sci. Technol., 2023, 13, 4895–4918 RSC.
- V.-H. Nguyen, S. M. Smith, K. Wantala and P. Kajitvichyanukul, Arabian J. Chem., 2020, 13, 8309–8337 CrossRef CAS.
- M. A. Ahmed, S. A. Mahmoud and A. A. Mohamed, Front. Environ. Sci. Eng., 2024, 18, 120 CrossRef CAS.
- M. A. Ahmed, M. A. Ahmed and A. A. Mohamed, J. Saudi Chem. Soc., 2023, 27, 101748 CrossRef CAS.
- S. Ahmad and N. Shoukat, J. Mater. Sci., 2024, 59, 86–94 CrossRef CAS.
- C. Gong, X. Lv, S. Liu, X. Chen, R. Weerasooriya and Z. Ding, J. Ind. Eng. Chem., 2025, 141, 340–350 CrossRef CAS.
- M. A. Ahmed, S. A. Mahmoud and A. A. Mohamed, RSC Adv., 2024, 14, 18879–18906 RSC.
- M. Kurnia, S. Suprapto and Y. L. Ni’mah, S. Afr. J. Chem. Eng., 2024, 47, 111–122 Search PubMed.
- D. Yanardağ and S. Edebali, Biomass Convers. Biorefin., 2024, 14, 5699–5710 CrossRef.
- M. A. Ahmed and A. A. Mohamed, J. Saudi Chem. Soc., 2024, 101923 CrossRef CAS.
- M. Adel, T. Nada, S. Amin, T. Anwar and A. A. Mohamed, Groundw. Sustainable Dev., 2022, 16, 100704 CrossRef.
- Q. Zhang, D. Zheng, B. Bai, Z. Ma and S. Zong, Chem. Eng. J., 2024, 157134 CrossRef CAS.
- M. A. Ahmed and A. A. Mohamed, iScience, 2024, 27, 108583 CrossRef CAS PubMed.
- K. Całus-Makowska, J. Dziubińska, A. Grosser and A. Grobelak, Desalination Water Treat., 2024, 100949 Search PubMed.
- K. Meera and M. Ramesan, J. Thermoplast. Compos. Mater., 2024, 37, 3036–3057 CrossRef CAS.
- M. Adel, M. A. Ahmed and A. A. Mohamed, Environ. Nanotechnol. Monit. Manag., 2021, 16, 100550 CAS.
- S. Naknonhan, S. Amnuaypanich, C. Randorn, W. Tanthanuch and S. Amnuaypanich, J. Environ. Chem. Eng., 2025, 115501 CrossRef CAS.
- Q. Wang, Y. Wan, Q. Liu, Y. Zhang, Z. Ma, Z. Xu, P. Sun, G. Wang, H.-L. Jiang and W. Sun, Sci. Bull., 2025, 70(7), 1118–1125 CrossRef CAS PubMed.
- D. Leybo, U. J. Etim, M. Monai, S. R. Bare, Z. Zhong and C. Vogt, Chem. Soc. Rev., 2024, 53, 10450–10490 RSC.
- F. Li, H. Yin, T. Zhu and W. Zhuang, Eco-Environ. Health, 2024, 3(1), 89–106 CrossRef PubMed.
- M. N. Lakhan, A. Hanan, Y. Wang, H. K. Lee and H. Arandiyan, Chem. Sci., 2024, 15(38), 15540–15564 RSC.
- K. Niu, Q. Liu, C. Liu, Z. Yu, Y. Zheng, Y. Su, Y. Zhao, B. Liu, S. Cui and G. Zang, Chem. Eng. J., 2024, 150714 CrossRef CAS.
- H. Li, H. Xu, C. Wang, X. Yang, L. Li, J. Sheng, Y. Jin, M. Wang, Y. Liu and Y. Zou, Chem. Eng. J., 2024, 479, 147597 CrossRef CAS.
- R. Medhi, M. D. Marquez and T. R. Lee, ACS Appl. Nano Mater., 2020, 3, 6156–6185 CrossRef CAS.
- K. H. Rahman, A. K. Kar and K.-C. Chen, Mater. Sci. Eng., B, 2024, 305, 117394 CrossRef.
- S. Saleem, M. N. Ashiq, S. Manzoor, U. Ali, R. Liaqat, A. Algahtani, S. Mujtaba, V. Tirth, A. M. Alsuhaibani and M. S. Refat, J. Mater. Res. Technol., 2023, 25, 6150–6166 CrossRef CAS.
- M. T. Uddin, M. E. Hoque and M. C. Bhoumick, RSC Adv., 2020, 10, 23554–23565 RSC.
- I. Manzoor and R. Vijayaraghavan, New J. Chem., 2024, 48(48), 20126–20139 RSC.
- R. Haounati, H. Ighnih, R. E. Malekshah, S. Alahiane, F. Alakhras, E. Alabbad, H. Alghamdi, H. Ouachtak, A. A. Addi and A. Jada, Mater. Today Commun., 2023, 35, 105915 CrossRef CAS.
- V. Kumar, D. Kumar, V. Singh, N. Kaushik, A. Kaushik, L. Purohit, N. K. Kaushik and S. K. Sharma, Colloids Surf., A, 2024, 698, 134460 CrossRef CAS.
- N. K. Shee and H.-J. Kim, Molecules, 2023, 28, 6481 CrossRef CAS PubMed.
- M. Kardes, H. C. Yatmaz and K. Ozturk, ACS Appl. Nano Mater., 2023, 6, 6605–6613 CrossRef CAS.
- X. Zhou, S. Fang, T. Zhang, Z. Wu, J. Li, W. Wang, J. Zhu, J. Wu, D. Ye and R. Han, Sep. Purif. Technol., 2025, 354, 129330 CrossRef CAS.
- C.-Y. Ma, X.-X. Li, M.-X. Du, W.-K. Dong and Y.-J. Ding, J. Mol. Struct., 2024, 1298, 137071 CrossRef CAS.
- M. A. Ahmed, S. A. Mahmoud and A. A. Mohamed, RSC Adv., 2024, 14, 25629–25662 RSC.
- H. M. Fernández, J. Gallenberger, C. Mempin Jr, I. Khalek, M. Neumann, S. Lotfi, S. M. Kim, M. Li, C. Tian and J. P. Hofmann, Electrochim. Acta, 2024, 498, 144626 CrossRef.
- A. Ali, S. Ahmad, M. Usman, N. Khan, M. Hashim, Y. Ali, R. Shah and N. U. Rahman, Adv. Condens. Matter Phys., 2024, 2024, 6645827 CrossRef.
- M. A. Ahmed and A. A. Mohamed, RSC Adv., 2023, 13, 421–439 RSC.
- Y. Xiao, X. Zhang, D. Yan, J. Deng, M. Chen, H. Zhang, W. Sun, J. Zhao and Y. Li, Nano Res., 2024, 17, 3043–3052 CrossRef CAS.
- G. Nabi, B. Atiq, H. Elsaeedy, M. Tanveer, W. Ali and A. Riaz, Inorg. Chem. Commun., 2023, 157, 111448 CrossRef CAS.
- P. Phogat, S. Shreya, R. Jha and S. Singh, MATEC Web of Conferences, 2024, vol. 393, p. 01001, DOI:10.1051/matecconf/202439301001.
- N. A. Joyner, J. G. F. Romeu, B. Kent and D. A. Dixon, Phys. Chem. Chem. Phys., 2024, 26, 19646–19657 RSC.
- C.-Y. Tsay, Y.-C. Chen, H.-M. Tsai and F.-H. Lu, Mater. Chem. Phys., 2023, 295, 127143 CrossRef CAS.
- Y. Da, Z. Tian, R. Jiang, G. Chen, Y. Liu, Y. Xiao, J. Zhang, S. Xi, W. Chen and X. Han, ACS Nano, 2023, 17, 18539–18547 CrossRef CAS PubMed.
- M. Bhattu, R. Acevedo and M. Alhadrawi, E3S Web of Conferences, 2024, vol. 588, p. 02013, DOI:10.1051/e3sconf/202458802013.
- V. Soni, P. Singh, A. A. P. Khan, A. Singh, A. K. Nadda, C. M. Hussain, Q. Van Le, S. Rizevsky, V.-H. Nguyen and P. Raizada, J. Nanostruct. Chem., 2022, 1–38 Search PubMed.
- S. Ananthi, M. Kavitha, A. Balamurugan, E. R. Kumar, G. Magesh, A. Abd El-Rehim, C. Srinivas, P. Anilkumar, J. Suryakanth and C. S. Rahale, Sens. Actuators, B, 2023, 387, 133742 CrossRef CAS.
- S. S. Nemati, M. H. S. Seresht, G. Dehghan and Y. Abdi, IEEE Sens. J., 2024, 24(19), 29651–29658 Search PubMed.
- T. Selema, T. Malevu, M. Mhlongo, S. Motloung and T. Motaung, Emergent Mater., 2024, 1–26 Search PubMed.
- D. R. Eddy, M. D. Permana, L. K. Sakti, G. A. N. Sheha, S. Solihudin, S. Hidayat, T. Takei, N. Kumada and I. Rahayu, Nanomaterials, 2023, 13, 704 CrossRef CAS PubMed.
- G. Chen, E. Xie, Q. Zhao, Z. Ye, S. Wang, Y. Zou, Y. Wang, J. Qiu, J. Ouyang and J. Wang, Appl. Surf. Sci., 2024, 673, 160818 CrossRef CAS.
- M. S. Rao, B. Rakesh, G. P. Ojha, R. Sakthivel, B. Pant and K. J. Sankaran, Molecules, 2024, 29, 4063 CrossRef CAS PubMed.
- Z. Hiroi, Prog. Solid State Chem., 2015, 43, 47–69 CrossRef CAS.
- Z. Zhao, X. Zhang, G. Zhang, Z. Liu, D. Qu, X. Miao, P. Feng and Z. Sun, Nano Res., 2015, 8, 4061–4071 CrossRef CAS.
- N. S. Allen, N. Mahdjoub, V. Vishnyakov, P. J. Kelly and R. J. Kriek, Polym. Degrad. Stab., 2018, 150, 31–36 CrossRef CAS.
- T. Y. Ahmed, O. G. Abdullah, S. M. Mamand and S. B. Aziz, Opt. Quantum Electron., 2024, 56, 1249 CrossRef CAS.
- T. Dagar, S. Sarkar, B. P. Choudhury and S. De, Nanotechnology for Environmental Management, 2024, vol. 285 Search PubMed.
- X. Bi, G. Du, A. Kalam, D. Sun, Y. Yu, Q. Su, B. Xu and A. G. Al-Sehemi, Chem. Eng. Sci., 2021, 234, 116440 CrossRef CAS.
- T. Nakajima, T. Nakamura and T. Tsuchiya, Catalysts, 2019, 9, 725 CrossRef CAS.
- H. Gulab, N. Fatima, U. Tariq, O. Gohar, M. Irshad, M. Z. Khan, M. Saleem, A. Ghaffar, M. Hussain and A. K. Jan, Nano-Struct. Nano-Objects, 2024, 39, 101271 CrossRef.
- J. S. Chang, J. Strunk, M. N. Chong, P. E. Poh and J. D. Ocon, J. Hazard. Mater., 2020, 381, 120958 CrossRef CAS PubMed.
- I. M. Kahura, K. Sharon and W. M. Mulwa, Am. J. Mater. Sci., 2024, 14, 31–44 Search PubMed.
- A. A. Lisachenko and B. V. Novikov, Photocatal.: Res. Potential, 2023, 1, 10005 Search PubMed.
- A. Phuruangrata, S. Thamsukhoa, S. Thungprasertb, T. Sakhonc, T. Thongtemd and S. Thongtemd, Synthesis, 2022, 8, 19–23 Search PubMed.
- Q. Zhang, X. Zhao, L. Duan, H. Shen and R. Liu, J. Photochem. Photobiol., A, 2020, 392, 112156 CrossRef CAS.
- J. Zhang and J. Li, Nanomaterials, 2022, 12, 433 CrossRef CAS PubMed.
- P. Okoye, S. Azi, T. Qahtan, T. Owolabi and T. Saleh, Mater. Today Chem., 2023, 30, 101513 CrossRef CAS.
- W. Lu, T. Gu, X. Jing, Y. Zhu, L. Yu, S. Hou, T. Pang, N. Lu and Z. Zhang, J. Alloys Compd., 2023, 968, 171864 CrossRef CAS.
- L. Arun, C. Karthikeyan, D. Philip and C. Unni, J. Phys. Chem. Solids, 2020, 136, 109155 CrossRef CAS.
- F. T. Joorabi, M. Kamali and S. Sheibani, Mater. Sci. Semicond. Process., 2022, 139, 106335 CrossRef.
- A. Khan, M. A. Gaikwad, J. H. Kim and A. Kadam, Tungsten, 2024, 1–16 Search PubMed.
- S. G. Kumar and K. K. Rao, Appl. Surf. Sci., 2015, 355, 939–958 CrossRef.
- M. Kang, J. Liang, F. Wang, X. Chen, Y. Lu and J. Zhang, Mater. Res. Bull., 2020, 121, 110614 CrossRef CAS.
- Y. Lu, J. Zhang, F. Wang, X. Chen, Z. Feng and C. Li, ACS Appl. Energy Mater., 2018, 1, 2067–2077 CrossRef CAS.
- H. Li, Q. Shen, H. Zhang, J. Gao, H. Jia, X. Liu, Q. Li and J. Xue, J. Adv. Ceram., 2022, 11, 1873–1888 CrossRef CAS.
- M. Karimi-Nazarabad and E. K. Goharshadi, Sol. Energy Mater. Sol. Cells, 2017, 160, 484–493 CrossRef CAS.
- V. Dutta, S. Sharma, P. Raizada, V. K. Thakur, A. A. P. Khan, V. Saini, A. M. Asiri and P. Singh, J. Environ. Chem. Eng., 2021, 9, 105018 CrossRef CAS.
- P. Umek, M. Dürrschnabel, L. Molina-Luna, S. Škapin, R. Cerc Korošec and C. Bittencourt, Molecules, 2023, 28(15), 5838 CrossRef CAS PubMed.
- K. Kalpana, M. Marimuthu and P. J. Rosy, Exploring the Electrochemical Potential of Egg in Wasp Nest-Like Ceo2/Pani Hollow Spheres from Morphology to Supercapacitor and Sensing Applications, available at SSRN: https://ssrn.com/abstract=4963894 or DOI:10.2139/ssrn.4963894.
- M. Bellardita, R. Fiorenza, L. Palmisano and S. Scirè, in Cerium Oxide (CeO2): Synthesis, Properties and Applications, Elsevier, 2020, pp. 109–167 Search PubMed.
- M. Bellardita, R. Fiorenza, L. D'Urso, L. Spitaleri, A. Gulino, G. Compagnini, S. Scirè and L. Palmisano, Catalysts, 2020, 10, 765 CrossRef CAS.
- O. Gorban, A. Shylo, S. Gorban and I. Danilenko, J. Alloys Compd., 2024, 1002, 175276 CrossRef CAS.
- Y. Li, Y. Zhang, H. Jiang, M. Qi, X. Zhang, B. Zhu and L. Han, Microchem. J., 2023, 195, 109467 CrossRef CAS.
- S. Kumar, G. Kaur, M. Rawat, Y. F. Tsang, K.-Y. Lin and K.-H. Kim, J. Cleaner Prod., 2022, 361, 132242 CrossRef CAS.
- X. Zhou, J. Liu and J. Sun, Adv. Compos. Hybrid Mater., 2024, 7, 239 CrossRef CAS.
- R. Bhargava, S. Khan, N. Ahmad and M. M. N. Ansari, AIP Conf. Proc., 2018, 1953(1), 030034 CrossRef.
- A. Akter, S. A. Razzaque, M. A. Haque, S. Ganguli, J. Khanam, A. S. Nur, M. A. Sabur and A. K. Chakraborty, Results Eng., 2022, 16, 100672 CrossRef CAS.
- X.-L. Xu, Z.-H. Chen, Y. Li, W.-K. Chen and J.-Q. Li, Surf. Sci., 2009, 603, 653–658 CrossRef CAS.
- R. Obodo, A. C. Nwanya, A. Ekwealor, I. Ahmad, T. Zhao, R. U. Osuji, M. Maaza and F. I. Ezema, Surf. Interfaces, 2019, 16, 114–119 CrossRef CAS.
- L. Bai, H. Huang, S. Yu, D. Zhang, H. Huang and Y. Zhang, J. Energy Chem., 2022, 64, 214–235 CrossRef CAS.
- T. Jan, S. Azmat, Q. Mansoor, H. Waqas, M. Adil, S. Z. Ilyas, I. Ahmad and M. Ismail, Microb. Pathog., 2019, 134, 103579 CrossRef CAS PubMed.
- R. Salomao, L. Milena, M. H. Wakamatsu and V. C. Pandolfelli, Ceram. Int., 2011, 37, 3063–3070 CrossRef CAS.
- M. Farahmandjou, M. Zarinkamar and T. Firoozabadi, Rev. Mex. Fis., 2016, 62, 496–499 CAS.
- L. Wolski, K. Grzelak, M. Muńko, M. Frankowski, T. Grzyb and G. Nowaczyk, Appl. Surf. Sci., 2021, 563, 150338 CrossRef CAS.
- S. D. Nagesh, D. Satheesh, R. D. Ravi, R. Pachaiappan, K. Manavalan, L. Cornejo-Ponce and T. Sethuramachandran, Surf. Interfaces, 2024, 105578 Search PubMed.
- S. Roguai and A. Djelloul, Solid State Commun., 2021, 334, 114362 CrossRef.
- E. Arulkumar, S. S. Shree and S. Thanikaikarasan, Results Chem., 2023, 6, 101169 CrossRef CAS.
- S. Mondal, S. A. Ayon, M. S. Islam, M. S. Rana and M. M. Billah, Heliyon, 2023, 9(10), e20948 CrossRef CAS PubMed.
- R. E. Nimshi, J. J. Vijaya, L. J. Kennedy, P. S. Selvamani, M. Bououdina and P. J. Sophia, Ceram. Int., 2023, 49, 13762–13773 CrossRef CAS.
- A. Kubiak, S. Żółtowska, E. Gabała, M. Szybowicz, K. Siwińska-Ciesielczyk and T. Jesionowski, Powder Technol., 2021, 386, 221–235 CrossRef CAS.
- C. Sanchez Tobon, I. Panžić, A. Bafti, G. Matijašić, D. Ljubas and L. Ćurković, Nanomaterials, 2022, 12, 3975 CrossRef CAS PubMed.
- A. Kubiak, Z. Bielan, M. Kubacka, E. Gabała, A. Zgoła-Grześkowiak, M. Janczarek, M. Zalas, A. Zielińska-Jurek, K. Siwińska-Ciesielczyk and T. Jesionowski, Appl. Surf. Sci., 2020, 520, 146344 CrossRef CAS.
- G. Meenakshi and A. Sivasamy, Colloids Surf., A, 2022, 645, 128920 CrossRef CAS.
- M. Arellano-Cortaza, E. Ramírez-Morales, U. Pal, G. Pérez-Hernández and L. Rojas-Blanco, Ceram. Int., 2021, 47, 27469–27478 CrossRef CAS.
- J. Yang, C. Shi, Y. Dong, H. Su, H. Sun, Y. Guo and S. Yin, J. Colloid Interface Sci., 2022, 605, 373–384 CrossRef CAS PubMed.
- F. Nazer, Z. Khakpour, A. Maghsoudipour and S. Hajati, Int. J. Hydrogen Energy, 2024, 78, 148–156 CrossRef CAS.
- N. Afza, M. Shivakumar, M. W. Alam, A. N. Kumar, A. S. Bhatt, H. A. Murthy, C. Ravikumar, M. Mylarappa and S. Selvanandan, Appl. Surf. Sci. Adv., 2022, 11, 100307 CrossRef.
- J. Madona and C. Sridevi, Inorg. Chem. Commun., 2022, 138, 109265 CrossRef CAS.
- M. A. Ahmed, M. A. Ahmed and A. A. Mohamed, React. Funct. Polym., 2023, 191, 105701 CrossRef CAS.
- M. Afaq, A. Sajid, Q. Manzoor, F. Imtiaz, A. Sajid, R. Javed, A. Ahmad, N. Alwadai, W. Mnif and M. Iqbal, Mater. Sci. Eng., B, 2025, 312, 117847 CrossRef CAS.
- S. Nie, J. Li, L. Tao, Y. He, D. Dastan, X. Meng, P. Poldorn and X. Yin, ACS Sens., 2023, 8, 4121–4131 CrossRef CAS PubMed.
- M. A. Ahmed, Inorg. Chem. Commun., 2025, 114641 CrossRef.
- K. P. Makhado, M. M. Mphahlele-Makgwane, N. Kumar, P. G. Baker and P. R. Makgwane, Mater. Today Sustainability, 2024, 100664 CrossRef.
- M. A. Ahmed, M. A. Ahmed and A. A. Mohamed, Opt. Mater., 2024, 151, 115339 CrossRef CAS.
- K. Dharmalingam, A. K. Bojarajan, R. Gopal, E. Thangavel, S. A. Burhan Al Omari and S. Sangaraju, Sci. Rep., 2024, 14, 14518 CrossRef CAS PubMed.
- Y. Li, H. Li, S. Li, M. Li, P. He, Y. Xiao, J. Chen, Y. Zhou and T. Ren, Appl. Surf. Sci., 2024, 642, 158622 CrossRef CAS.
- M. A. Ahmed, S. A. Mahmoud and A. A. Mohamed, FlatChem, 2025, 100825 CrossRef CAS.
- P. Latifian, S. F. Hosseini, M. S. S. Dorraji and M. H. Rasoulifard, J. Mol. Liq., 2023, 376, 121445 CrossRef CAS.
- J. Li, H. Liu, Z. Liu, D. Yang, M. Zhang, L. Gao, Y. Zhou and C. Lu, Arabian J. Chem., 2022, 15, 103513 CrossRef CAS.
- T. Xu, Y. Wang, X. Zhou, X. Zheng, Q. Xu, Z. Chen, Y. Ren and B. Yan, Appl. Surf. Sci., 2017, 403, 564–571 CrossRef CAS.
- E. M. Hashem, M. A. Hamza, A. N. El-Shazly, S. A. Abd El-Rahman, E. M. El-Tanany, R. T. Mohamed and N. K. Allam, Chemosphere, 2021, 277, 128730 CrossRef CAS PubMed.
- S. Ke, M. Naghizadeh, L. Sun, H. Jin, S. Dong and T. Huang, Chem. Eng. Sci., 2025, 121361 CrossRef CAS.
- H. Yu, M. Zhao, C. Xue, J. Huang, N. Zhao and L. Kong, J. Mol. Struct., 2024, 1300, 137272 CrossRef CAS.
- S. Devi, S. Kumar, R. Kumar, V. Kumar, A. Kumar, O. Singh and P. Kumar, Phys. B, 2025, 696, 416649 CrossRef CAS.
- L. Che, J. Pan, K. Cai, Y. Cong and S.-W. Lv, Sep. Purif. Technol., 2023, 315, 123708 CrossRef CAS.
- S. Wang, C.-Y. Huang, L. Pan, Y. Chen, X. Zhang and J.-J. Zou, Catal. Today, 2019, 335, 151–159 CrossRef CAS.
- S. Wang, L. Pan, J.-J. Song, W. Mi, J.-J. Zou, L. Wang and X. Zhang, J. Am. Chem. Soc., 2015, 137, 2975–2983 CrossRef CAS PubMed.
- L. Xiao, Z. Yang, H. Zhu and G. Yan, Inorg. Chem. Commun., 2022, 146, 110167 CrossRef CAS.
- Q. Meng, W. Liu, J. Jiang and X. Zhang, Ceram. Int., 2021, 47, 19402–19413 CrossRef CAS.
- M. Aadil, T. Kousar, M. Hussain, H. Somaily, A. K. Abdulla, E. R. Muhammad, E. A. Al-Abbad, M. A. Salam, S. M. Albukhari and D. F. Baamer, Ceram. Int., 2023, 49, 4846–4854 CrossRef CAS.
- Y. Che, B. Lu, Q. Qi, H. Chang, J. Zhai, K. Wang and Z. Liu, Sci. Rep., 2018, 8, 1–12 CAS.
- Y. Liu, H. Liu, H. Zhou, T. Li and L. Zhang, Appl. Surf. Sci., 2019, 466, 133–140 CrossRef CAS.
- S. Chen, L. Zhang, D. A. Alshammari, M. M. Hessien, W. Yu, L. Cui, J. Ren, Z. M. El-Bahy and Z. Guo, Sep. Purif. Technol., 2025, 354, 129414 CrossRef CAS.
- K. Michalec, B. Mozgawa, A. Kusior, P. Pietrzyk, Z. Sojka and M. Radecka, J. Phys. Chem. C, 2024, 128, 5011–5029 CrossRef CAS.
- X. Zhou, X. Wang, T. Tan, H. Ma, H. Tang, F. Dong and Y. Yang, Chem. Eng. J., 2023, 470, 143933 CrossRef CAS.
- Z. Jiang, B. Cheng, Y. Zhang, S. Wageh, A. A. Al-Ghamdi, J. Yu and L. Wang, Journal of Materials Science & Technology, 2022, 124, 193–201 Search PubMed.
- J. Hu, J. Li, Z. Pu, W. Xiao, H. Yu, Z. Zhang, F. Yu, C. Liu and Q. Zhang, J. Colloid Interface Sci., 2024, 665, 780–792 CrossRef CAS PubMed.
- R. Tahawy, M. Esmat, H. El-Hosainy, F. E. Farghaly, E.-S. A. Abdel-Aal, F. I. El-Hosiny and Y. Ide, Bull. Chem. Soc. Jpn., 2024, 97, uoae079 CrossRef CAS.
- Y. Liu, T. Zheng, Y.-T. Xu, A. Li, H. Xu, Y. Gao, X.-F. Wang, R. Fujii and S.-i. Sasaki, Appl. Surf. Sci., 2025, 684, 161943 CrossRef CAS.
- A. Y. Kurenkova, S. Kharina, E. Aydakov and E. Kozlova, Kinet. Catal., 2024, 65, 703–709 CrossRef CAS.
- X. Ma, D. Li, Y. Jiang, H. Jin, L. Bai, J. Qi, F. You and F. Yuan, J. Colloid Interface Sci., 2022, 628, 768–776 CrossRef CAS PubMed.
- W. Raza, K. Ahmad, R. A. Khan and H. Kim, Int. J. Hydrogen Energy, 2023, 48, 29071–29081 CrossRef CAS.
- R. Kumar, S. Janbandhu, G. Sukhadeve and R. Gedam, J. Mater. Res., 2023, 38, 557–570 CrossRef CAS.
- R. Kumar, S. Y. Janbandhu, G. K. Sukhadeve and R. S. Gedam, Environ. Sci. Pollut. Res., 2023, 30, 98619–98631 CrossRef CAS PubMed.
- S. A. Mahyoub, A. Hezam, F. A. Qaraah, K. Namratha, M. B. Nayan, Q. A. Drmosh, D. Ponnamma and K. Byrappa, ACS Appl. Energy Mater., 2021, 4, 3544–3554 CrossRef CAS.
- A. Hezam, J. Wang, Q. Drmosh, P. Karthik, M. A. Bajiri, K. Namratha, M. Zare, T. Lakshmeesha, S. Shivanna and C. Cheng, Appl. Surf. Sci., 2021, 541, 148457 CrossRef CAS.
- Y. Guo, X. Fu, Y. Xie, L. Zhu, R. Liu and L. Liu, Opt. Mater., 2022, 133, 112980 CrossRef CAS.
- B. Kholikov, J. Hussain, S. Hayat and H. Zeng, J. Chin. Chem. Soc., 2021, 68, 1908–1915 CrossRef CAS.
- M. F. Afrin, M. Furukawa, I. Tateishi, H. Katsumata, M. Uzzaman and S. Kaneco, J. Compos. Sci., 2025, 9, 68 CrossRef CAS.
- A. Sampieri, K. López, G. Che-Galicia, R. Zanella and J. F. Guayaqiuil-Sosa, Influence of Cu on Au/Al2o3 Catalyst for Co Oxidation: Mitigating Deactivation for Sintering and Carbonate Formation, available at SSRN: https://ssrn.com/abstract=5128879 or DOI:10.2139/ssrn.5128879.
- A. H. Jawhari, N. Hasan, I. A. Radini, M. A. Malik and K. Narasimharao, Fuel, 2023, 344, 127998 CrossRef CAS.
- J. A. Okwako, S. H. Song, S. Park, H. Van Tran, B. O. Aduda, S. Waita, Y.-S. Hong, S. Hong and C.-H. Han, Electrochem. Commun., 2024, 165, 107762 CrossRef.
- S. Bhardwaj, D. Dogra, B. Pal and S. Singh, Sol. Energy, 2019, 194, 618–627 CrossRef CAS.
- A. E. Noua, D. Kaya, G. Sigircik, T. Tuken, F. Karadag and A. Ekicibil, J. Mater. Sci.: Mater. Electron., 2024, 35, 1220 CrossRef CAS.
- R. Sonkar, N. J. Mondal, B. Boro, M. P. Ghosh and D. Chowdhury, J. Phys. Chem. Solids, 2024, 185, 111715 CrossRef CAS.
- Y. Davoodi and M. S. Shafeeyan, Water Resour. Ind., 2024, 31, 100239 CrossRef CAS.
- S. M. Y. Qattali, J. Nasir, C. Pritzel, T. Kowald, Y. Sakalli, S. F. K. Moni, J. Schmedt auf der Günne, C. Wickleder, R. H. Trettin and M. S. Killian, Constr. Mater., 2024, 4, 315–328 Search PubMed.
- N. Khalid, S. Ilyas, F. Ali, T. Iqbal, M. Rafique, M. Imran and M. A. Assiri, Electron. Mater. Lett., 2024, 20, 85–94 CrossRef CAS.
- M. A. M. Khan, M. Pawar, A. A. Ansari, M. Ahamed, S. Kumar and S. Rani, J. Mater. Sci.: Mater. Electron., 2024, 35, 1565 CrossRef CAS.
- Q. Gao, Y. Dai, C. Li, K. Wang and X. Li, Mater. Sci. Semicond. Process., 2024, 181, 108637 CrossRef CAS.
- S. Karidas, B. K. Veena, N. Pujari, P. Krishna and V. Chunduru, Sādhanā, 2020, 45, 1–9 CrossRef.
- M. Sharma, K. Behl, S. Nigam and M. Joshi, Vacuum, 2018, 156, 434–439 CrossRef CAS.
- J. Zhang, Y. Hu, H. Zheng and P. Zhang, Catal. Sci. Technol., 2020, 10, 3603–3612 RSC.
- L. Zhao, T. Yu, B. Yang, H. Guo and L. Liu, Catalysts, 2022, 12(10), 1250 CrossRef CAS.
- M. A. Mannaa, K. F. Qasim, F. T. Alshorifi, S. M. El-Bahy and R. S. Salama, ACS Omega, 2021, 6, 30386–30400 CrossRef CAS PubMed.
- X. Hong, Y. Li, X. Wang, J. Long and B. Liang, J. Alloys Compd., 2022, 891, 162090 CrossRef CAS.
- L. Meng, Z. T. How, P. Chelme-Ayala, C. Benally and M. G. El-Din, J. Hazard. Mater., 2023, 454, 131441 CrossRef CAS PubMed.
- W. He, L. Liu, T. Ma, H. Han, J. Zhu, Y. Liu, Z. Fang, Z. Yang and K. Guo, Appl. Catal., B, 2022, 306, 121107 CrossRef CAS.
- J. Tang, Y. Xue, C. Ma, S. Zhang and Q. Li, New J. Chem., 2022, 46, 13010–13020 RSC.
- L. Zhu, H. Li, P. Xia, Z. Liu and D. Xiong, ACS Appl. Mater. Interfaces, 2018, 10, 39679–39687 CrossRef CAS PubMed.
- Z. Li, Z. Li, J. Liang, W. Fan, Y. Li, Y. Shen, D. Huang, Z. Yu, S. Wang and Y. Hou, Sep. Purif. Technol., 2023, 310, 123197 CrossRef CAS.
- V. Subhiksha, A. A. Alatar, M. K. Okla, I. A. Alaraidh, A. Mohebaldin, M. Aufy, M. A. Abdel-Maksoud, L. L. Raju, A. M. Thomas and S. S. Khan, Chemosphere, 2022, 303, 135177 CrossRef CAS PubMed.
- B. Shirdel and M. A. Behnajady, J. Mol. Liq., 2020, 315, 113633 CrossRef CAS.
- Z. Jichao, W. Yuan, H. Jie, H. Lifang and D. Rui, J. Phys. Chem. Solids, 2019, 126, 33–42 CrossRef.
- T. Jiang, K. Wang, T. Guo, X. Wu and G. Zhang, Chin. J. Catal., 2020, 41, 161–169 CrossRef CAS.
- H. Bian, Z. Zhang, X. Xu, Y. Gao and T. Wang, Phys. E, 2020, 124, 114236 CrossRef CAS.
- T. Gul, S. Ahmad, I. Khan, I. Khan, M. Almehmadi, A. A. Alsaiari, M. Allahyani and K. Saeed, J. Saudi Chem. Soc., 2023, 27, 101654 CrossRef CAS.
- S. Chen, Y. Rong, L. Tu, Z. Yu, H. Zhu, S. Wang and Y. Hou, Process Saf. Environ. Prot., 2022, 166, 328–340 CrossRef CAS.
- H. Boulahbel, M. Benamira, F. Bouremmad, N. Ahmia, S. Kiamouche, H. Lahmar, A. Souici and M. Trari, Inorg. Chem. Commun., 2023, 154, 110921 CrossRef CAS.
- H. Nezzal, S. Rahmane, E. G. Temam, M. Al-Abri, H. H. Kyaw, B. Gasmi, M. Althamthami, H. B. Temam and J. Hu, J. Alloys Compd., 2025, 1010, 177331 CrossRef CAS.
- T. Wang, Q. Zhu, C. Huo, Z. Yin, Q. Shi, J. Tao, F. Su and S. Cao, J. Alloys Compd., 2023, 950, 169889 CrossRef CAS.
- S. Ghattavi and A. Nezamzadeh-Ejhieh, J. Mol. Liq., 2021, 322, 114563 CrossRef CAS.
- S. Ghattav and A. Nezamzadeh-Ejhieh, Desalin. Water Treat., 2019, 166, 92–104 CrossRef.
- S. R. Amelia, Y. Rohmatulloh, P. Listiani, M. J. Devi, Y. Ichikawa, M. Honda, N. Nurrosyid, I. Isnaeni, T. Sudiarti and A. L. Ivansyah, Ceram. Int., 2024, 50, 11216–11235 CrossRef.
- S. A. Kumar, T. Govindhan, K. Selvakumar, K. Yusuf, S. Mahalingam, T. H. Oh, S. Ramasundaram and J. Kim, Mater. Sci. Semicond. Process., 2025, 186, 109052 CrossRef CAS.
- S. Borthakur, R. Das, P. Basyach, K. Sonowal and L. Saikia, RSC Adv., 2024, 14, 1156–1168 RSC.
- S. Demarema, M. Nasr, S. Ookawara and A. Abdelhaleem, Chemosphere, 2024, 349, 140840 CrossRef CAS PubMed.
- A. Taufik, R. Saleh, T. Sekino and S. Yin, Mater. Sci. Semicond. Process., 2025, 189, 109299 CrossRef CAS.
- L. T. Pérez-Poyatos, S. Morales-Torres, L. M. Pastrana-Martínez and F. J. Maldonado-Hódar, Catal. Today, 2024, 115115 Search PubMed.
- Z. K. Baboukani, A. N. Chermahini and H. Farrokhpour, J. Alloys Compd., 2024, 1002, 175478 CrossRef.
- A. Vijeta, C. Casadevall and E. Reisner, Angew. Chem., 2022, 134, e202203176 CrossRef.
- M. A. Ahmed and A. A. Mohamed, Int. J. Biol. Macromol., 2023, 242, 124787 CrossRef CAS PubMed.
- D. Zheng, H. Zhao, S. Wang, J. Hu and Z. Chen, Catalysts, 2021, 11, 1427 CrossRef CAS.
- W. Ge, K. Liu, S. Deng, P. Yang and L. Shen, Appl. Surf. Sci., 2023, 607, 155036 CrossRef CAS.
- P. Qiu, J. Xiong, M. Lu, L. Liu, W. Li, Z. Wen, W. Li, R. Chen and G. Cheng, J. Colloid Interface Sci., 2022, 622, 924–937 CrossRef CAS PubMed.
- M. S. Hamdy, H. S. Abd-Rabboh, M. Benaissa, M. G. Al-Metwaly, A. Galal and M. Ahmed, Opt. Mater., 2021, 117, 111198 CrossRef CAS.
- Y. R. Girish, N. M. Byrappa, G. Alnaggar, A. Hezam, G. Nagaraju, K. Pramoda and K. Byrappa, J. Hazard. Mater. Adv., 2023, 9, 100230 CAS.
- Z. Li, D. Jin and Z. Wang, Int. J. Hydrogen Energy, 2021, 46, 6358–6368 CrossRef CAS.
- S. Farhan, A. H. Raza, S. Yang, Z. Yu and Y. Wu, J. Colloid Interface Sci., 2024, 669, 430–443 CrossRef CAS PubMed.
- C. Zhu, Q. He, W. Wang, F. Du, F. Yang, C. Chen, C. Wang, S. Wang and X. Duan, J. Colloid Interface Sci., 2022, 620, 253–262 CrossRef CAS PubMed.
- G. Li, Y. Sun, Q. Zhang, Z. Gao, W. Sun and X. Zhou, Chem. Eng. J., 2021, 410, 128397 CrossRef CAS.
- L. Fang, L.-W. Bai, D. Wu, H.-T. Che, Y. Jiang, Y. Wang, H. Dong and F.-M. Zhang, Chem. Eng. J., 2025, 161820 CrossRef CAS.
- Z. Jiang, X. Zhang, Z. Yuan, J. Chen, B. Huang, D. D. Dionysiou and G. Yang, Chem. Eng. J., 2018, 348, 592–598 CrossRef CAS.
- G. Chen, Z. Zhou, B. Li, X. Lin, C. Yang, Y. Fang, W. Lin, Y. Hou, G. Zhang and S. Wang, J. Environ. Sci., 2024, 140, 103–112 CrossRef CAS PubMed.
- W. Chen, J. Xiong, Z. Wen, R. Chen and G. Cheng, Mol. Catal., 2023, 542, 113138 CrossRef CAS.
- J.-C. Wang, L. Zhang, W.-X. Fang, J. Ren, Y.-Y. Li, H.-C. Yao, J.-S. Wang and Z.-J. Li, ACS Appl. Mater. Interfaces, 2015, 7, 8631–8639 CrossRef CAS PubMed.
- K.-L. Bae, J. Kim, C. K. Lim, K. M. Nam and H. Song, Nat. Commun., 2017, 8, 1156 CrossRef PubMed.
- T. Zhao, W. Zhang, J. Xiong, W. Li and G. Cheng, Sep. Purif. Technol., 2025, 360, 130849 CrossRef CAS.
- X. Tong, W. Zhang, G. Cheng and J. Xiong, Mol. Catal., 2024, 555, 113877 CrossRef CAS.
- H. Zhang, H. Bian, F. Wang, L. Zhu, S. Zhang and D. Xia, Colloids Surf., A, 2023, 674, 131989 CrossRef CAS.
- C. Ban, Y. Wang, J. Ma, Y. Feng, X. Wang, S. Qin, S. Jing, Y. Duan, M. Zhang and X. Tao, Chem. Eng. J., 2024, 488, 150845 CrossRef CAS.
- J. Wu, K. Li, S. An, S. Yan, J. Liu, C. Song and X. Guo, Appl. Catal., B, 2024, 347, 123822 CrossRef CAS.
- M. Adel, M. A. Ahmed, M. A. Elabiad and A. A. Mohamed, Environ. Nanotechnol. Monit. Manag., 2022, 18, 100719 CAS.
- M. Adel, M. A. Ahmed and A. A. Mohamed, J. Phys. Chem. Solids, 2021, 149, 109760 CrossRef CAS.
- M. Adel, M. A. Ahmed and A. A. Mohamed, FlatChem, 2021, 26, 100233 CrossRef CAS.
- M. Adel, M. A. Ahmed and A. A. Mohamed, Compos. Commun., 2020, 22, 100450 CrossRef.
- M. A. Ahmed, M. A. Ahmed and A. A. Mohamed, Inorg. Chem. Commun., 2022, 144, 109912 CrossRef CAS.
- N. A. Nahyoon, M. Mehdi, K. H. Thebo, N. Mahar, A. A. Memon, N. Memon and N. Hussain, Opt. Mater., 2023, 135, 113260 CrossRef.
- L. Zhu, Y. Zhou, L. Fei, X. Cheng, X. Zhu, L. Deng and X. Ma, Chemosphere, 2022, 309, 136721 CrossRef CAS PubMed.
- M. Shkir, S. Aldirham, S. AlFaify and A. M. Ali, Chemosphere, 2024, 357, 141934 CrossRef CAS PubMed.
- M. A. Ahmed, M. A. Ahmed and A. A. Mohamed, RSC Adv., 2023, 13, 5337–5352 RSC.
- M. A. Ahmed, M. Farag, M. Ahmed, S. A. Mahmoud and A. A. Mohamed, Surf. Interfaces, 2025, 106321 CrossRef.
- Z. Zainal, L. K. Hui, M. Z. Hussein and A. H. Abdullah, J. Hazard. Mater., 2009, 164, 138–145 CrossRef CAS PubMed.
- A. Jbeli, Z. Hamden, S. Bouattour, A. Ferraria, D. Conceição, L. V. Ferreira, M. Chehimi, A. B. do Rego, M. R. Vilar and S. Boufi, Carbohydr. Polym., 2018, 199, 31–40 CrossRef CAS PubMed.
- A. Hamdi, S. Boufi and S. Bouattour, Appl. Surf. Sci., 2015, 339, 128–136 CrossRef CAS.
- S. Rajabi, Z. Derakhshan, A. Nasiri, M. Feilizadeh, A. Mohammadpour, M. Salmani, S. H. Kochaki, H. Shouhanian and H. Hashemi, Environ. Technol. Innovat., 2024, 35, 103724 CrossRef CAS.
- J. Chen, H. Li, L. Ma, G. Jiang, D. Li, Y. Wu, X. Shi, X. Wang and H. Deng, Carbohydr. Polym., 2021, 273, 118559 CrossRef CAS PubMed.
- M. A. Ahmed and A. A. Mohamed, Nutrients and Colored Compounds in Wastewater, 2025, pp. 393–431 Search PubMed.
- X. Feng, X. Li, B. Su and J. Ma, Colloids Surf., A, 2022, 648, 129114 CrossRef CAS.
- N. Dineshbabu, R. Jayaprakash, P. Karuppasamy, T. Arun, J. J. Vijaya, R. E. Nimshi, M. S. Pandian, S. M. Packiam and P. Ramasamy, J. Environ. Chem. Eng., 2022, 10, 107368 CrossRef CAS.
- F. Maloofi and A. D. Koohi, Int. J. Biol. Macromol., 2024, 138226 Search PubMed.
- P. Veerakumar, A. Sangili, S.-M. Chen, R. S. Kumar, G. Arivalagan, M. J. Firdhouse, K. S. Hameed and S. Sivakumar, Chem. Eng. J., 2024, 489, 151127 CrossRef CAS.
- L. Tan, H. Feng, L. Li, H. Lin and J. Xiong, J. Environ. Chem. Eng., 2024, 12, 112020 CrossRef CAS.
- T. Lerdwiriyanupap, A. Waehayee, T. Choklap, J. Prachanat, H. Nakajima, T. Chankhanittha, T. Butburee and T. Siritanon, Ceram. Int., 2024, 50, 52723–52732 CrossRef CAS.
- F. Xu, L. Zhang, B. Cheng and J. Yu, ACS Sustain. Chem. Eng., 2018, 6, 12291–12298 CrossRef CAS.
- S. Wang, B. Zhu, M. Liu, L. Zhang, J. Yu and M. Zhou, Appl. Catal., B, 2019, 243, 19–26 CrossRef CAS.
- C.-H. Shen, X.-J. Wen, Z.-H. Fei, Z.-T. Liu and Q.-M. Mu, J. Colloid Interface Sci., 2020, 579, 297–306 CrossRef CAS PubMed.
- F. He, A. Meng, B. Cheng, W. Ho and J. Yu, Chin. J. Catal., 2020, 41, 9–20 CrossRef CAS.
- Q. Xiang, J. Yu and M. Jaroniec, Nanoscale, 2011, 3, 3670–3678 RSC.
- C. Chen, Y. Huang and S. Huo, Vacuum, 2022, 205, 111467 CrossRef CAS.
- A. Yadav, Y. Hunge and S.-W. Kang, Surf. Interfaces, 2021, 24, 101075 CrossRef CAS.
- A. Alhafez, E. Aytar and A. Kilic, J. CO2 Util., 2022, 63, 102129 CrossRef CAS.
- X. Meng, S. Ouyang, T. Kako, P. Li, Q. Yu, T. Wang and J. Ye, Chem. Commun., 2014, 50, 11517–11519 RSC.
- D. Huang, S. Chen, G. Zeng, X. Gong, C. Zhou, M. Cheng, W. Xue, X. Yan and J. Li, Coord. Chem. Rev., 2019, 385, 44–80 CrossRef CAS.
- J. Low, S. Qiu, D. Xu, C. Jiang and B. Cheng, Appl. Surf. Sci., 2018, 434, 423–432 CrossRef CAS.
- Y. Wang, X. Shang, J. Shen, Z. Zhang, D. Wang, J. Lin, J. C. Wu, X. Fu, X. Wang and C. Li, Nat. Commun., 2020, 11, 3043 CrossRef CAS PubMed.
- A. Hayat, Z. Ajmal, A. Y. A. Alzahrani, S. B. Moussa, M. Khered, N. Almuqati, A. Alshammari, Y. Al-Hadeethi, H. Ali and Y. Orooji, Coord. Chem. Rev., 2025, 522, 216218 CrossRef CAS.
- Z. Chen, D. Yao, C. Chu and S. Mao, Chem. Eng. J., 2023, 451, 138489 CrossRef CAS.
- Y. Hong, Y. Cho, E. M. Go, P. Sharma, H. Cho, B. Lee, S. M. Lee, S. O. Park, M. Ko and S. K. Kwak, Chem. Eng. J., 2021, 418, 129346 CrossRef CAS.
- H. Jiang, L. Wang, X. Yu, L. Sun, J. Li, J. Yang and Q. Liu, Chem. Eng. J., 2023, 466, 143129 CrossRef CAS.
- J. Wang, J. Wang, S. Zuo, J. Pei, W. Liu and J. Wang, Chin. Chem. Lett., 2023, 34, 108157 CrossRef CAS.
- T. Zhang, T. Zhang, H. Fu, L. Feng, Q. Zhang, S. Ren, J. Cheng, Q. Liang and X. Xiao, J. Solid State Chem., 2024, 331, 124490 CrossRef CAS.
- L. Qin, P. She, P. Si, S. Liu, G. Dai and C. Pan, Environ. Res., 2025, 264, 120309 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |