
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New sugar-derived compounds as inhibitors of carbon steel against corrosion in acid solutions: experimental analyses and theoretical approaches†
M. Rbaa *abc,
A. Barrahic,
R. Seghiride,
Konstantin P. Katinf,
Elyor Berdimurodovghi,
Hatem A. Abuelizzj,
C. Jamak,
F. Bentisskl,
B. Lakhrissib and
A. Zarrouk*c
*abc,
A. Barrahic,
R. Seghiride,
Konstantin P. Katinf,
Elyor Berdimurodovghi,
Hatem A. Abuelizzj,
C. Jamak,
F. Bentisskl,
B. Lakhrissib and
A. Zarrouk*c
aThe Higher Institute of Nursing Professions and Health Techniques of Casablanca, P.O. Box 20250, Casablanca, Morocco. E-mail: mohamed.rbaa10@gmail.com; Tel: +212702099423
bLaboratory of Organic Chemistry, Catalysis and Environment, Faculty of Sciences, Ibn Tofail University, PO Box 133, 14000, Kenitra, Morocco
cLaboratory of Materials, Nanotechnology, and Environment, Faculty of Sciences, Mohammed V University in Rabat, P.O. Box 1014, Agdal, Rabat, Morocco. E-mail: azarrouk@gmail.com; Tel: +212665201397
dEcole Nationale Supérieure de Chimie de Kenitra (ENSCK), Ibn Tofail University, Kenitra, Morocco
eLaboratory of Advanced Materials and Process Engineering, Faculty of Sciences, Ibn Tofail University, Kenitra, Morocco
fNational Research Nuclear University “MEPhI”, KashirskoeShosse 31, Moscow 115409, Russian Federation
gFaculty of Chemistry, National University of Uzbekistan, Tashkent, 100034, Uzbekistan
hDepartment of Pharmacy and Chemistry, Alfraganus University, Tashkent, 100190, Uzbekistan
iChemistry and Physics, Western Caspian University, Baku, AZ-1001, Azerbaijan
jDepartment of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, PO Box 2457, Riyadh, 11451, Saudi Arabia
kUniv. Lille, CNRS, INRAE, Centrale Lille, UMR 8207, UMET – Unité Matériaux et Transformations, F-59000 Lille, France
lLaboratory of Catalysis and Corrosion of Materials, Faculty of Sciences, Chouaib Doukkali University, PO Box 20, M-24000 El Jadida, Morocco
First published on 19th May 2025
Abstract
The purpose of this study is to investigate the acid corrosion inhibition efficiency on carbon steel (CS) by utilizing two novel quinoxaline derivatives obtained from the reaction of recently synthesized D-mannose (MR1 and MR2) via nucleophilic substitution (SN1). The synthesized compounds were recently characterized by 13C-NMR and 1H-NMR spectroscopy. Electrochemistry testing was employed to evaluate their protective efficiency, whereas the surface was investigated using X-ray photoelectron spectroscopy (XPS) and scanning electron microscopy (SEM). The results indicate that the two inhibitors MR1 and MR2 exhibit inhibition efficiencies of 95.3% and 94.8% at 10−3 M for MR1 and MR2, respectively. The impedance results indicated that the incorporation of MR1 and MR2 into the corrosive medium reduces charge capacitance, hence systematically enhancing the interface charge/discharge function and creating an adsorbed layer on the metal surface. Moreover, SEM, water contact angle, and XPS techniques corroborated the formation of a protective coating on the carbon steel substrate surface following the incorporation of MR1 and MR2. The chemical interaction mechanisms at the atomic scale were analysed using theoretical calculations, DFT calculations and MD simulations.
1. Introduction
Heterocycles are organic compounds characterized by a ring structure primarily composed of carbon atoms.1 Other elements, like oxygen (O),2 nitrogen (N),3 sulfur (S),4 phosphorus (P), and silicon (Si)5 may also be integrated into their structure as well. These compounds are widely found in nature, particularly in biological molecules such as amino acids,6 nucleotides,7 and vitamins.8 Furthermore, they are also used in scientific and industrial fields, including pharmacology,9 agrochemistry,10 materials science,11 and corrosion inhibition.12–21Environmentally friendly organic compounds, also known as non-toxic compounds, are molecules derived from renewable natural raw materials or synthetically designed through specific processes, to reduce their environmental impact.22,23 These compounds are widely utilized across various industries,24 including cleaning agents, personal care products,25 food products,26 and building materials.27 The grafting of D-mannose onto organic compounds involves the formation of a covalent bond between a D-mannose molecule and an organic substrate, such as quinoline or quinoxaline.28 This chemical synthesis alters the structure of the original molecule or generates new compounds with biological or pharmaceutical properties.28
This study focuses on the synthesis of novel organic compounds derived from D-mannose and their characterization using various spectroscopic techniques, particularly, 1H-NMR and 13C-NMR nuclear magnetic resonance. The primary objective of synthesizing the MR1 and MR2 compounds is to explore their potential as corrosion inhibitors in an acidic environment (1.0 M HCl). To achieve this, a series of electrochemical analyses, including EIS as well as analytical investigations such as XPS, contact angle measurements and SEM, were conducted on both the corrosive solution and the metallic substrate surface of the steel.
2. Experimental details
2.1. Synthesis of compounds
All materials and reagents necessary for the synthesis of MR1 and MR2, used as corrosion inhibitors in this study, were procured from Sigma-Aldrich Chemical Company in France. The synthesized MR1 and MR2 compounds characterized through nuclear magnetic resonance spectroscopy (JNM-ECZ500R/S1-FT NMR System de JEO, DMSO-d6 reference at δppm). Reaction progress was observed by thin-layer chromatography on silica gel (TLC, 60 F254). Melting points (MP) was determined using a Kofler bench. The organic solvents employed for the reaction and subsequent product purification were purified by distillation.2.2. Surface characterization
The morphology of CS was analyzed using scanning electron microscopy (SEM) on a JEOL 5300 microscope with a 5 kV, enabling the examination of the substrate in the absence and presence of MR1 and MR2 inhibitors.Surface wettability was assessed by measuring water contact angles using a DSA100 drop shape analyzer (Krüss, Germany). Samples measuring of 2 × 2 × 0.3 cm3 were tested with 2 μL drops. The surface free energy (SFE) was determined using three polar solvents: pure deionized water, formamide, and diiodomethane (purity > 99%, Sigma-Aldrich).13,29
X-ray photoelectron spectroscopy (XPS) was conducted on a KRATOS AXIS Ultra DLD spectrometer equipped with a monochromated Al Kα X-ray source (hv = 1486.6 eV) and a 1 mm beam diameter. Measurements were performed at a constant pass energy of 40 eV over an analysis area of 700 μm × 300 μm under an ultra-high vacuum 10−10 torr. Charge compensation was applied to mitigate electrostatic effects. Spectral deconvolved was carried out using a non-linear least-squares fitting approach with a Shirley baseline and a Gaussian–Lorentzian peak shape function. Data processing was performed using CasaXPS software.
2.3. Electrochemical measurements
The steel used in this study had a chemical composition that is composed of 0.370% (C), 0.059% (Ni), 0.230% (Si), 0.016% (S), 0.077% (Cr), 0.011% (Ti), and 0.680% (Mn), with the remaining mass percentage consisting of iron (Fe). Prior to each experiment, the steel samples were abraded using silicon carbide (SiC) abrasive paper with grit sizes ranging from 180 to 1200. The samples were then polished, rinsed with distilled water, and allowed to air dry at room temperature. Analytical-grade hydrochloric acid (HCl, 37% purity) was diluted with distilled water to prepare corrosive solutions with a concentration of 1 M HCl. The evaluated concentrations of the MR1 and MR2 inhibitors varied from 0.1 to 0.4 g L−1.The electrochemical measurements were performed using a PGZ/100 potentiostat and Volta Master 4.0 software and three-electrode cell consisting of a carbon steel working electrode, a saturated calomel reference electrode (SCE) and a platinum counter electrode. The carbon steel reached a steady open circuit potential after 1800 seconds of immersion in the test solutions. Electrochemical impedance spectroscopy (EIS) was then performed at the open circuit potential (EOCP) with an AC signal amplitude of 10 mV frequency range of 100 kHz to 10 mHz. The data obtained were analyzed using the Zview software. Carbon steel, both with and without inhibitors, was then tested by potentiodynamic polarization experiments in 1 M HCl solutions. Potentials ranging from −800 mV/SCE to −100 mV/SCE were scanned at a rate of 0.5 mV s−1. The linear segments of the anodic and cathodic Tafel curves were extrapolated to obtain the current densities and associated electrochemical parameters. Data processing for polarization measurements was carried out using EC-Lab software.
2.4. Computational details
Two inhibitor molecules, MR1 and MR2, were analyzed computationally using DFT and MD methods. Both the neutral (MR1 and MR2) and protonated (MR1-H, MR2-H) forms of the inhibitors were considered. The protonated forms include two additional hydrogen atoms, attached to the nitrogen and to![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group. DFT calculations were performed using the B3LYP hybrid functional and 6-311G* basis set. Geometry optimization was conducted without symmetry constrains using the GAMESS-US program.30 The influence of solvent (water) was incorporated through the polarized continuum model in GAMESS-US.31
O group. DFT calculations were performed using the B3LYP hybrid functional and 6-311G* basis set. Geometry optimization was conducted without symmetry constrains using the GAMESS-US program.30 The influence of solvent (water) was incorporated through the polarized continuum model in GAMESS-US.31
The behavior of MR1 and MR2 on the steel surface in a corrosive media was simulated using a realistic ReaxFF interatomic potential suitable for corrosion modeling.32 The surface Fe (110) was represented by 6-layers cell (approximately 1.2 nm thickness) consisting of 1440 Fe atoms (3 nm × 5 nm). The corrosive media was explicitly represented by 1000 H2O and 18 dissociated HCl molecules.32 Various initial orientations of the inhibitor molecule were tested to determine the most stable adsorption position. Molecular dynamics simulations were done employing the NVT ensemble at room temperature (300 K) with the LAMMPS program.33 The total simulation time was 1 ns, with a time step of 0.2 fs for Verlet integration of the equations motion.
3. Results and discussions
3.1. Organic synthesis
The organic compounds used in this study were synthesized through a three-step process. The initial step involved the synthesis of epoxy D-mannose, which has been previously detailed in a separate publication.13 The next step focused on the preparation of quinoxaline derivatives, a method that has also been documented in another manuscript.34The final step involved grafting epoxy D-mannose with quinoxaline derivatives. This reaction was conducted by adding 0.85 eq. of quinoxalinone to a solution of the activated substrate in anhydrous DMF (33 g L−1) at 110 °C, in the presence of 1.2 eq. of K2CO3 and 0.1 eq. of BTBA. The resulting mixture was filtered, and then, the solvent was evaporated. Finally, the product was moved to silica gel chromatography for purification.
The progress of the reaction was monitored by TLC (eluent: hexane–acetone, 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v, 12 hours). The compounds were purified through silica gel chromatography (eluent: hexane–acetone, 9.5:0.5, v/v), and their purity was purified by chromatographic. Their structures were confirmed by 13C-NMR, 1H-NMR (Fig. 1).
:1, v/v, 12 hours). The compounds were purified through silica gel chromatography (eluent: hexane–acetone, 9.5:0.5, v/v), and their purity was purified by chromatographic. Their structures were confirmed by 13C-NMR, 1H-NMR (Fig. 1).
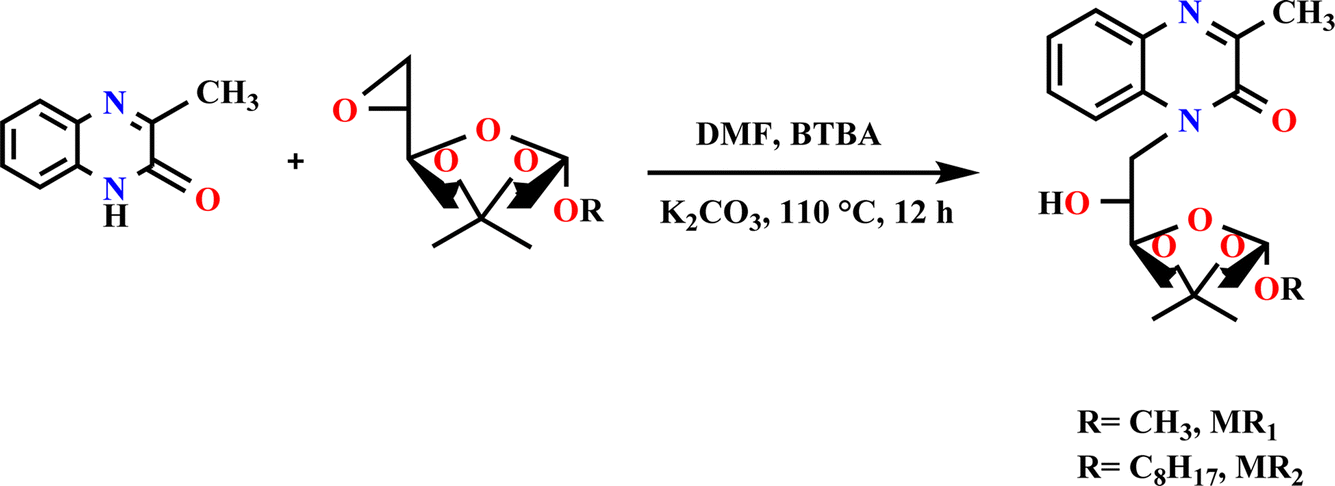 | ||
| Fig. 1 Preparation method for MR1 and MR2 compounds. | ||
The mechanism of the two compounds is illustrated in Fig. 2.
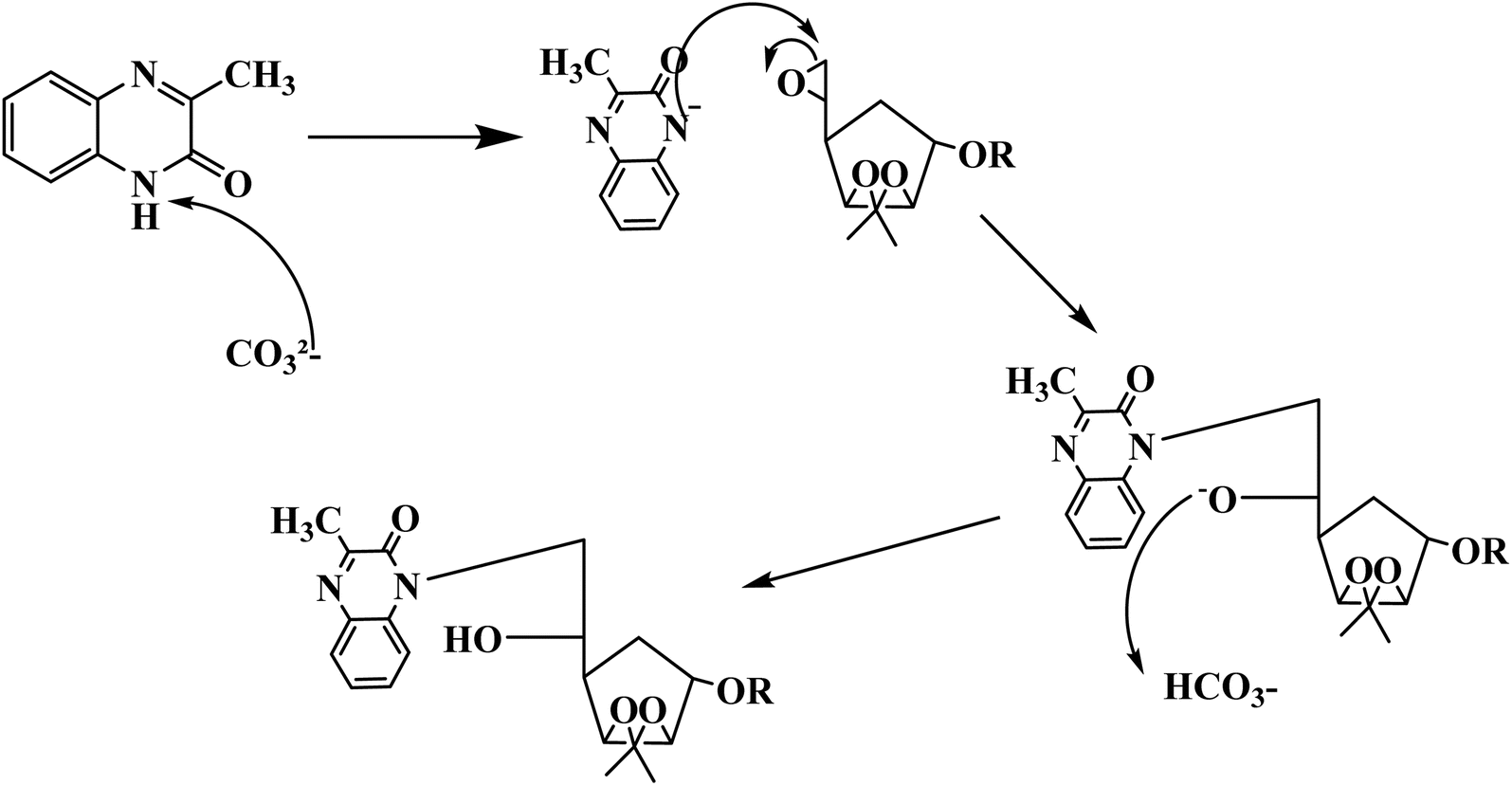 | ||
| Fig. 2 Proposed synthetic mechanism of the two compounds MR1 and MR2. | ||
3.2. Spectroscopic analysis of MR1 and MR2
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3-glucose), 28.248 (O–H3-glucose), 27.082 (C–CH3-quinoxaline), 57.853–111.267 (H–H2-glucose), 120.987–130.732 (Ar–ArHquinoxaline), 154.709 (–Nar), 154.235 (–O).H3-glucose), 19.327 (O–H3-glucose), 39.305 (C–CH3-quinoxaline), 61.315–82.007 (H–H2-glucose), 97.439–111.265 (Ar–ArHquinoxaline), 163.580 (–O).
H3-glucose), 28.248 (O–H3-glucose), 27.082 (C–CH3-quinoxaline), 57.853–111.267 (H–H2-glucose), 120.987–130.732 (Ar–ArHquinoxaline), 154.709 (–Nar), 154.235 (–O).H3-glucose), 19.327 (O–H3-glucose), 39.305 (C–CH3-quinoxaline), 61.315–82.007 (H–H2-glucose), 97.439–111.265 (Ar–ArHquinoxaline), 163.580 (–O).3.3. OCP analysis
Fig. 3 illustrates the variation in open circuit potential (EOCP) of carbon steel in a 1.0 M HCl solution with and without different concentrations of MR1 and MR2 inhibitors at 303 K. The curves exhibit a shift toward more positive potentials, indicating that the addition of MR1 and MR2 inhibitors significantly influences the electrochemical behavior of carbon steel. The open circuit potential shifted anodically, suggesting a slowdown in the anodic dissolution of iron. This finding implies that MR1 and MR2 form a protective layer on the metal surface, effectively mitigating corrosion in hydrochloric acid. Furthermore, after approximately 30 minutes of immersion, the potential stabilized, indicating that oxidation and reduction processes had reached equilibrium at the electrode–solution interface. This immersion time was used as a reference for subsequent electrochemical experiments. | ||
| Fig. 3 The time-dependent variation of OCP for carbon steel in 1 M HCl at 303 K with and without the different concentrations of MR1 and MR2. | ||
3.4. EIS analysis
The EIS is a technique employed for observing the deterioration of steel, particularly carbon steel. In this study, we applied this approach while examining the impact of organic inhibitors obtained from 8-hydroxyquinoline grafted by D-mannose MR1 and MR2 at 0.1, 0.2, 0.3 and 0.4 g L−1, both with and without their presence.Fig. 4 illustrates the Nyquist impedance electrochemical spectra for the two organic inhibitors obtained from 8-hydroxyquinoline grafted by D-mannose MR1 and MR2 in comparison with the control (1 M HCl). The recorded spectra exhibit slightly diminished semicircles, suggesting that the corrosion of steel in 1 M HCl occurs via a charge transfer mechanism.35 The semicircles diameter expands following the introduction of the two organic inhibitors, derived from 8-hydroxyquinoline grafted by D-mannose MR1 and MR2, compared to the solution only hydrochloric acid (HCl). At lower frequencies, the capacitive loop surpasses the Nyquist semicircles, indicating surface heterogeneity on the carbon steel (CS) due to the adsorption of MR1 and MR2 onto the metallic surface (CS).
 | ||
| Fig. 4 The Nyquist, Bode and phase plots were constructed for CS after in 1 M HCl, with and without different concentrations of MR1 and MR2 and the equivalent circuit used. | ||
Moreover, the obtained curves exhibit similarity upon the addition of MR1 and MR2, suggesting that the corrosion mechanism remains unchanged.35
In Fig. 4, the examination of the Bode and phase diagrams reveals a shift in the phase angle values. The phase angle value peaks at a moderate frequency, signifying the presence of a single charge transfer process.36
A singular time constant and a streamlined comparable circuit were utilized to examine the EIS data. The electrochemical values obtained from this approach are detailed in Table 1. A constant phase element (CPE) representing the double layer capacitance (Cdl) on a heterogeneous surface is one of these attributes, alongside Rp (polarization resistance) and Rs (solution resistance). Additionally, the equations presented below (1) (ref. 37 and 38) were employed to quantify the impedance of the constant-phase element (ZCPE).
 | (1) |
| Cdl=(A × RP1−n)1/n | (2) |
| Inhibitor | Conc. (g L−1) | Rs (Ω cm2) | Rp (Ω cm2) | 106 × A (Ω−1 sn−1 cm−2) | ndl | Cdl (μF cm−2) | χ2 | ηEIS% |
|---|---|---|---|---|---|---|---|---|
| 1 M HCl | — | 0.83 | 21.57 | 293.9 | 0.845 | 116.2 | 0.002 | — |
| MR2 | 0.1 | 1.8 | 126.2 | 170.9 | 0.83 | 77.8 | 0.006 | 82.9 |
| 0.2 | 2.0 | 231.1 | 113.2 | 0.83 | 53.7 | 0.005 | 90.7 | |
| 0.3 | 2.1 | 279.6 | 96.8 | 0.84 | 48.7 | 0.006 | 92.3 | |
| 0.4 | 1.9 | 343.5 | 71.4 | 0.85 | 37.1 | 0.009 | 93.7 | |
| MR1 | 0.1 | 1.9 | 109.9 | 149.8 | 0.82 | 60.8 | 0.004 | 80.3 |
| 0.2 | 2.3 | 183.4 | 121.8 | 0.83 | 55.9 | 0.009 | 88.2 | |
| 0.3 | 2.4 | 244.5 | 97.2 | 0.84 | 47.7 | 0.008 | 91.2 | |
| 0.4 | 3.0 | 319.5 | 80.7 | 0.84 | 40.2 | 0.007 | 93.2 |
The inhibition efficiency (ηEIS (%)) was determined using the subsequent formula(3):41
 | (3) |
The fitted values are displayed in Table 1, according to the circuit equivalent illustrated in Fig. 4. This circuit comprises solution resistance (Rs), polarization resistance (Rp), and the constant and exponent of the constant phase element (CPE), denoted as A and n.
The findings in Table 1 demonstrate that when the concentration of the inhibitors MR1 and MR2, which are derived from 8-hydroxyquinoline grafted onto D-mannose, increases, the polarization resistance (Rp) gradually increases. This increase in Rp is accompanied by a significant decrease in double layer capacitance (Cdl), which reflects the replacement of the water molecules initially adsorbed by the MR1 and MR2 inhibitor molecules on the metal surface.43 This substitution reduces the dielectric constant and promotes the formation of an electrical double layer, enhancing inhibitor adsorption. MR2 outperforms MR1 with a maximal inhibitory efficiency (ηEIS) of 93.7% vs. 93.2%.
The Rp values increase as a result of the inhibitor molecules adhering to the surface exposed to the corrosive environment and blocking active corrosion sites. This interaction also leads to an increase in the deflection parameter (n), which is associated with a reduction in surface defects, as well as a significant decrease in the admittance (Q) of protected substrates compared to untreated ones. Increasing surface coverage (θ = η (%)/100) leads to a linear decrease in Cdl, indicating effective molecular adsorption and limiting metal dissolution. Furthermore, the strong bonds between the active components (MR1 and MR2) and the metal are made possible by the presence of free electron pairs on the heteroatoms and the d-orbitals, which enhance inhibitor–molecule interactions with the metal surface. This process slows down the rate of corrosion of the metal by blocking the active corrosion sites. Finally, the reliability of the equivalent circuit used to describe the electrochemical interactions is confirmed by the chi-square (χ2) values, which are around 10−3.
3.5. Potentiodynamic polarization studies
In these studies, key Tafel parameters were found. Additionally, this method allows for the classification of the inhibitor as either anodic, cathodic, or a combination of both, as illustrated in Table 2 and Fig. 5. Inhibitory efficacy was calculated using the following relationship:
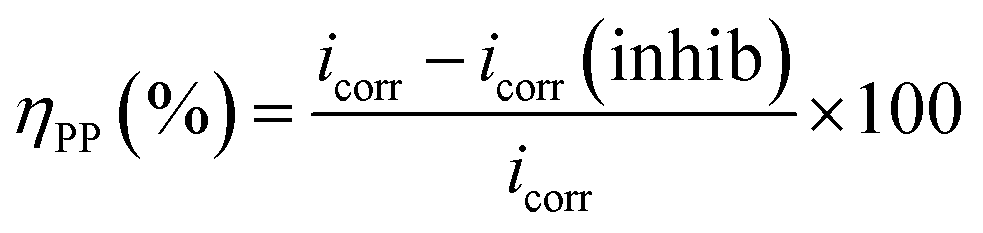 | (4) |
| Inhibitor | Conc. (g L−1) | Ecorr (mV vs. SCE) | icorr (μA cm−2) | −βc (mV dec−1) | βa (mV dec−1) | ηpp (%) |
|---|---|---|---|---|---|---|
| 1 M HCl | — | 456.3 | 1104 | 155.4 | 112.8 | — |
| MR2 1800 | 0.1 | −415.2 | 161.1 | 135 | 98 | 85.4 |
| 0.2 | −394.7 | 97.6 | 128 | 85 | 91.2 | |
| 0.3 | −400.1 | 72.4 | 121 | 84 | 93.4 | |
| 0.4 | −447.8 | 51.4 | 118 | 68 | 95.3 | |
| MR1 | 0.1 | −429.2 | 191.0 | 138 | 83 | 82.7 |
| 0.2 | −428.4 | 128.5 | 129 | 76 | 88.4 | |
| 0.3 | −457.0 | 75.8 | 123 | 78 | 93.1 | |
| 0.4 | −461.1 | 57.2 | 105 | 75 | 94.8 |
 | ||
| Fig. 5 Tafel plots of MR1 and MR2. | ||
Fig. 5 shows a shift in the Tafel curves towards lower potentials when the D-mannose derivatives derived from quinoxaline MR1 and MR2 were introduced into the corrosive media. This shift indicates that the inhibitors were effective in reducing the electrochemical processes involved in corrosion. Specifically, MR1 and MR2 inhibited both the anodic dissolution of iron and the cathodic hydrogen evolution reaction when added to a 1 M HCl solution.
Table 2 shows that the inclusion of D-mannose derivatives derived from quinoxaline MR1 and MR2 resulted in a significant drop in corrosion current density (icorr), which is consistent with the polarization curves. The inhibition efficiency (ηpp) reached 95.3% for MR2 and 94.8% for MR1 at 0.4 g L−1, indicating successful inhibition of electrochemical processes and the formation of a durable protective coating on the steel surface.44
Furthermore, the variation in corrosion potential (Ecorr) after the addition of the inhibitors remained minimal, at less than 85 mV. This suggests that MR1 and MR2 act as mixed inhibitors, affecting both anodic and cathodic processes.45 The cathodic polarization curves show parallel lines and slight variations in the cathodic Tafel slope (βc), indicating that the hydrogen evolution reaction is controlled but less intense due to inhibitor coverage on the steel surface. This inhibition is generated by the adsorption of MR1 and MR2 molecules on the substrate surface of carbon steel, which prevents the active sites from engaging in corrosion reactions. As a result, the decreases H+ ion access to the metal surface is restricted without affecting the cathodic reaction process. The surface coverage reduces the number of reactive sites available for H+ ions, slowing the cathodic reaction. In addition, the lower current densities observed in the anodic region indicate a slower rate of decrease in corrosion rate. The inhibitors MR1 and MR2 form a protective coating that reduces the severity of anodic iron dissolution, thereby enhancing the steel's resistance to corrosion.
3.6. Adsorption isotherm
To better understand the adsorption behaviour of D-mannose derived compounds MR1 and MR2 on the studied steel surface, some models were explored, including Langmuir, Frumkin, Freundlich, and Temkin. These isotherms serve to characterize the interaction behavior between the inhibitors and the surface, distinguishing between electrostatic adsorption (physisorption) or chemical adsorption (chemisorption).46The association between the derivatives of D-mannose MR1 and MR2 and the metal surface is presented in Fig. 6. The visual representation in the figure clearly validates that the Langmuir model is the most suitable for these inhibitors. The values of the adsorption constant (K), free enthalpy (ΔGads), and correlation factors (R2) derived from the Langmuir isotherm for the D-mannose derivatives MR1 and MR2 are presented in Table 3.
 | ||
| Fig. 6 Langmuir model of MR1 and MR2. | ||
| Inhibitor | K × 104 (L mol−1) | ΔGads (kJ mol−1) | Slope | R2 |
|---|---|---|---|---|
| MR2 | 5.74 | −37.7 | 1.02332 | 0.9999 |
| MR1 | 4.29 | −37.0 | 1.01614 | 0.9999 |
The Langmuir isotherm was used to calculate the inhibitors adsorption coefficient K, which was found to be 5.74 × 104 L mol−1 for MR2 and 4.29 × 104 L mol−1 for MR1. These high values indicate a strong affinity between the inhibitors. Effective adsorption is shown by a high adsorption coefficient, signifying that the inhibitor molecules cover a large surface area and keep corrosive species like Cl− ions from getting to the steel surface.
The good adsorption of the inhibitors can be attributed to the displacement of adsorbed water molecules by the MR1 and MR2 leading to the formation of a stable protective layer. This layer serves as a barrier to the diffusion of corrosive species, significantly decreasing the corrosion rate. Furthermore, the presence of free electron pairs on the heteroatoms (nitrogen and oxygen) promotes chemical interactions, along with interactions with the metal's d-orbitals, resulting in strong bonds between the inhibitors and the surface. The difference in Kads values between MR2 and MR1 indicates that MR2 exhibits a stronger affinity for the metal surface, resulting in superior inhibitory efficiency (95.3% vs. 94.8%).
The calculated ΔGads values are negative, ranging from −37.7 kJ mol−1 to −37.0 kJ mol−1. According to the literature, a free enthalpy value equal to or exceeding −40 kJ mol−1 an adsorption mechanism involving a both physisorption and chemisorption nature of the MR1 and MR2.47,48
The outcomes confirm the strong adhesion of the glucose-derived inhibitors MR1 and MR2 to the steel surface, providing long-term corrosion prevention, even in demanding industrial conditions.29
The findings we obtained are in agreement with literature values with respect to both inhibition efficiencies and adsorption behaviors. For example, Lei Guo et al. have looked at a newly synthesized triazolopyrimidine derivative as a corrosion inhibitor for toward mild steel in molar HCl. In their study, the reported inhibition efficiency was around 96.2% for the optimum concentration measured by impedance spectroscopy, and 93.7% at the optimal concentration using potentiodynamic polarization measurements.
In comparison, we obtained inhibition efficiencies via impedance spectroscopy of 93.7% for the MR2 and 93.2% for the MR1, and using potentiodynamic polarization measurements, we obtained 95.3% and 94.8% inhibition efficiencies for MR2 and MR1, respectively.
Both our compounds and those of by Lei Guo et al. adsorb to the steel surface via the Langmuir isotherm, which would suggest a similar adsorption mechanism.49
3.7. Surface analysis
To validate the adsorption of D-mannose derivatives on metal surfaces, a series of surface characterization analyses were carried out. These analyses involved the use of SEM, contact angle measurements and XPS.50 | ||
| Fig. 7 SEM images of M-steel surface were captured at different stages: (A) untreated metal surface, (B) immersed surface, with (C) MR1, and (D) MR2 at 400 ppm. | ||
 | ||
| Fig. 8 Images of the surface contact angle for selected metal: (A) before, (B) after corrosion, (C) with MR1, and (D) with MR2. | ||
When the metal surface interacted with the HCl solution, this value was decreased to around 46°, which can be attributed to the formation of more hydrophobic compounds on the surface due to the metal–oxidation processes, such as the formation of Fe2O3 and Fe3O4. These oxidation products can reduce hydrophilic nature. These products develop the surface's efficiency for the corrosive medium, promoting the spread of droplets and accelerating the corrosion process.
The introduction of MR1 and MR2, resulted in changes to the contact angle, which increased to 121.2° for MR1 and 129.5° for MR2 (Fig. 8). This increase indicates a higher degree of surface hydrophobization, suggesting the formation of an effective organic barrier that protects from the water droplets and corrosion attacks.
The surface hydrophobicity increases with the adsorption of MR1 and MR2, which linked with the surface through the –OH, –CO, –N and aromatic groups. These functional groups are more hydrophilic and formed the rigid covalent bonds between surface and MR1 and MR2. In contrast, the hydrophobic alkyl chains of MR1 and MR2, especially the longer chain in MR2, enhance the surface hydrophobicity. The MR2 based protective film is more hydrophobic than that of MR1 due to the long alkaline chain of MR2, which supports the hydrophobicity of surface, providing better surface coverage and more effective blocking of corrosive ions.52–54
 | ||
| Fig. 9 XPS spectrum of MR1/treated CS surface. | ||
 | ||
| Fig. 10 XPS spectrum of MR2/treated CS surface. | ||
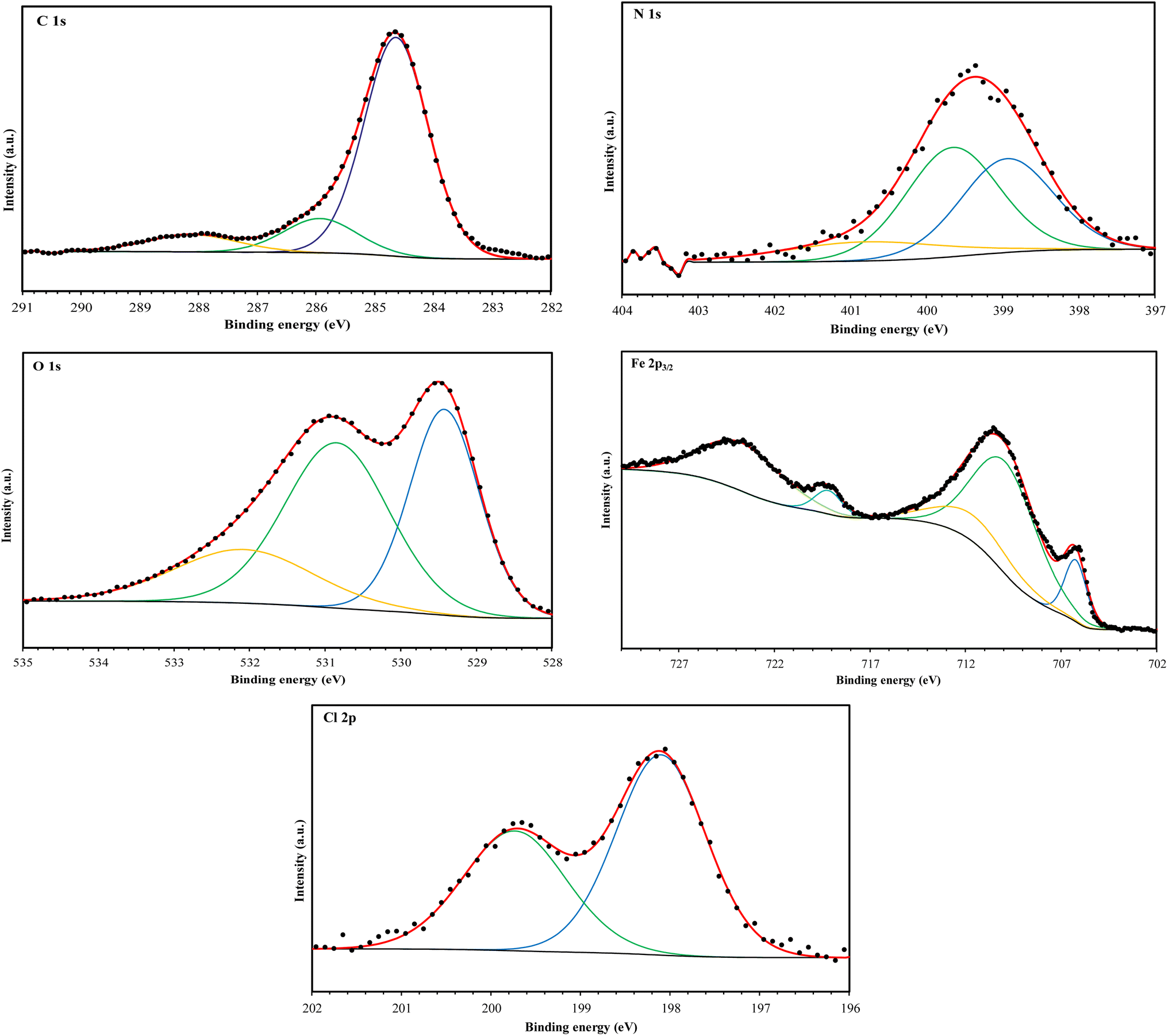
The high-resolution C 1s spectra (Fig. 9–12) exhibits three main peaks, corresponding to different C positions within the MR1 and MR2 structures. The first peak appears at 284.7 eV for MR1 and 284.6 eV for MR2. This dominant peak, accounting for 67% (MR1) and 77% (MR2) of the C 1s signal, is attributed to carbon atoms in C–C, CC, and C–H bonds, which are characteristic of the aromatic and aliphatic hydrocarbons in the inhibitor structures. Secondly, the next peaks were appeared at 285.6 eV for MR1 and 285.9 eV for MR2, contributes 25% (MR1) and 14% (MR2) of the C 1s signal. These peaks are associated with C atoms bonded to electronegative atoms such as N and O, indicating the presence of C–N, CN, and C–O bonds within the inhibitor structures. These functional groups play a crucial role in facilitating interaction between the metallic surface and Fe. The third peak is appeared at 288.2 eV for MR1 and 288.1 eV for MR2, corresponding to 8% (MR1) and 9% (MR2) of the C 1s signal, respectively. This peak is attributed to carbon atoms involved in CO bonds and possibly C–N+ bonds. The presence of C–N+ suggests protonation of N atoms in the acidic environment, enhancing the adsorption of the inhibitors through electrostatic interactions.
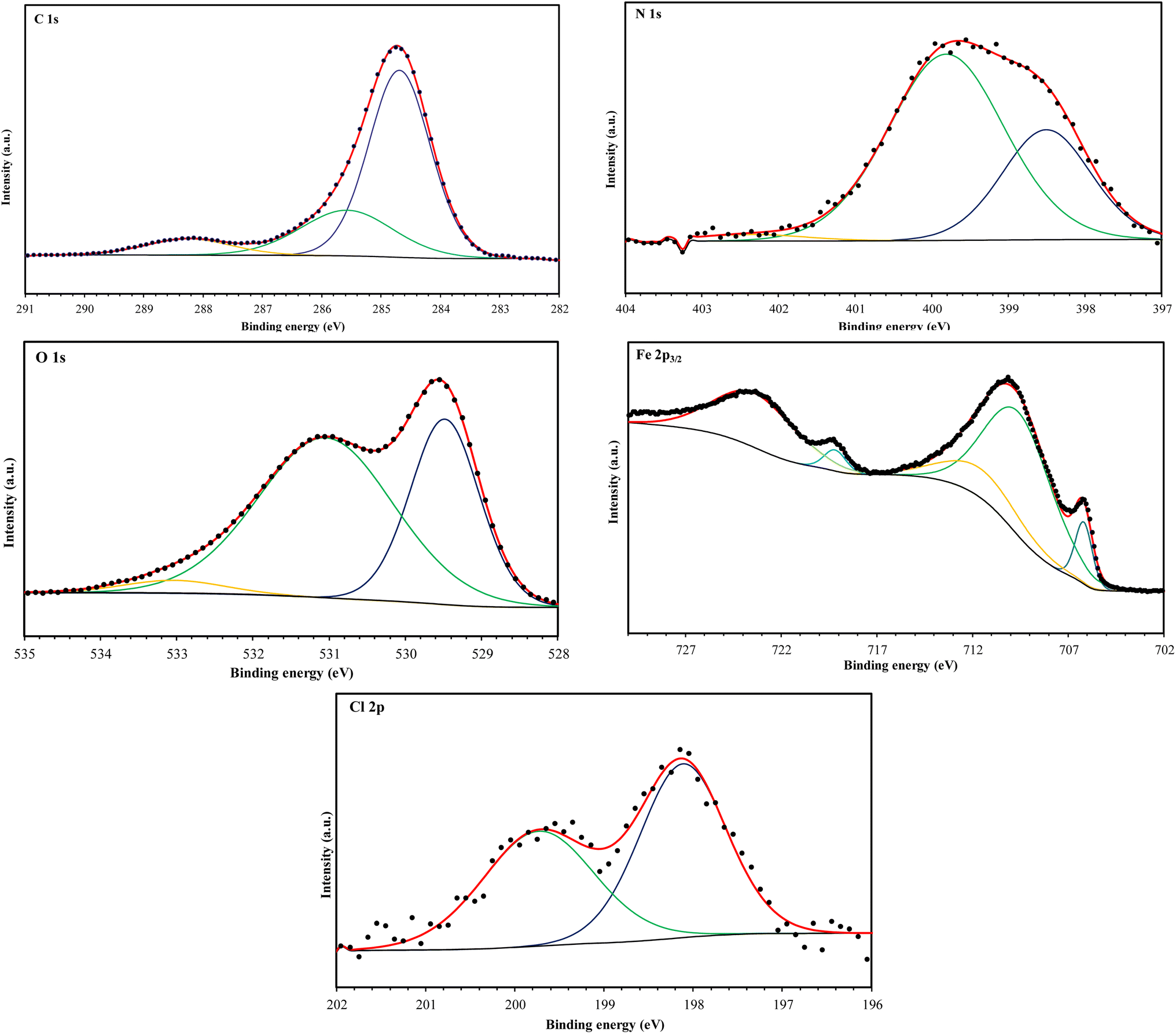 | ||
| Fig. 11 C 1s, N 1s, O 1s, Fe 2p, and Cl 2p XPS signals for MR1. | ||
 | ||
| Fig. 12 C 1s, N 1s, O 1s, Fe 2p, and Cl 2p XPS signals for MR2. | ||
The second XPS spectra for N 1s spectra (Fig. 9–12) reveal three N 1s peaks, showing their various positions within the inhibitor structures. The first peak, signaled at 398.5 eV for MR1 and 398.9 eV for MR2 is attributed to non-protonated N atoms in C–N and N– bonds. This indicates that some N atoms remain non-protonated and are available for coordination with the metal surface. The second set of peaks, appearing at 399.8 eV for MR1 and 399.7 eV for MR2, confirms the presence of N–Fe bonds, signifying a direct interaction between N atoms with Fe. These signals suggest the chemisorption of the MR1 and MR2 via N atoms, thereby contributing to the formation of a protective layer on the metal. Finally, the presence of protonated N atoms (N+H–) is evidenced by the signals at the 401.4 eV for MR1, and 401.0 eV for MR2, enhancing their positive charge and interaction with negatively charged Cl− ions on the surface.
The peak of O 1s spectra at 529.5 eV for MR1 and 529.4 eV for MR2 is associated with O2− ions in iron oxides such as Fe2O3 and Fe3O4, indicating the formation of oxide layers during the corrosion process. Next O 1s peak observed at 531.1 eV for MR1 and 530.8 eV for MR2 corresponds to OH− ions present in hydrous iron oxides like FeOOH and oxygen atoms involved in CO, O–H, and C–O bonds within the inhibitors. This peak thus reflects both corrosion products and the adsorbed MR1 and MR2 onto the metal surface. Lastly, the O of adsorbed water molecules on the steel surface is detected at 533.0 eV for MR1 and 532.1 eV for MR2, confirming the presence of H2O on the surface.
The oxidation states of Fe at the surface were also analyzed. The Fe 2p spectrum reveals a peak at 706.2 eV, corresponding to metallic iron (Fe0), indicating regions that remain unoxidized. The second peak at 710.1 eV confirms the presence of Fe3+ species in iron oxides (Fe2O3) and oxyhydroxides (FeOOH), which are typical corrosion products forming a passive layer on the underlying metal. Finally, peaks at 713.9 eV for MR1 and 714.1 eV for MR2 are also characteristic of Fe3+ and may suggest the presence of FeCl3, indicating a potential interaction between iron and chloride ions from the HCl solution.
The chlorine (Cl 2p) spectra exhibit a peak at 198.1 eV (Cl 2p3/2) indicating that the Cl− ions have interacted with iron to form FeCl3. The relatively low intensity of Cl peaks (0.34% for MR1 and 1.12% for MR2, as shown in Table 4) suggests that the inhibitors effectively reduce chloride adsorption, thereby mitigating its corrosive effect.
| Element | Position (eV) | Assignment |
|---|---|---|
| MR1 | ||
| C 1s | 288.2 (8%) | C–N+/CO |
| 285.6 (25%) | C–O/CN/C–N |
|
| 284.7 (67%) | CC/C–C/C–H |
|
| N 1s | 402.4 (2%) | –N+H |
| 399.8 (67%) | N–Fe | |
| 398.5 (31%) | –N/N–C structure |
|
| O 1s | 529.5 (36%) | O2− in Fe2O3 |
| 531.1 (60%) | OH− in FeOOH/OH/O–C/OC |
|
| 533.0 (4%) | Adsorbed H2O | |
| Cl 2p | 199.7 (46%) | Cl 2p1/2 |
| 198.1 (54%) | Cl 2p3/2 | |
| Fe 2p3/2 | 706.2 (9%) | Fe0 |
| 710.1 (88%) | Fe3+ in Fe2O3 and in FeOOH | |
| 713.9 (3%) | Satellite of Fe3+/FeCl3 | |
|
||
| MR2 | ||
| C 1s | 284.6 (77%) | CC/C–C/C–H/ |
| 285.9 (14%) | C–N/CN/C–O |
|
| 288.1 (9%) | CO/C–N+ |
|
| N 1s | 401.0 (14%) | –N+H |
| 399.7 (46%) | N–Fe | |
| 398.9 (40%) | N–C/–N structure |
|
| O 1s | 529.4 (37%) | O2− in Fe2O3 |
| 530.8 (44%) | OH− in FeOOH/OH/O–C/OC |
|
| 532.1 (19%) | Adsorbed H2O | |
| Cl 2p | 199.7 (41%) | Cl 2p1/2 |
| 198.1 (59%) | Cl 2p3/2 | |
| Fe 2p3/2 | 706.2 (12%) | Fe0 |
| 710.1 (86%) | Fe3+ in Fe2O3 and in FeOOH | |
| 714.1 (2%) | Satellite of Fe3+/FeCl3 | |
Table 5 shows a higher C content on MR2-treated steel (56.07%) compared to MR1 (49.51%) suggesting a more extensive inhibitor coverage and, consequently, enhanced protective performance. The low content of O implies less formation of iron oxides, possibly due to a more effective inhibition of the oxidation process by MR2. Lastly, the presence of N confirms the Fe–N interactions.55–57
| Element | MR1 treated-steel | MR2 treated-steel |
|---|---|---|
| C | 49.51 | 56.07 |
| O | 35.46 | 31.77 |
| N | 1.54 | 1.38 |
| Cl | 0.34 | 1.12 |
| Fe | 13.14 | 9.66 |
3.8. Theoretical studies
 | ||
| Fig. 13 DFT results for (MR1, left) and protonated (MR1-H, right) forms. | ||
 | ||
| Fig. 14 DFT results for (MR2, left) and protonated (MR2-H, right) forms. | ||
| Parameter | MR1 | MR1-H | MR2 | MR2-H |
|---|---|---|---|---|
| EHOMO | −6.36 | −4.47 | −6.34 | −4.55 |
| ELUMO | −1.98 | −0.09 | −1.95 | −0.17 |
| Gap | 4.38 | 4.38 | 4.39 | 4.38 |
| DM | 3.84 | 5.41 | 3.57 | 5.47 |
| IP | 6.93 | 4.50 | 7.14 | 4.54 |
| EA | 2.05 | −0.45 | 2.06 | −0.67 |
| χ | 4.49 | 2.03 | 4.60 | 1.93 |
| η | 2.19 | 2.19 | 2.20 | 2.19 |
| S | 0.46 | 0.46 | 0.46 | 0.46 |
| ΔN | 0.08 | 0.64 | 0.05 | 0.66 |
| Ebd | −0.55 | −0.55 | −0.55 | −0.55 |
Based on IP and EA, it was derived other relevant chemistry descriptors of inhibitors reactivity, following the methodology outlined in.59 Electronegativity (χ), global hardness (η) and softness (S), electron transfer ΔN and back-donation energy Ebd were also obtained and are presented in Table 6. The energy levels of the frontier molecular orbitals (EHOMO and ELUMO) reveal that protonation raises the HOMO and LUMO levels, denoting greater chemical reactivity for MR1-H and MR2-H but unchanged HOMO–LUMO energy gaps (∼4.38–4.39 eV) for each protonated form showing similar chemical stability. The dipole moment, which is related to the molecular polarity of the selected compounds, increases from protonation (MR1: 3.84 to 5.41 D; MR2: 3.57 to 5.47 D) and indicates that the protonated forms can interact further with the polar surface of steel, thus creating stronger adsorption. Protonation significantly lowers both the IP and the EA, with the EA exhibiting negative values, which illustrates a greater tendency for these protonated molecules to donate electrons to the Fe surface.
Moreover, MR1-H and MR2-H show superior charge transfer interactions with the Fe surface, which is essential when considering the coating's effectiveness in corrosion inhibition. The electronegativity of neutral forms of both molecules nearly equals that of the steel surface (4.82 eV (ref. 59)) which can suggest little to no transfer of electrons.
Comparing the calculated electronegativity of the protonated forms demonstrates an enlivened ΔN due to the lowers approximating ∼1.9–2.0 eV MR1: 0.08 to 0.64; MR2: 0.05 to 0.66.
These results that the protonated forms of MR1-H and MR2-H exhibit a stronger tendency to donate electrons to the Fe surface, thereby forming stronger coordination complexes on this surface. The nearly identical HOMO–LUMO gaps of all the molecules lead to similar values of η and Ebd, as shown in Table 6. The values of η and S values remain consistent across all molecules, suggesting comparable reactivity. Furthermore, the Ebd remains stable at −0.55 eV, indicating stable interactions involving electron back-donation from the metal to the inhibitors' antibonding orbitals, further stabilizing the inhibitor-metal complex.
To reveal the impact of protonation on the electronic structures of the inhibitors, we have depicted the electronic density of states (DOS) for both the neutral and protonated forms of the inhibitors, as shown in Fig. 15. It is evident that protonation induces significant changes in the DOS, particularly near the gap and gap shifting. To assess local reactivity, Fukui indices were calculated for all inhibitor atoms. The Fukui indices for nucleophilic (f+) and electrophilic (f−) attacks were defined as (f+ = D(M−) − D(M0); f− = D(M0) − D(M+)).
 | ||
| Fig. 15 DOS for MR1 (a) and MR2 (b) inhibitors in both neutral and protonated forms (FWHM = 0.2 eV). | ||
The electron density at the considered nucleus of the neutral, positively charged, and negatively charged inhibitor molecule is denoted as D(M0), D(M+) and D(M−). Atoms with f+ and f− values greater than 0.1 e bohr−3 are highlighted in Fig. 16.
 | ||
| Fig. 16 Fukui results for MR1 and MR2 molecules in neutral (a) and protonated forms (b) (f+ and f− are higher than 0.1 e bohr−3). | ||
 | ||
| Fig. 17 Metal-inhibitor interactions: MR1 (a), MR1-H (b), MR2 (c) and MR2-H (d). | ||
 | ||
| Fig. 18 Metal-inhibitor interactions: MR1 (a) and MR2 (b) in a corrosive media. RDF plots: MR1 (c) and MR2 (d). | ||
At parallel positions, this parallel increases surface contact giving maximum adsorption strength and overall coverage of the surface which is expected to be a good corrosion inhibition. The principal adsorption sites of these molecules are centered on heteroatoms such as nitrogen (–N and –NH–) and oxygen (CO, –OH) as well as the conjugated π-systems of the quinoxaline aromatic rings. Representatives of these functional groups are a rich source of electron, making them suitable for establishing coordination with the iron surface. The presence of lone pair electrons on N and O atoms also favors coordination bond formation or donor–acceptor interactions with the vacant d-orbitals of Fe atoms. In addition, the alkyl chains of MR1 and MR2 form a hydrophobic protective film that adds additional shielding to the metal surface from the corrosive environment. The two hexagonal rings contained in the quinoxaline core portion of MR1 and MR2 perform vital functions to immobilize each molecule metal surface. They are in the region of frontier molecular orbitals (HOMO and LUMO) that are largely sitting on aromatic systems and surrounding nitrogen atoms. The overlapping spatial overlap interactions facilitate closer electronic coupling at adsorption interface.
3.9. Comparative study
Table 7 presents a comparison between the inhibitors studied here and other inhibitors reported in literature.


Table 7 that our inhibitors display intensive corrosion inhibition efficiency for triazolopyridine and ionic liquid in the same concentration level and at ambient temperature. Nevertheless, our compounds found an efficiency almost that is almost similar to 8-hydroxyquinoline. The potential functionalities computed for the organic compounds MR1 and MR2 would allow them, as it has molecular structure,65 with two quinoxalines a attached together through D-mannose derived. As sited above, this configuration results in many hydroxyl groups per molecule and capable of forming a stronger adsorption onto steel surface.
4. Conclusion
The key findings of this study show that two quinoxaline derivatives, MR1 and MR2, were synthesized from D-mannose, and their structures were confirmed by 13C-NMR, and 1H-NMR spectroscopy. At 0.4 g per L concentration of these inhibitors exhibited significant corrosion inhibition efficiencies of 95.3% for MR1 and 94.8% for MR2. Both inhibitors demonstrated a mixed inhibition mechanism, leading to a substantial reduction in corrosion current density (icorr), with a minimum value of 51.4 μA cm−2 for MR2. Electrochemical impedance spectroscopy (EIS) analysis revealed an increase in polarization resistance (Rp), reaching a maximum of 343.5 Ω cm−2 for MR2, indicating improved surface protection. The slight shift in corrosion potential (Ecorr) (<22 mV) suggests that the inhibition mechanism is primarily mixed-type, with minimal influence on anodic and cathodic processes. Additionally, the decrease in double-layer capacitance (Cdl) with increasing inhibitor concentration suggests a moderate displacement of water molecules by the inhibitor at the metal surface.The adsorption of MR1 and MR2 inhibitors on the steel surface follows the Langmuir adsorption isotherm model, with an excellent fit (R2 = 0.9999). The calculated ΔGads values of −37.7 kJ mol−1 for MR2 and -37 kJ mol−1 for MR1 indicate spontaneous adsorption. These results, along with XPS data, highlight strong interactions between the inhibitors and the steel surface, suggesting that the adsorption mechanism involves both physisorption and chemisorption. The formation of a protective inhibitor layer led to a noticeable smoothing of the steel surface treated with MR2, as observed by scanning electron microscopy (SEM). The contact angles for MR1 and MR2 increased to 121.2° and 129.5°, respectively, indicating enhanced surface hydrophobicity, which is further reinforced by the alkyl chains present in the inhibitors. XPS analysis confirmed the formation of protective layers, with significant shifts in Fe, N, and O peaks, indicating effective adsorption.
XPS data also revealed the presence of Fe, C, N, and O on the treated surfaces, with a reduction in oxygen concentration to 31.77% for MR2, providing optimal protection. The HOMO–LUMO gap of approximately 4.38 eV for both MR1 and MR2 attests to their chemical stability and adequate reactivity. Although their electrical characteristics are similar, the longer alkyl chain of MR2 promotes enhanced physical adsorption. Molecular dynamics simulations confirmed stable adsorption of MR1 and MR2 on the Fe(110) surface, with RDF peaks indicating a proximity of 2 Å. A significant reduction in the corrosion rate of steel in 1.0 M HCl solution validated the effectiveness of both inhibitors. Corrosion inhibition by D-mannose derivatives presents an eco-friendly and environmentally benign approach. Furthermore, the protonated forms of MR1 and MR2 exhibit high electron transfer capacity (ΔN close to 0.66), further strengthening their interaction with the metal surface.
Data availability
All cited data can be found in the manuscript and the ESI files.†Conflicts of interest
The authors state that the publication of this paper is not influenced by any conflicts of interest.Acknowledgements
The authors extend their appreciation to the Researchers Supporting Project, King Saud University, Riyadh, Saudi Arabia for funding this work through grant number RSPD2025R566.References
- G. Prakash, N. Paul, G. A. Oliver, D. B. Werz and D. Maiti, Chem. Soc. Rev., 2022, 51, 3123–3163 Search PubMed.
- F. Meng, M. Song, Y. Wei and Y. Wang, Environ. Sci. Pollut. Res., 2019, 26, 7195–7204 Search PubMed.
- Y. Lv, D. Ma, K. Song, S. Mao, Z. Liu, D. He, X. Zhao, T. Yao and J.-W. Shi, J. Mater. Chem. A, 2023, 11, 800–808 Search PubMed.
- T. A. Saleh, J. Mol. Liq., 2022, 359, 119340 Search PubMed.
- Y.-M. Cui, Y. Lin and L.-W. Xu, Coord. Chem. Rev., 2017, 330, 37–52 Search PubMed.
- D. V. Grishin, D. D. Zhdanov, M. V. Pokrovskaya and N. N. Sokolov, All Life, 2020, 13, 11–22 Search PubMed.
- M. F. Turchinsky and L. P. Shershneva, Anal. Biochem., 1973, 54, 315–318 Search PubMed.
- H. Göring, Biochemistry, 2018, 83, 1350–1357 Search PubMed.
- I. Szabó and C. Varga, Int. J. Biometeorol., 2020, 64, 989–995 Search PubMed.
- M. Rani, D. Utreja and S. Sharma, Curr. Org. Chem., 2022, 26, 651–678 Search PubMed.
- J.-P. Lellouche, R. R. Koner and S. Ghosh, Rev. Chem. Eng., 2013, 29(6), 413–437 Search PubMed.
- E. Berdimurodov, I. Eliboyev, K. Berdimuradov, A. Kholikov, K. Akbarov, O. Dagdag, M. Rbaa, B. El Ibrahimi, D. K. Verma, R. Haldhar and N. Arrousse, Carbohydr. Polym., 2022, 292, 119719 Search PubMed.
- M. Rbaa, P. Dohare, A. Berisha, O. Dagdag, L. Lakhrissi, M. Galai, B. Lakhrissi, M. E. Touhami, I. Warad and A. Zarrouk, J. Alloys Compd., 2020, 833, 154949 Search PubMed.
- M. Alahiane, R. Oukhrib, Y. Ait Albrimi, H. Abou Oualid, R. Idouhli, A. Nahlé, A. Berisha, N. Z. Azzallou and M. Hamdani, Appl. Surf. Sci., 2023, 612, 155755 Search PubMed.
- M. El Faydy, F. Benhiba, B. Lakhrissi, M. Ebn Touhami, I. Warad, F. Bentiss and A. Zarrouk, J. Mol. Liq., 2019, 295, 111629 Search PubMed.
- H. Zarrok, A. Zarrouk, R. Salghi, M. Assouag, B. Hammouti, H. Oudda, S. Boukhris, S. S. Al Deyab and I. Warad, Der Pharm. Lett., 2013, 5(2), 43–53 Search PubMed.
- H. Zarrok, A. Zarrouk, R. Salghi, M. Ebn Touhami, H. Oudda, B. Hammouti, R. Touir, F. Bentiss and S. S. Al-Deyab, Int. J. Electrochem. Sci., 2013, 8, 6014–6032 Search PubMed.
- H. Zarrok, A. Zarrouk, R. Salghi, H. Oudda, B. Hammouti, M. Assouag, M. Taleb, M. Ebn Touhami, M. Bouachrine and S. Boukhris, J. Chem. Pharm. Res., 2012, 4(12), 5056–5066 Search PubMed.
- H. Zarrok, R. Salghi, A. Zarrouk, B. Hammouti, H. Oudda, Lh. Bazzi, L. Bammou and S. S. Al-Deyab, Der. Pharma Chem., 2012, 4(1), 407–416 Search PubMed.
- A. Zarrouk, B. Hammouti, R. Touzani, S. S. Al-Deyab, M. Zertoubi, A. Dafali and S. Elkadiri, Comparative Study of New Quinoxaline Derivatives Towards Corrosion of Copper in Nitric Acid, Int. J. Electrochem. Sci., 2011, 6, 4939–4952 Search PubMed.
- F. Benhiba, N. K. Sebbar, H. Bourazmi, M. E. Belghiti, R. Hsissou, T. Hokelek, A. Bellaouchou, A. Guenbour, I. Warad, H. Oudda, A. Zarrouk and E. M. Essassi, Int. J. Hydrogen Energy, 2021, 46(51), 25800–25818 Search PubMed.
- C. Grundke, J. Groß, N. Vierengel, J. Sirleaf, M. Schmitz, L. Krieger and T. Opatz, Org. Biomol. Chem., 2023, 21, 644–650 Search PubMed.
- Y. Pourshojaei, M.-H. Jadidi, K. Eskandari, A. Foroumadi and A. Asadipour, Res. Chem. Intermed., 2018, 44, 4195–4212 Search PubMed.
- A. Pecquet, D. McAvoy, C. Pittinger and K. Stanton, Integr. Environ. Assess. Manage., 2019, 15, 621–632 Search PubMed.
- N. Mohammad and R. B. Baharun, Adv. Sci. Lett., 2017, 23, 7367–7369 Search PubMed.
- J. S. Barrett and P. R. Gibson, Ther. Adv. Gastroenterol., 2012, 5, 261–268 Search PubMed.
- E. A. Nurdiah, Procedia Soc. Behav. Sci., 2016, 216, 30–38 Search PubMed.
- K. W. Y. Chan, M. T. McMahon, Y. Kato, G. Liu, J. W. M. Bulte, Z. M. Bhujwalla, D. Artemov and P. C. M. Van Zijl, Magn. Reson. Med., 2012, 68, 1764–1773 Search PubMed.
- I. Nadi, M. Bouanis, F. Benhiba, K. Nohair, A. Nyassi, A. Zarrouk, C. Jama and F. Bentiss, J. Mol. Liq., 2021, 342, 116958 Search PubMed.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045–1052 Search PubMed.
- H. Li, C. S. Pomelli and J. H. Jensen, Theor. Chem. Acc., 2003, 109, 71–84 Search PubMed.
- E. B. Kalika, K. P. Katin, A. I. Kochaev, S. Kaya, M. Elik and M. M. Maslov, J. Mol. Liq., 2022, 353, 118773 Search PubMed.
- J. Wang, Y. Liu, Y. He, X. Liu, Z. Lin, B. Zhang and R. Chai, in 58th U.S. Rock Mechanics/Geomechanics Symposium, ARMA, Golden, Colorado, USA, 2024, p. D022S019R001 Search PubMed.
- M. Rbaa, M. Galai, M. El Faydy, Y. Lakhrissi, M. EbnTouhami, A. Zarrouk and B. Lakhrissi, J. Mater. Environ. Sci., 2018, 9, 172–188 Search PubMed.
- W. Choi, H.-C. Shin, J. M. Kim, J.-Y. Choi and W.-S. Yoon, J. Electrochem. Sci. Technol., 2020, 11, 1–13 Search PubMed.
- X. Li, L. Wang, L. Fan, M. Zhong, L. Cheng and Z. Cui, Corros. Sci., 2021, 192, 109812 Search PubMed.
- M. El Faydy, F. Benhiba, A. Berisha, Y. Kerroum, C. Jama, B. Lakhrissi, A. Guenbour, I. Warad and A. Zarrouk, J. Mol. Liq., 2020, 317, 113973 Search PubMed.
- A. Berrissoul, A. Ouarhach, F. Benhiba, A. Romane, A. Guenbour, B. Dikici, F. Bentiss, A. Zarrouk and A. Dafali, Ind. Crops Prod., 2022, 187, 115310 Search PubMed.
- G. J. Brug, A. L. G. van den Eeden, M. Sluyters-Rehbach and J. H. Sluyters, J. Electroanal. Chem. Interfacial Electrochem., 1984, 176, 275–295 Search PubMed.
- A. Saxena, D. Prasad, R. Haldhar, G. Singh and A. Kumar, J. Mol. Liq., 2018, 258, 89–97 Search PubMed.
- H. Rahmani, K. I. Alaoui, M. EL Azzouzi, F. Benhiba, A. El Hallaoui, Z. Rais, M. Taleb, A. Saady, B. Labriti, A. Aouniti and A. Zarrouk, Chem. Data Collect., 2019, 24, 100302 Search PubMed.
- G. Laadam, F. Benhiba, M. El Faydy, A. Titi, A. S. Al-Gorair, M. Alshareef, H. Hawsawi, R. Touzani, I. Warad, A. Bellaouchou, A. Guenbour, M. Abdallah and A. Zarrouk, Inorg. Chem. Commun., 2022, 145, 109963 Search PubMed.
- Z. Rouifi, M. Rbaa, A. S. Abousalem, F. Benhiba, T. Laabaissi, H. Oudda, B. Lakhrissi, A. Guenbour, I. Warad and A. Zarrouk, Surf. Interfaces, 2020, 18, 100442 Search PubMed.
- A. Naseef Jasim, B. A. Abdulhussein, S. Mohammed Noori Ahmed, W. K. Al-Azzawi, M. M. Hanoon, M. K. Abbass and A. A. Al-Amiery, Prog. Color, Color. Coat., 2023, 16, 319–329 Search PubMed.
- M. El Faydy, F. Benhiba, H. About, Y. Kerroum, A. Guenbour, B. Lakhrissi, I. Warad, C. Verma, E. M. Sherif, E. E. Ebenso and A. Zarrouk, J. Colloid Interface Sci., 2020, 576, 330–344 Search PubMed.
- A. Kokalj, Corros. Sci., 2023, 217, 111112 Search PubMed.
- A. Ghazoui, A. Zarrouk, N. Bencaht, R. Salghi, M. Assouag, M. El Hezzat, A. Guenbour and B. Hammouti, J. Chem. Pharm. Res., 2014, 6(2), 704–712 Search PubMed.
- A. Zarrouk, B. Hammouti, S. S. Al-Deyab, R. Salghi, H. Zarrok, C. Jama and F. Bentiss, Int. J. Electrochem. Sci., 2012, 7, 5997–6011 Search PubMed.
- L. Guo, Y. E. Bakri, R. Yu, J. Tan and E. M. Essassi, J. Mater. Res. Technol., 2020, 9, 6568–6578 Search PubMed.
- S. J. Garcia, T. A. Markley, J. M. C. Mol and A. E. Hughes, Corros. Sci., 2013, 69, 346–358 Search PubMed.
- M. H. Fekri, F. Omidali, M. M. Alemnezhad and A. Ghaffarinejad, Mater. Chem. Phys., 2022, 286, 126150 Search PubMed.
- S. K. Behera, A. Kumar P, N. Dogra, M. Nosonovsky and P. Rohatgi, Langmuir, 2019, 35, 16120–16129 Search PubMed.
- T. Umasankareswari, R. Dorothy, J. Jeyasundari, G. Singh, S. Rajendran, A. Subramania, A. Al-Hashem and J. Aslam, in Electrochemical and Analytical Techniques for Sustainable Corrosion Monitoring, Elsevier, 2023, pp. 141–153 Search PubMed.
- N. S. Abdelshafi, A. A. Farag, F. E.-T. Heakal, A.-S. Badran, K. M. Abdel-Azim, A.-R. Manar El Sayed and M. A. Ibrahim, J. Mol. Struct., 2024, 1304, 137638 Search PubMed.
- M. Tabyaoui, M. Tourabi, H. Zarrok, C. Jama, F. Benhiba, A. Zarrouk and F. Bentiss, Environ. Sci. Pollut. Res., 2024, 31, 43757–43780 Search PubMed.
- M. T. Saeed, M. Saleem, S. Usmani, I. A. Malik, F. A. Al-Shammari and K. M. Deen, J. King Saud Univ., Sci., 2019, 31, 1344–1351 Search PubMed.
- Y. Kharbach, F. Z. Qachchachi, A. Haoudi, M. Tourabi, A. Zarrouk, C. Jama, L. O. Olasunkanmi, E. E. Ebenso and F. Bentiss, J. Mol. Liq., 2017, 246, 302–316 Search PubMed.
- N. V. Bondarev, K. P. Katin, V. B. Merinov, A. I. Kochaev, S. Kaya and M. M. Maslov, Phys. Status Solidi RRL, 2022, 16, 2100191 Search PubMed.
- S. Kaya, F. Siddique, D. O. Isin, K. P. Katin, V. Asati and A. Berisha, Res. Surf. Interfaces, 2024, 14, 100184 Search PubMed.
- G. Bahlakeh, A. Dehghani, B. Ramezanzadeh and M. Ramezanzadeh, J. Mol. Liq., 2019, 293, 111559 Search PubMed.
- C. Verma, H. Lgaz, D. K. Verma, E. E. Ebenso, I. Bahadur and M. A. Quraishi, J. Mol. Liq., 2018, 260, 99–120 Search PubMed.
- N. B. Iroha, V. C. Anadebe, N. J. Maduelosi, L. A. Nnanna, L. C. Isaiah, O. Dagdag, A. Berisha and E. E. Ebenso, Colloids Surf., A, 2023, 660, 130885 Search PubMed.
- F. El-Hajjaji, M. Messali, A. Aljuhani, M. R. Aouad, B. Hammouti, M. E. Belghiti, D. S. Chauhan and M. A. Quraishi, J. Mol. Liq., 2018, 249, 997–1008 Search PubMed.
- D. Douche, H. Elmsellem, E. H. Anouar, L. Guo, B. Hafez, B. Tüzün, A. El Louzi, K. Bougrin, K. Karrouchi and B. Himmi, J. Mol. Liq., 2020, 308, 113042 Search PubMed.
- J. Fang and J. Li, J. Mol. Struct.–THEOCHEM, 2002, 593, 179–185 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00835b |
| This journal is © The Royal Society of Chemistry 2025 |