
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vivo determination of analgesic and anti-inflammatory activities of isolated compounds from Cleome amblyocarpa and molecular modelling for the top active investigated compounds†
Mayada M. El-Ayouty‡
 a,
Nermeen A. Eltahawy‡b,
Ahmed M. Abd EL-sameaaa,
Ahmed M. Badawya,
Khaled M. Darwishc,
Sameh S. Elhadyde,
Mostafa M. Shokrf and
Safwat A. Ahmed*b
a,
Nermeen A. Eltahawy‡b,
Ahmed M. Abd EL-sameaaa,
Ahmed M. Badawya,
Khaled M. Darwishc,
Sameh S. Elhadyde,
Mostafa M. Shokrf and
Safwat A. Ahmed*b
aDepartment of Pharmacognosy, Faculty of Pharmacy, Sinai University, El-Arish 45511, Egypt. E-mail: miada.mohamed@su.edu.eg; ahmed.abdelsameaa@su.edu.eg; ahmed.badawy@su.edu.eg
bDepartment of Pharmacognosy, Faculty of Pharmacy, Suez Canal University, Ismailia 41522, Egypt. E-mail: Nermeenazmy25@gmail.com; nermeen.azmy@pharm.suez.edu.eg; safwat_aa@yahoo.com; safwat_ahmed@pharm.suez.edu.eg; Fax: +20-064-323074; Tel: +20-010-92638387
cDepartment of Medicinal Chemistry, Faculty of Pharmacy, Suez Canal University, Ismailia 41522, Egypt. E-mail: Khaled_darwish@pharm.suez.edu.eg
dKing Abdulaziz University Herbarium, Faculty of Science, King Abdulaziz University, Jeddah 21589, Saudi Arabia. E-mail: ssahmed@kau.edu.sa
eDepartment of Biological Sciences, Faculty of Science, King Abdulaziz University, Jeddah 21589, Saudi Arabia
fDepartment of Pharmacology and Toxicology, Faculty of Pharmacy, Sinai University – Arish Branch, Arish, 45511, Egypt. E-mail: mostafa.mohsen@su.edu.eg
First published on 5th August 2024
Abstract
Cleome amblyocarpa Barr. and Murb. from the family Cleomaceae is used in folk medicine as it has analgesic, anti-inflammatory, antibacterial and antioxidant activities. In this study, ten compounds from the whole plant of C. amblyocarpa, a wild plant that grows in the Sinai Peninsula of Egypt, were isolated. Six compounds, β-sitosterol 3-O-β-D-glucoside 2, calycopterin 5, rhamnocitrin 6, 17α-hydroxycabraleahy-droxylactone 7, cleogynol 8, and β-sitosterol 10 were first isolated from this species. In addition, four previously reported compounds, kaempferol-3, 7-dirhamnoside 1, 15α-acetoxycleomblynol A 3, and 11-α-acetylbrachy-carpone-22(23)-ene 4, as well as cleocarpanol 9, were isolated and identified. Isolated compounds were evaluated to determine their analgesic properties utilizing a hot-plate test method, and their anti-inflammatory effects utilizing rat paw edema. In a hot-plate test, compounds 3, 4, 7, 8, and 9 showed significant pain inhibition in latency time as compared to the normal group. Compounds 3–9 exhibited a significant inhibition of carrageenan-induced inflammation. According to the results of this work, compounds 3 and 4 (Dammarane triterpenoid) have the strongest analgesic/anti-inflammatory activity as compared to the other tested compounds. These results give support to the medicinal benefits of the plant as an analgesic along with an anti-inflammatory agent in traditional therapy. Molecular modelling studies of the isolated compounds 3 and 4 assessed the molecular affinity and binding interaction patterns for these compounds towards COX-2 as compared to specific COX-2 inhibitors and in relation to COX-1 isozyme. Compound 3 revealed extended accommodation across COX-2's hydrophobic sub-pockets and preferential thermodynamic stability across molecular dynamics simulations.
1. Introduction
Inflammation can be simply defined as the localized reaction of living mammalian tissues to damage.1 An inflammatory reaction can have a number of different components that might lead to tissue damage and related symptoms, including granuloma development, leukocyte infiltration, and edema. However, it serves as a defense mechanism.2 Multiple reactions can be triggered or exacerbated by the intricate processes and mediators associated with an inflammatory response.3An important class of pharmaceuticals that have numerous medicinal applications in the management of different cases including fever, inflammation, and pain are called non-steroidal anti-inflammatory drugs (NSAIDs).4
According to different reports, the generation of prostaglandins via arachidonic acid by the usage of non-selective suppression of the COX cyclooxygenase enzymes (COX-1 and COX-2),5 leading to down regulation of inflammatory mediators that consist of tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β) alongside with glutathione (GSH) which is an antioxidant parameter, is the mechanism behind NSAID-associated gastrointestinal side effects.6
The World Health Organization reports that between 70 and 80% of people in the developing world receive their primary care through complementary medicine, primarily from herbal sources.7 Anti-inflammatory medications from natural sources are used to treat pain from a number of illnesses, such as muscular diseases, and arthritis.8 There are many inflammatory disorders, and controlling their occurrence and outcome is a challenge for conventional medication treatments.9 As many plant extracts and isolates are proved to have anti-inflammatory and analgesic activities,10,11 the purpose of the current study was to assess the anti-inflammatory and analgesic activities of compounds isolated from a natural source.
As of right now, Cleomaceae family includes 25 genera, with more to be described.12 Over 200 species of Cleome can be recognized in various regions of the world. It belongs to the largest genus within the family and possesses numerous conventional and medicinal uses.13 in traditional medicine, Cleome species are utilized as analgesic, anti-inflammatory, antibacterial, and antioxidant agents.14
C. amblyocarpa Barr. & Murb. is a species of herbaceous plant that belongs to the Cleomaceae family.15 The plant, which has offensive odor and can be annual or short-lived perennial, is found in North and East Africa, Sinai, Ethiopia, Sudan, Saudi Arabia, Palestine, Iran, and Iraq.16
Many bioactive substances have been isolated from C. amblyocarpa, such as triterpenoids,17 flavonoids, and saponins.18 Consequently, many biological activities of this plant have been described, such as anti-leishmanial,19 antifungal,20 cytotoxic and antioxidant.21 According to Khlifi et al.22 the leaves of C. amblyocarpa were proved to have analgesic and anti-inflammatory activities and could be used as a source of phytochemicals which may be utilized for the development of novel pharmaceutical combinations. As a result, in our study, we focused on isolating the active components from their total MeOH extract of the whole plant of C. ambylocarpa and the identification of these isolated compounds using several spectroscopic techniques. The isolated compounds (3–9) were screened for their analgesic, and anti-inflammatory effects. The current research has the purpose to examine the influence of isolated compounds (3–9) on the inflammation caused by carrageenan in albino-rats, and the nociception caused by hot-plate in mice. Additionally, molecular modelling studies have been carried out to improve a better understanding about the molecular mechanism behind the activity of the isolated compounds 3 and 4 (Dammarane triterpenoid).
2. Experimental
2.1 Extraction, isolation and identification of isolated compounds from C. amblyocarpa
In this study, four fractions (50% EtOAc in hexane), (75% EtOAc in hexane), (95% EtOAc in hexane) and (95% EtOAc in MeOH) were subjected to further chromatographic study according to their promising TLC.
2.1.3.1 Isolation of secondary metabolites of the fraction F-3 (50% EtOAc in hexane). 24.5 g of the fraction F3 (50% EtOAc in hexane) was applied at the top of the stationary phase of a silica gel column (3 × 120 cm). First, n-hexane was used for elution, then gradient systems of n-hexane and EtOAc. Two promising subfractions were subjected to additional purification:
Subfraction (F3-A) (20% EtOAc in hexane) 8 g was re-chromatographed with gradient elution with a normal phase silica gel 60 Å column (230–400 mesh, Merck, KGaA, Darmstadt, Germany) utilizing (n-hexane/EtOAc). The eluate was gathered in 5 ml of successive fractions. This resulted in obtaining one pure compound 10 (25 mg).
Subfraction (F3-B) (30% EtOAc in hexane) 10 g was further purified using Sephadex LH-20 column with (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture of CHCl3 and MeOH to isolate two pure substances, were compound 9 (50 mg), and compound 8 (40 mg).
:1) mixture of CHCl3 and MeOH to isolate two pure substances, were compound 9 (50 mg), and compound 8 (40 mg).
2.1.3.2 Isolation of secondary metabolites of the fraction F-4 (75% EtOAc in hexane). 18 g of the fraction F4 (75% EtOAc in hexane) was adsorbed on silica gel powder and put towards the top of the stationary phase using the silica gel column (3 × 120 cm). The initial elution was conducted utilizing gradient systems of (n-hexane/EtOAc). Two promising sub-fractions were obtained:
Sub-fraction (F4-A) (40% EtOAc in hexane) 8 g was submitted to further purification using a MeOH:CHCl3 (1:1) over a Sephadex LH-20 column to give two pure substances, were compound 7 (30 mg), and compound 6 (25 mg).
Sub-fraction (F4-B) (50% EtOAc in hexane) 6 g was re-chromatographed via gradient elution over a normal phase silica gel column with (n-hexane/EtOAc). Sub-fractions with matching TLC patterns were mixed, yielding one pure compound 5 (30 mg).
2.1.3.3 Isolation of secondary metabolites of the fraction F-5 (95% EtOAc in hexane). 15 g of the fraction F5 (95% EtOAc in hexane) was adsorbed on silica gel powder and put towards the top of the stationary phase using the silica gel column (3 × 120 cm). The initial elution was conducted utilizing gradient systems of (n-hexane/EtOAc/MeOH). Two promising sub-fractions were obtained:
Sub-fraction (F5-A) (60% EtOAc in hexane) 7 g was re-chromatographed on a normal phase silica gel column via gradient elution (n-hexane/EtOAc/MeOH). Subfractions with identical TLC patterns were mixed, yielding one pure compound 4 (40 mg).
Sub-fraction (F5-B) (70% EtOAc in hexane) 6 g was submitted to further purification using a MeOH:CHCl3 (1:1) over a Sephadex LH-20 column to produce a single pure compound 3 (30 mg).
2.1.3.4 Isolation of secondary metabolites of the fraction F-6 (95% EtOAc in MeOH). 12 g of the fraction F6 (95% EtOAc in MeOH) was adsorbed on the silica gel and put towards the top of the stationary phase of a silica gel column (3 × 120 cm). The initial elution was conducted utilizing gradient systems of (n-hexane/EtOAc/MeOH). Two sub-fractions promising were obtained:
Sub-fraction (F6-A) (90% EtOAc in hexane) 5 g was re-chromatographed on a normal phase silica gel column via gradient elution (n-hexane/EtOAc/MeOH). Subfractions with identical TLC patterns were mixed, obtaining one pure compound 2 (30 mg).
Subfraction (F6-B) (97.5% EtOAc in MeOH) 4 g was submitted to further purification using a MeOH:CHCl3 (1:1) over a Sephadex LH-20 column to produce a single pure compound 1 (20 mg).
2.2 In vivo anti-inflammatory study for isolated compounds from C. amblyocarpa
The animals were kept under normal environments, which included a 12 hour light–dark cycle, a temperature of 25 ± 2 °C and a relative humidity of 55 ± 5%. The animals had free access to water and a regular diet.
Suez Canal University's ethical committee (No. 202205M3) approved the experimental investigation. The Committee for the Purpose of Control and Supervision of Experimental Animals (CPCSEA) guidelines were followed and was authorized by the Institutional Animal Ethical Committee (IAEC). Also, the study was performed in conformance with the Guide for Laboratory Animals the Care and Use [NIH Publication, No. 8023, revised (1996)]. The experimental research was performed at Faculty of Pharmacy, Sinai University, Egypt.
:9, v/w). Subsequently, the supernatants were eliminated, and the levels of TNF-α, GSH, and COX-2 were measured using ELISA technique.27The other inflammatory mediator (IL-1β) was determined using RT-PCR technique and the primer sequence used for evaluation of IL-1β by RT-PCR (catalogue no. RP300022, Sinobiological, USA) is Forward: ATGGCAACTGTCCCTGAACT, Reverse: AGTGACACTGCCTTCCTGAA, using GADPH as the standard gene, following the manufacturer's instructions.28
2.3 Molecular modelling investigation
Top-active investigated triterpenoids, compounds 3 and 4, were 3D-constructed as well as COX-2 (PDB: 3mqe) COX-1 (PDB: 6y3c) were prepared, and docking protocol was done via Auto Dock Vina V.1.2.0 (Scripps Research, La Jolla, CA, United States) as per reported studies.29,30 Binding site was defined as endorsing co-crystallized ligand, 2H-chromene-3-carboxylic acid derivative, and also refined to include key binding residues for small molecule ligands.31 Docking was proceeded under Vina Forcefield and Lamarckian_Genetics with biological target center as the docking box center.32 Global search exhaustiveness 100 kcal mol−1 and poses' maximum energy differences 3 kcal mol−1 were set.33 Visualizing poses and compound/COX-2 binding interactions were performed via PyMol V2.0.6 (Schrödinger, NY, USA).3 Results and discussion
3.1 Extraction, isolation and identification of active constituent from C. amblyocarpa
By using column chromatographic isolation techniques, ten compounds were isolated of MeOH extract of C. amblyocarpa which were identified as kaempferol-3,7-dirhamnoside 1, β-sitosterol, 3-O-β-D-glucoside 2, 15α-acetoxycleomblynol A 3, 11-α-acetylbrachy-carpone-22(23)-ene 4, calycopterin 5, rhamnocitrin 6, 17α-hydroxycabraleahy-droxylactone 7, cleogynol 8, cleocarpanol 9, and β-sitosterol 10.Compound 1: Yellow needles; Rf = 0.52 (87:13 CHCl3/MeOH); MP = 203 °C; LC-MS/MS (m/z: 579.1666 [M + H]+.34 It was identified as kaempferol-3, 7-dirhamnoside (Fig. 1) by comparing 1H-NMR (300 MHz; DMSO-d6) and 13C-NMR (75 MHz; DMSO-d6) analyses with the spectral published data,34 see (Table S.1†).
 | ||
| Fig. 1 Molecular structure of isolated compounds 1–10. | ||
Compound 2: Pale white powder; Rf = 0.43 (90:10 CHCl3/MeOH); MP = 283 °C; it was identified as β-sitosterol 3-O-β-D-glucoside (Fig. 1) by comparing 1H-NMR (400 MHz; DMSO-d6) and 13C-NMR (100 MHz; DMSO-d6) analyses with the spectral published data,35 see (Table S.2†).
Compound 3: white powder; Rf = 0.52 (80:20 EtOAc/n-hexane); MP = 259.5–262 °C; it was identified as 15α-acetoxycleomblynol A (Fig. 1) by comparing 1H-NMR (300 MHz; CDCl3-CD3OD) and 13C-NMR (75 MHz; CDCl3-CD3OD) analyses with the spectral published data,17 see (Table S.3†).
Compound 4: white powder; Rf = 0.45 (60:40 EtOAc/n-hexane); MP = 269–273 °C; it was identified as 11-α-acetylbrachy-carpone-22(23)-ene (Fig. 1) by comparing 1H-NMR (300 MHz; CDCl3) and 13C-NMR (75 MHz; CDCl3) analyses with the spectral published data,36 see (Table S.4†).
Compound 5: crystalline yellow solid; Rf = 0.52 (50:50 EtOAc/n-hexane); MP = 223–225 °C; It was identified as calycopterin (Fig. 1) by comparing 1H-NMR (300 MHz; CDCl3-CD3OD) and 13C-NMR (75 MHz; CDCl3-CD3OD) analyses with the spectral published data,36 see (Table S.5†).
Compound 6: yellow solid; Rf = 0.43 (50:50 EtOAc/n-hexane); MP = 214–216 °C; EI-MS (m/z: 299.97);37 UV spectrum (MeOH, λmax nm): 269; 349, plus AcONa: 272; 354 confirms the absence of OH in C7, plus AlCl3 276 and 391 OH free in C5 and C3 (ref. 38); it was identified as rhamnocitrin (Fig. 1) by comparing 1H-NMR (300 MHz; CD3OD) analyses with the spectral published data,37 see (Table S.6†).
Compound 7: white powder; Rf = 0.45 (50:50 EtOAc/n-hexane); MP = 250–252 °C; LC-MS/MS (m/z: 433.3283 [M + H]+;39 it was identified as 17α-hydroxycabraleahy-droxylactone (Fig. 1) by comparing 1H-NMR (300 MHz; CDCl3) and 13C-NMR (75 MHz; CDCl3) analyses with the spectral published data,39 see (Table S.7†).
Compound 8: colorless crystals; Rf = 0.43 (35:65 EtOAc/n-hexane); MP = 108–110 °C; LC-MS/MS (m/z: 475.3754 [M + H]+; It was identified as cleogynol (Fig. 1) by comparing 1H-NMR (300 MHz; CDCl3) and 13C-NMR (75 MHz; CDCl3) analyses with the spectral published data,39,40 see (Table S.8†).
Compound 9: white powder; Rf = 0.39 (30:70 EtOAc/n-hexane); MP = 251 °C; EI-MS m/z: 474 [M]+, 456 [M − H2O]+, 441 [456-Me]+, 410, 370, 143, 142 (100), and 85;41 it was identified as cleocarpanol (Fig. 1) by comparing 1H-NMR (300 MHz; CDCl3) and 13C-NMR (75 MHz; CDCl3) analyses with the spectral published data,39,41 see (Table S.9†).
Compound 10: white powder; Rf = 0.41 (20:80 EtOAc/n-hexane); MP = 136 °C; It was identified as β-sitosterol (Fig. 1) by comparing 1H-NMR (300 MHz; CDCl3) and 13C-NMR (75 MHz; CDCl3) analyses with the spectral published data,36 see (Table S.10†).
3.2 In vivo anti-inflammatory study for isolated compounds from C. amblyocarpa
 | ||
| Fig. 2 Hot plate test results. Analgesic activity of indomethacin and the tested compounds on hot plate. Means ± SEM (n = 6/group), p ≤ 0.0001, are used to express the data (a) comparison to the normal group. One-way ANOVA and Tukey's post hoc test for numerous comparisons were used in the statistical analysis, which was conducted using GraphPad Prism. | ||
 | ||
| Fig. 3 Rat paw thickness results (A); the anti-inflammatory effect of indomethacin and the tested compounds against carrageenan induced paw edema. Data are expressed as means ± SEM (n = 6/group), p ≤ 0.0001. Statistical analysis was carried out by using GraphPad prism, two-way ANOVA followed by Dunnett post hoc test for multiple comparison. (B) Different photographs demonstrating the rat paw thickness in different groups. A Normal group, B; carrageenan group, C; indomethacin + carrageenan, D; compound 3 + carrageenan, E; compound 4 + carrageenan, F; compound 5 + carrageenan, G; compound 6 + carrageenan, H; compound 7 + carrageenan, I; compound 8 + carrageenan, J; compound 9 + carrageenan. | ||
3.2.2.1 The Effect of indomethacin and the tested compounds on the inflammatory mediators (IL-1β and TNF-α). The results obtained from RT-PCR data on mRNA expression of IL-1β showed that carrageenan injected group showed a considerable buildup in expression in comparison to normal group by 1.6-fold, while indomethacin treated group showed a significant downregulation by 51.1% in comparison to carrageenan treated group. In the same line, the tested compounds (compounds 3–9) showed a significant reduction by (47.7%, 45.4%, 29.7%, 33.5%, 42.1%, 45%, and 41.8%) in comparison to carrageenan injected group, respectively (Fig. 4A).
 | ||
| Fig. 4 The effect of indomethacin and the tested compounds on the inflammatory mediators (IL-1β and TNF-α). (A) Effect of indomethacin and the tested compounds on pro-inflammatory mediator IL-1β, (B); effect of indomethacin and the tested compounds on pro-inflammatory mediator TNF-α. Data are expressed as means ± SEM (n = 6/group), p ≤ 0.0001. (a) As compared to normal group, (b) as compared to carrageenan injected group. Statistical analysis was carried out by using GraphPad prism, one-way ANOVA followed by Tukey's post hoc test for multiple comparison. | ||
The results obtained from ELISA technique demonstrated a significant increase of TNF-α levels in carrageenan treated group by 75.6% in comparison to normal group, while indomethacin treated group showed a significant reduction by 33.9% in comparison to carrageenan treated group. In the same line, the tested compounds (compounds 3–9) showed a significant reduction by (23.4%, 22.3%, 14.6%, 18.6%, 22.3%, 22.3%, and 21.9%) in comparison to carrageenan injected group, respectively (Fig. 4B).
3.2.2.2 The Effect of indomethacin and the tested compounds on COX-2 and GSH. The results obtained showed that carrageenan injected group had a noteworthy increase in the levels of COX-2 as compared to normal group by 79.9%, while indomethacin treated group showed a significant reduction by 34.9% in comparison to carrageenan treated group. In the same line, the tested compounds (compounds 3–9) showed a significant reduction by (30.1%, 29.6%, 11.2%, 14.9%, 25.4%, 28.4%, and 24.9%) in comparison to carrageenan injected group, respectively (Fig. 5A).
 | ||
| Fig. 5 The effect of indomethacin and the tested compounds on COX-2 and GSH (A); effect of indomethacin and the tested compounds on COX-2, (B); effect of indomethacin and the tested compounds on GSH. Data are expressed as means ± SEM (n = 6/group), p ≤ 0.0001. a as compared to normal group, b as compared to carrageenan injected group. Statistical analysis was carried out by using GraphPad prism, one-way ANOVA followed by Tukey's post hoc test for multiple comparison. | ||
And the results obtained showed that carrageenan injected group had a considerable downregulation in the levels of GSH by 42.9% as compared to normal group, while indomethacin treated group showed a significant increase by 68.4% as compared to carrageenan treated group. In the same line, the tested compounds (compounds 3–9) showed a significant reduction by (47.4%, 45.9%, 37.8%, 44.6%, 33.7%, 34.1%, and 37.1%) as compared to carrageenan injected group, respectively (Fig. 5B).
3.3 Triterpenoids derivatives showed significant molecular affinity to COX-2 catalytic site
For gaining further insights regarding the molecular aspects for the anti-inflammatory activity of the top-active triterpenoids, compounds 3 and 4, a computational approach has been adopted. Molecular docking simulation for compounds 3 and 4 was performed to assess the molecular affinity and binding interaction patterns for these compounds towards COX-2, the key mediator of pro-inflammatory activities.42 The later would provide valuable physicochemical requirements in terms of efficient interaction for these compounds to possess potential inhibitory activity against COX-2 biotarget. Typically, COX-2 coexist as homo-A-dimer comprising of ∼580 amino acid sequence with each monomeric unit at ∼70 kDa in size.43,44 The protein's tertiary architecture harbors triple structural domains; (i) carboxy-terminus catalytic domain comprising the protein bulk, (ii) membrane-bound α-helical domain, and (iii) amino terminal epidermal growth factor domain (Fig. 6A).45 The catalytic/cyclooxygenase domain encompasses the deep wide substrate binding pocket being highly hydrophobic in nature mediating relevant van der Waals interactions via its non-polar lining residues; Leu338, Phe367, Leu370, Tyr371, Trp373, Met508, Gly512, and Ala513 (Pocket-I). Later residues, in addition to a second vicinal hydrophobic pocket-II (Val102, Val335, Tyr341, Leu345, and Leu517), have been reported important for binding of several small molecule COX-2 inhibitors.46–49 Presence of less sterically hindered residue Val509 and outward orientation of Phe504 provide access to the hydrophilic selectivity side pocket with Arg499 at its base to provide preferential polar (H-bond) anchoring of sulphonamide/methyl sulphone-based scaffolds of diaryl heterocyclic COX-2 selective inhibitors.43 Typical sulphonamide COX-2 inhibitors show preferential polar interaction with lining residues; Arg499/Gln178 side chains and Leu338/Ser339 main chains.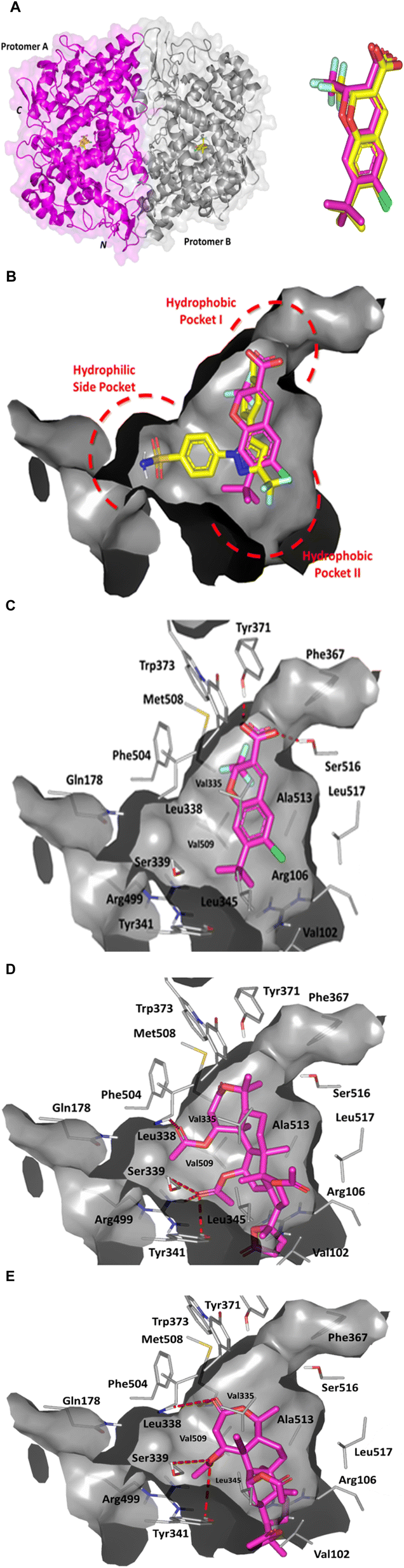 | ||
| Fig. 6 Architecture of COX-2 enzyme and depicted molecular docking poses. (A) Left panel; cartoon/surface 3D-representation of COX-2 enzyme (PDB: 3mqe) showing the dimeric structure bounded to two co-crystallized (chromene; yellow sticks). Bold C and N letters denote the carboxy and amino-terminals. Right panel; aligned redocked chromene reference control (magenta) over its co-crystalline state (yellow). (B) Superimposed redocked chromene (magenta) over celecoxib (yellow) sticks as co-crystalline ligands within the COX-2 binding pockets (PDB: 3mqe and 3ln1, respectively). Right panel: (C) predicted binding mode of chromene co-crystallized ligand as reference ligand at COX-2 binding site. (D and E) Binding modes of compound 3 (D) and compound 4 (E) at COX-2 binding site. Only surrounding residues within 5 Å radius as lines are shown and polar interactions are illustrated as red-dash lines. | ||
The COX-2 protein (PDB: 3mqe) employed within our docking simulation harbors the co-crystallized structure of 2H-chromene-3-carboxylic acid derivative (D72) within the protein's canonical binding site. The co-crystallized compound was developed by Pfizer Global R&D and introduced as orally active long acting (t1/2 = 34 h) benzopyran-based compounds with great COX-2 selectivity (IC50 = 0.062 μM at COX-2 versus 1.02 μM at COX-1). Further, the reported compound was illustrated with relevant pharmacokinetic profile besides superior air pouch model efficacy and advanced chronic/acute in vivo studies including edema, hyperalgesia, and arthritis animal models. Despite lacking the methyl sulphone/sulphonamide-based scaffold and typical diaryl heterocyclic skeleton, this chromene-based compound depicted superior anti-inflammatory actions over NSAIDs and other selective COX-2 inhibitors under clinical investigation. Adopting the chromene-COX-2 complex as positive reference control within our computational simulation was highly rationalized since our compounds similarly lacked the sulphone-based moiety. The later would predict a differential binding orientation/conformation at COX-2 pocket for our compounds in relation to typical sulphonamide COX-2 inhibitors the thing that was confirmed with the chromene compound. Moreover, our investigated triterpenoids illustrated comparable pharmacophoric features, to a certain extent, with those of the chromene reference compound. Starting with the carboxylate groups being incorporated within both structures and ending with the hydrophobic core units, the chromene and triterpenoid rings. In these regards, guiding the docking algorithm for the compounds exhibiting relevant structural closeness with the co-crystallized ligand would provide validity for the docking findings. This has been rationalized since a guided docking approach would increase the likeness of predicting the compounds' binding mode at a significant biological relevance.50
Further validation for our docking approach was highlighted through the successful achievement of great superimposition for the redocked positive chromene control over its co-crystalline reference orientation and conformation states (aligned root-mean square; RMSD = 0.52 Å) (Fig. 6A). Being able to replicate the binding mode of the co-crystallized ligand at RMSD below 2.00 Å would generally confer the validity of the docking procedure/protocol to obtain both the binding energies and modes within high predicted biological significance.51–56 Redocked chromene reference standard showed relevant anchoring at the COX-2 binding site with partial accommodation of the whole wide pocket owing to the almost linear conformation of its chromene central scaffold and substituted functional groups (COOH, CH3, and chloride) [Fig. 6B and C]. The ligand further lacked relevant anchoring into the COX-2's hydrophilic selectivity side pocket (unlike selective COX-2 inhibitor, celecoxib; IC50 = 40 nM.57 Stability of chromene ring was highlighted through polar interaction with Tyr371 and Ser516 (H-bond distance/angle = 2.0 Å/157.9° and 2.2 Å/133.0°, respectively). Substituted chloride atom depicted preferential proximity towards the Arg at the other side of the pocket. Close hydrophobic contacts (<4.5 Å) with both hydrophobic pockets (I and II) were also depicted important for chromene/COX-2 stability. To our delight, the furnished chromene's pharmacodynamic binding profile has successfully replicated the reported ligand-residue patterns the thing that would rationalize the furnished docking binding energy (−10.97 kcal mol−1) being highly correlated to chromene's reported in vitro results (IC50 = 0.062 μM).
Molecular docking of the investigated triterpenoid-based compounds revealed interesting findings. Both compounds revealed extended accommodation across both COX-2's hydrophobic pockets with their 5-membered lactone rings being anchored at the hydrophobic pocket II (Fig. 6C and D). Compound 3 showed partial accommodation of the enzyme's selectivity side pocket where its C1 acetyl side arm showed preferential orientation into this hydrophilic pocket mediating good hydrogen bonding with Ile338 mainchain (3.1 Å/127.0°). Further ligand stability was mediated via C12 acetyl substitution through extended polar contacts with the sidechains of the selectivity pocket's gatekeeper and comprising residues; Ser339 (3.0 Å/125.6°), Tyr341 (3.0 Å/128.2°), and Arg499 (2.6 Å/141.1°). On the other hand, compound 4 could not achieve relevant anchoring at the enzyme's selectivity pocket. However, the compound's C1 acetyl side chain predicted several polar contacts with the pocket's gatekeeper and vicinal residues; Ser339 (2.8 Å/126.8°) and Tyr341 (2.9 Å/128.1°). Further stability of the compound was driven via its Ring A lactone carbonyl group towards both Met508 (3.0 Å/126.4°) and Ile338 (2.6 Å/155.3°) mainchains. The differentially depicted orientation/conformation between both triterpenoid-based compounds could be reasoned for the different number of the incorporated acetyl sidechain. Having three acetyl sidechains, one being extra on C16 of the triterpenoid ring, compound 3 exhibited relevant steric clashes at the hydrophobic pocket II. This would have forced the compound to anchor further towards the hydrophobic I where the two other acetyl arms being flexed with C1 acetyl sidechain being anchored into the enzyme's selectivity pocket. Contrarily, compound 4 lacked C16 acetyl arm the thing that made the compound to free accommodate the enzyme's second hydrophobic pocket (site-II). The later was suggested to cause the rest of the compound to be partially retracted from an anchoring at pocket-I the thing that was translated into minimal binding at the selectivity side pocket. The more extended polar contacts and favored docking of compound 3 towards the three COX-2 pockets over compound 4 was translated into higher docking binding energy for the earlier ligand (−12.60 kcal mol−1 versus −10.44 kcal mol−1).
Preferential affinity of the investigated compounds towards COX-2 over its constitutional isoform, COX-1, was suggested relying on the compound structures and enzyme's differential pocket architecture. Typically, COX isozymes were reported with more than 60% amino acid sequence similarity mediating comparable mechanisms of arachidonic acid metabolic pathway.58,59 Nonetheless, significant structural difference between the isozymes' pockets based on their respective lining residues have been highlighted to mediate inhibitor specificity.59,60 Presence of the secondary side pocket has been reported to significantly contribute to a larger inhibitor binding cavity (almost 25%) at COX-2 as compared to COX-1.60 The amino acid variation at the secondary pocket-II border (Val509 in COX-2 versus bulkier Ile509 in COX-1) makes this side pocket only accessible for inhibitor binding at COX-2. Moreover, the central channel of the binding pocket shows nearly 17% bigger size at COX-2 in relation to its isozyme (Fig. 7A).61 The later has been correlated to other key residue substitutions, including His499 → Arg and Ile420 → Val in COX-2 at the active site and secondary shell. Further altered orientation/conformation of the COX-2 membrane binding domain terminal α-helix (Helix D) caused significant Arg106 shift mediating larger solvent accessible surface area (SASA) at the active site.62
 | ||
| Fig. 7 Comparative binding site analysis for COX isozymes predicting differential molecular docking findings. (A) Upper panel; overlaid surface representation binding sites of COX isozymes showing differential orientation/conformation of key pocket residues (COX-2 red lines; COX-1 green lines) contributing to the shape and size of the binding site at each target enzyme. Terminal α-helix of the membrane binding domain (Helix D) is shown as gray cartoon. Lower panel; side-to-side comparison between the shape and size of the isozyme binding sites. COX-2 showed wider channel and extra accessible secondary pocket (hydrophobic pocket-II) being fundamental for inhibitor specificity. (B and C) Binding modes of compound 3 (B) and compound 4 (C) at COX-1 binding site. Compounds are demonstrated as magenta sticks and binding site is shown as gray-green surface representation. | ||
Owing to the large and extended architecture of our investigated compounds, it is postulated that lower affinity towards COX-1 binding would be depicted. As expected, molecular docking simulation for our compounds at COX-1 pocket was conducted to comparatively assess the compounds' binding affinity across both inflammatory isozymes. No deep anchoring was depicted for both compounds at COX-1 binding site (Fig. 7B). Both docked ligands illustrated high solvent exposure near the pocket entrance lacking relevant contacts with the key binding residues reported crucial for enzyme inhibition. All have been translated into fair docking scores at COX-1 (−5.54 kcal mol−1 and −4.03 kcal mol−1 for compounds 3 and compound 4, respectively) the thing that would confer trivial ligand-target binding affinities and stabilities.
Further docking validation and top-active compound thermodynamic stability evaluation at COX-2 active site were proceeded through explicit molecular dynamics simulations as compared to reference selective COX-2 inhibitors, chromene and celecoxib. The adopted investigation has been considered relevant to provide near-physiological analysis of the compound-COX-2 stability and time-evolution conformation changes at more accurate context than any sophisticated mechanics energy minimization and flexible docking approaches.63 Exploring the root mean-squared deviations (RMSDs) of the protein Cα-atoms showed higher values/fluctuations for the apo-unliganded COX-2 in relation to its ligand-bound states across the time plateau from 50 to 100 ns (Fig. 8A). It has been agreed that low steady RMSDs confer significant conformational stability,64 that is why obtained RMSDs would confer protein's stability upon ligand binding. Notably, all simulated proteins showed typical thermodynamic behavior since RMSDs increased gradually owing to relaxation and removal of all constrains till RMSDs reached an equilibrium and respective convergence for more than half the simulation run redeeming no further simulation time extensions.65,66
 | ||
| Fig. 8 Molecular dynamics stability analysis of compound-COX-2 complexes. (A) COX-2 Cα-atom RMSDs; (B) compound RMSDs; (C) overlaid conformation/orientation of simulated complexes at start and final extracted frames. COX-2 (cartoon) and compounds (sticks) are colored with respect to extracted frame (start → green and final → red). (D) Cα-atom ΔRMSF trajectories as per amino acid sequence (amino-terminus Ala18–Gln569 carboxy-terminus). | ||
Great COX-2 pocket accommodation was highlighted almost for all of the simulated compounds as depicted throughout monitoring the RMSDs of bounded ligands in reference to their respective initial orientation at the protein pocket Cα-atoms (Fig. 8B). Higher pocket accommodation with minimal conformation changes were set for the two control molecules, chromene and celecoxib, followed by compound 3. Contrarily, compound 4 depicted quite conformation/orientation shift as seen with its higher RMSD tones. Differential ligand's pocket accommodations were further highlighted through the overlaid snapshots at the start and end of the simulation runs (Fig. 8C).
Exploring the residue-wise stability of the simulated proteins was further done through estimating the RMS Fluctuation (RMSF) as indicative to the mobility and fluctuating profiles of the constituting amino acids.67 Further RMSF analytical tool allow to pinpoint the residue being impacted by the compound's anchoring.68,69 Difference RMSFs have been reported as better estimations for protein's local flexibility where ΔRMSFs = apo's RMSFs – holo's RMSFs. Adopting a ΔRMSF threshold of 0.30 Å is generally significant for considering residues with higher values (>0.30) to be of limited flexibility.70 Interestingly, all simulated COX-2 enzymes showed typical thermodynamic behaviours since terminal residues were assigned with higher mobility as compared to the protein's core amino acids which has been coherent with reported data and B-factor analysis.71,72 The estimated ΔRMSF for the simulated proteins were mostly entirely of positive values with several residues regions depicting significant stability and rigidity profile (Fig. 8D). Almost the entire catalytic domain (Ile111–Gln569) exhibited positive-value ΔRMSF tones, except for limited regions around Gly150, Leu210, and Ile503 residues. To our interest, the membrane-bound domain residues (Thr56–Leu109) were assigned with the highest stability/immobility characteristics with ΔRMSF values up to 2.70 Å. The latter thermodynamic behavior further highlights the significant stabilizing impact of ligand binding on the global and secondary COX-2 protein structure being extended far from the canonical binding site. On the contrarily, the epidermal growth factor domain (Ala18–Thr55) showed high mobility patterns with ΔRMSFs of negative values. This could be reasoned to the inherited flexibility of this peculiar domain comprising of lower intramolecular hydrogen bonding between just two anti-parallel β-sheet and singular short α-helix.71
Trajectory-driven Molecular Mechanics_Poisson Boltzmann Surface Area (MM_PBSA) binding-free energy calculation was used to estimate the compounds' affinity towards COX-2 pocket. MM_PBSA is generally of comparable accuracy to the Free-Energy Perturbation techniques, yet with much reduced computational costs.73,74 Calculated total binding energies (ΔGTotal) were depicted at high negative-values (better affinity) for compound 3 and celecoxib (−121.75 ± 14.61 kJ mol−1 and −127.74 ± 18.74 kJ mol−1) followed by that of chromene (−114.23 ± 29.04 kJ mol−1), whereas compound 4 was at fair value (−127.74 ± 15.16 kJ mol−1) (Fig. 9A). Preferential affinity patterns for compound 3 were in great translation for the above described RMSDs data and conformational analysis. Dissecting ΔGTotal demonstrated preferential energy contributions for the non-polar van der Waal (ΔGvdW) energies over the polar/electrostatic (ΔGelectrostatic) potentials for both isolated compounds and celecoxib owing to the hydrophobic cage-like triterpenoid skeleton and triple aromatic structure of these respective compounds the thing being complementary to COX-2 non-polar sub-pockets. Contrarily, chromene reference compound showed higher ΔGelectrostatic energy contribution being highly related to its ionizable carboxylic group. Nonetheless, chromene's high-negative electrostatic contribution was further associated with much greater polar solvation penalties (189.89 ± 30.02 kJ mol−1) the thing that might disfavour ligand's anchoring since binding process is a solvent-displacement approach.75 On the other hand, the incorporation of chromene aromatic scaffold could partially compensate for the predicted solvation entropies through mediating high hydrophobic potentials and so the furnished reasonable ΔGTotal.
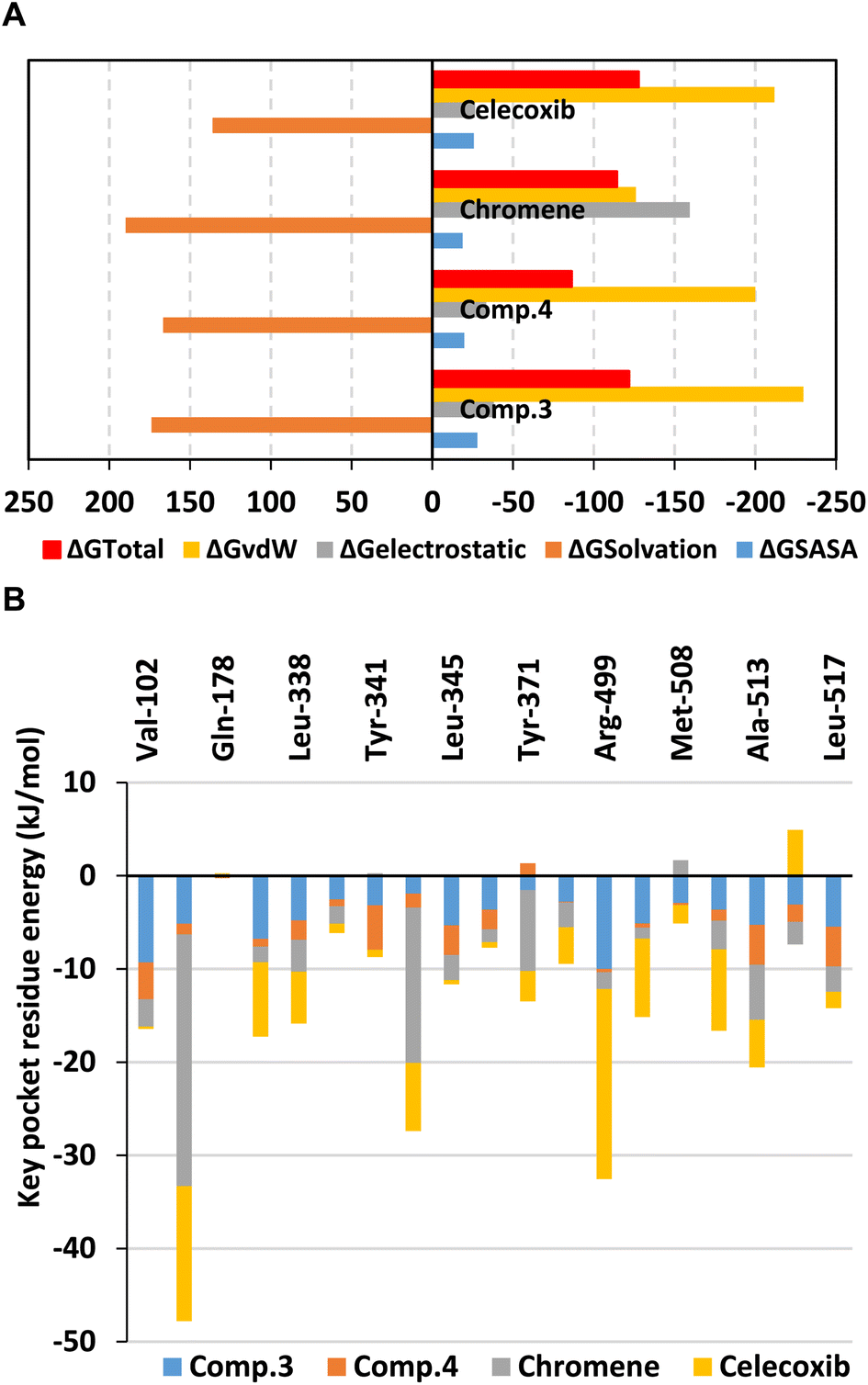 | ||
| Fig. 9 Binding-free energy MM_PBSA calculations for compound-COX-2 complexes. (A) Total free binding energies with their contributing energy terms; (B) residue-directed energy contributions. | ||
Significant residue-wise energy contributions were highlighted to several non-polar pocket residues including Val102, Val335, Leu338, Tyr371, Phe504, Val509, Ala513, and Leu517. Polar amino acids such as Arg106 and Lys344 were depicted significant for chromene binding whereas Arg499 has been highly associated with that of celecoxib (Fig. 9B). Relying on the depicted MM_PBSA energy contributions, it would be beneficial to introduce polar yet lower ionized functional group at our investigated compounds for prospective lead optimization and development. Such strategy would satisfy the high COX-2's pocket hydrophobic nature while maintaining favoured affinity to key polar amino acids through reduced solvation penalty and suggested improved pharmacokinetics.76 Exemplary for such functionalities could be the carboxylate isosteres including the tetrazole ring.77,78
4 Conclusion
In the present investigation, the phytochemical examination of the MeOH extract of C. amblyocarpa resulted in the isolation of six compounds, β-sitosterol glucopyranoside 2, calycopterin 5, rhamnocitrin 6, 17α-hydroxycabraleahy-droxylactone 7, cleogynol 8, and β-sitosterol 10 for the first time from this species. Additionally, four previously reported compounds, Kaempferol-3, 7-dirhamnoside 1, 15α-acetoxycleomblynol A 3, 11-α-acetylbrachy-carpone-22(23)-ene 4, and cleocarpanol 9, were isolated. Based on the given data and the statistical analyses, it was proved that compound 3 and 4 has the strongest analgesic/anti-inflammatory activities as compared to the other tested compounds. Molecular modelling investigation of top-active isolated compounds 3 and 4 highlighted the promising affinity and selective binding interaction patterns for the earlier compound towards COX-2 showing extended accommodation across COX-2's hydrophobic pockets compared to celecoxib.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization, S. A. A., M. M. E., N. A. E., A. M. A.; methodology, S. A. A., M. M. E., N. A. E., A. M. A., A. M. B., M. M. S., S. S. E.; supervision, S. A. A., M. M. E., N. A. E., data curation, S. A. A., M. M. E., N. A. E., A. M. A., K. M. D., S. S. E., M. M. S.; software, K. M. D., S. S. E.; resources, S. A. A.; writing – original draft, S. A. A., M. M. E., N. A. E., A. M. A., K. M. D., S. S. E., M. M. S.; writing, review, and editing, all authors. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The simulations in this work were performed at King Abdulaziz University's High Performance Computing Center (Aziz Supercomputer) (https://hpc.kau.edu.sa, accessed on 1 July 2024). The authors, therefore, acknowledge with thanks the center for technical support.Notes and references
- X. Dai, M. Ding, W. Zhang, Z. Xuan, J. Liang, D. Yang, Q. Zhang, B. Su, H. Zhu and X. M. Jia, Int. J. Exp. Clin. Res., 2019, 25, 7958 CAS.
- S. Amdekar, P. Roy, V. Singh, A. Kumar, R. Singh and P. Sharma, Int. J. Inflammation, 2012, 2012, 752015 CrossRef PubMed.
- C. Novoa, P. Salazar, P. Cisternas, C. Gherardelli, R. Vera-Salazar, J. M. Zolezzi and N. C. Inestrosa, Biol. Res., 2022, 55, 39 CrossRef CAS PubMed.
- S. A. Pathan, B. Mitra and P. A. Cameron, Eur. Urol., 2018, 73, 583–595 CrossRef CAS PubMed.
- A. Zarghi and S. Arfaei, Iran. J. Pharm. Res., 2011, 10, 655–683 CAS.
- J. M. Wright, CMAJ, 2002, 167, 1131–1137 Search PubMed.
- M. Ekor, Front. Pharmacol, 2014, 4, 177 Search PubMed.
- J. C. Maroon, J. W. Bost and A. Maroon, Surg. Neurol. Int., 2010, 1, 80 CrossRef PubMed.
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch, T. International Natural Product Sciences and C. T. Supuran, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS PubMed.
- K. I. R. Notarte, M. T. J. Quimque, I. T. Macaranas, A. Khan, A. M. Pastrana, O. B. Villaflores, H. C. P. Arturo, D. Y. i. H. Pilapil IV, S. M. M. Tan and D.-Q. Wei, ACS Omega, 2023, 8, 5377–5392 CrossRef CAS PubMed.
- Y. Tamrat, T. Nedi, S. Assefa, T. Teklehaymanot and W. Shibeshi, BMC Complementary Altern. Med., 2017, 17, 1–10 Search PubMed.
- S. Bayat, M. E. Schranz, E. H. Roalson and J. C. Hall, Trends Plant Sci., 2018, 23, 808–821 CrossRef CAS PubMed.
- H. Singh, A. Mishra and A. K. Mishra, Biomed. Pharmacother., 2018, 101, 37–48 CrossRef CAS PubMed.
- L. V. Monroy, J. C. Cauich, A. M. Ortega and M. S. Campos, in Oncological Functional Nutrition, Elsevier, 2021, vol. 2021, pp. 161–194 Search PubMed.
- A. M. Abd-ElGawad, A. M. Elgamal, Y. A. Ei-Amier, T. A. Mohamed, A. E.-N. G. El Gendy and A. I. Elshamy, Plants, 2021, 10, 1294 CrossRef CAS PubMed.
- W. M. Kamel, M. M. Abd El-Ghani and M. El-Bous, Plant Sci. Biotechnol., 2010, 4, 11–16 Search PubMed.
- A. A. Ahmed, A. M. Kattab, S. G. Bodige, Y. Mao, D. E. Minter, M. G. Reinecke, W. H. Watson and T. J. Mabry, J. Nat. Prod., 2001, 64, 106–107 CrossRef CAS PubMed.
- A. A. Zaki, A. A. Al-Karmalawy, Y. A. El-Amier and A. Ashour, New J. Chem., 2020, 44, 16752–16758 RSC.
- I. Al Nasr, J. Acta Parasitol., 2020, 65, 696–703 CrossRef CAS PubMed.
- M. Hashem, J. Mycopathol., 2011, 172, 37–46 CrossRef PubMed.
- A. Khlifi, A. B. Chrifa, J. B. Lamine, A. Thouri, K. Adouni, G. Flamini, W. Oleszek and L. Achour, J. Environ. Sci. Pollut. Res. Int., 2020, 27, 22670–22679 CrossRef CAS PubMed.
- A. Khlifi, Ł. Pecio, J. C. Lobo, D. Melo, S. B. Ayache, G. Flamini, M. B. P. Oliveira, W. Oleszek and L. Achour, J. Ethnopharmacol., 2021, 269, 113739 CrossRef CAS PubMed.
- M. Y. Mahat, N. M. Kulkarni, S. L. Vishwakarma, F. R. Khan, B. S. Thippeswamy, V. Hebballi, A. A. Adhyapak, V. S. Benade, S. M. Ashfaque, S. Tubachi and B. M. Patil, Eur. J. Pharmacol., 2010, 642, 169–176 CrossRef CAS PubMed.
- B. M. Wangusi, L. W. Kanja, I. M. Ole-Mapenay and J. M. Onyancha, J. Evidence-Based Complementary Altern. Med., 2021, 2021, 3121785 Search PubMed.
- H. P. Maciel, L. G. Cardoso, L. R. Ferreira, F. F. Perazzo and J. C. Carvalho, Inflammopharmacology, 2004, 12, 203–210 CrossRef CAS PubMed.
- J. R. Deuis, L. S. Dvorakova and I. Vetter, Front. Mol. Neurosci., 2017, 10, 284 CrossRef PubMed.
- X. Dai, M. Ding, W. Zhang, Z. Xuan, J. Liang, D. Yang, Q. Zhang, B. Su, H. Zhu and X. Jia, Int. J. Exp. Clin. Res., 2019, 25, 7958–7965 CAS.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- M. Y. Behairy, R. A. Eid, H. M. Otifi, H. M. Mohammed, M. A. Alshehri, A. Asiri, M. Aldehri, M. S. A. Zaki, K. M. Darwish and S. S. Elhady, J. Pers. Med., 2023, 13, 1648 CrossRef PubMed.
- M. A. Soltan, M. A. Eldeen, B. H. Sajer, R. F. Abdelhameed, F. A. Al-Salmi, E. Fayad, I. Jafri, H. E. M. Ahmed, R. A. Eid and H. M. Hassan, Biology, 2023, 12, 613 CrossRef CAS PubMed.
- J. L. Wang, D. Limburg, M. J. Graneto, J. Springer, J. R. Hamper, S. Liao, J. L. Pawlitz, R. G. Kurumbail, T. Maziasz, J. J. Talley, J. R. Kiefer and J. Carter, Bioorg. Med. Chem. Lett., 2010, 20, 7159–7163 CrossRef CAS PubMed.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed.
- Q. Xue, X. Liu, P. Russell, J. Li, W. Pan, J. Fu and A. Zhang, Ecotoxicol. Environ. Saf., 2022, 233, 113323 CrossRef CAS PubMed.
- A. A. Gohara and M. Elmazar, Phytother. Res.: Int. J. Plants Res., 1997, 11, 564–567 CrossRef.
- T. Ibrahim, A. El-Hela, G. Dawoud and M. Zhran, Indian J. Pharm. Sci., 2019, 81, 651–660 CAS.
- A. Ladhari, F. Omezzine, M. DellaGreca, A. Zarrelli, S. Zuppolini and R. Haouala, S. Afr. J. Bot., 2013, 88, 341–351 CrossRef CAS.
- R. E. Barrientos, M. J. Simirgiotis, J. Palacios, A. Paredes, J. Bórquez, A. Bravo and F. Cifuentes, Molecules, 2020, 25, 3105 CrossRef CAS PubMed.
- J. Rodriguez, L. M. Gómez, A. C. Gutierrez, G. Mendez-Callejas, A. I. Reyes, L. C. Tellez, A. O. E. Rodriguez and G. RubÃ, Indian J. Pharm. Sci., 2018, 1–7 Search PubMed.
- H. Nagaya, Y. Tobita, T. Nagae, H. Itokawa, K. Takeya, A. F. Halim and O. B. Abdel-Halim, Phytochemistry, 1997, 44, 1115–1119 CrossRef CAS PubMed.
- P. C. Das, A. Patra, S. Mandal, B. Mallick, A. Das and A. Chatterjee, J. Nat. Prod., 1999, 62, 616–618 CrossRef CAS PubMed.
- F. Tsichritzis, M. Abdel-Mogib and J. Jakupovic, Phytochemistry, 1993, 33, 423–425 CrossRef CAS.
- C. Chen, Nat. Chem. Biol., 2010, 6, 401–402 CrossRef CAS PubMed.
- W. L. Smith, D. L. DeWitt and R. M. Garavito, Annu. Rev. Biochem., 2000, 69, 145–182 CrossRef CAS PubMed.
- C. Yuan, C. J. Rieke, G. Rimon, B. A. Wingerd and W. L. Smith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6142–6147 CrossRef CAS PubMed.
- C. A. Rouzer and L. J. Marnett, J. Lipid Res., 2009, 50(Suppl), S29–S34 CrossRef PubMed.
- S. Xu, D. J. Hermanson, S. Banerjee, K. Ghebreselasie, G. M. Clayton, R. M. Garavito and L. J. Marnett, J. Biol. Chem., 2014, 289, 6799–6808 CrossRef CAS PubMed.
- S. W. Rowlinson, J. R. Kiefer, J. J. Prusakiewicz, J. L. Pawlitz, K. R. Kozak, A. S. Kalgutkar, W. C. Stallings, R. G. Kurumbail and L. J. Marnett, J. Biol. Chem., 2003, 278, 45763–45769 CrossRef CAS PubMed.
- A. J. Vecchio and M. G. Malkowski, J. Struct. Biol., 2011, 176, 254–258 CrossRef CAS PubMed.
- M. J. Uddin, B. C. Crews, S. Xu, K. Ghebreselasie, C. K. Daniel, P. J. Kingsley, S. Banerjee and L. J. Marnett, ACS Chem. Biol., 2016, 11, 3052–3060 CrossRef CAS PubMed.
- B. J. Bender, S. Gahbauer, A. Luttens, J. Lyu, C. M. Webb, R. M. Stein, E. A. Fink, T. E. Balius, J. Carlsson, J. J. Irwin and B. K. Shoichet, Nat. Protoc., 2021, 16, 4799–4832 CrossRef CAS PubMed.
- M. Kontoyianni, L. M. McClellan and G. S. Sokol, J. Med. Chem., 2004, 47, 558–565 CrossRef CAS PubMed.
- S. O. Albuquerque, T. G. Barros, L. R. S. Dias, C. Lima, P. Azevedo, L. A. P. Flores-Junior, E. G. Dos Santos, H. F. Loponte, S. Pinheiro, W. B. Dias, E. M. F. Muri and A. R. Todeschini, Eur. J. Pharm. Sci., 2020, 154, 105510 CrossRef CAS PubMed.
- A. S. de Souza, B. D. C. Pacheco, S. Pinheiro, E. M. F. Muri, L. R. S. Dias, C. H. S. Lima, R. Garrett, M. B. M. de Moraes, B. E. G. de Souza and L. Puzer, Bioorg. Med. Chem. Lett., 2019, 29, 1094–1098 CrossRef CAS PubMed.
- A. M. El-Naggar, A. M. A. Hassan, E. B. Elkaeed, M. S. Alesawy and A. A. Al-Karmalawy, Bioorg. Chem., 2022, 123, 105770 CrossRef CAS PubMed.
- S. S. Elhady, R. F. A. Abdelhameed, R. T. Malatani, A. M. Alahdal, H. A. Bogari, A. J. Almalki, K. A. Mohammad, S. A. Ahmed, A. I. M. Khedr and K. M. Darwish, Biology, 2021, 10, 389 CrossRef CAS PubMed.
- M. Maher, A. E. Kassab, A. F. Zaher and Z. Mahmoud, Anticancer Agents Med. Chem., 2019, 19, 1368–1381 CrossRef CAS PubMed.
- T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha and J. M. Miyashiro, J. Med. Chem., 1997, 40, 1347–1365 CrossRef CAS PubMed.
- D. A. Jones, D. P. Carlton, T. M. McIntyre, G. Zimmerman and S. Prescott, J. Biol. Chem., 1993, 268, 9049–9054 CrossRef CAS PubMed.
- M. D. Percival, M. Ouellet, C. J. Vincent, J. A. Yergey, B. P. Kennedy and G. P. Oneill, Biophys. Biochem. Arch., 1994, 315, 111–118 CrossRef CAS PubMed.
- R. G. Kurumbail, A. M. Stevens, J. K. Gierse, J. J. McDonald, R. A. Stegeman, J. Y. Pak, D. Gildehaus, J. M. Iyashiro, T. D. Penning and K. Seibert, Nature, 1996, 384, 644–648 CrossRef CAS PubMed.
- C. Luong, A. Miller, J. Barnett, J. Chow, C. Ramesha and M. F. Browner, Nat. Struct. Mol. Biol., 1996, 3, 927–933 CrossRef CAS PubMed.
- A. L. Blobaum and L. J. Marnett, J. Med. Chem., 2007, 50, 1425–1441 CrossRef CAS PubMed.
- M. Karplus and G. A. Petsko, Nature, 1990, 347, 631–639 CrossRef CAS PubMed.
- M. Arnittali, A. N. Rissanou and V. Harmandaris, Procedia Comput. Sci., 2019, 156, 69–78 CrossRef.
- A. A. Al-Karmalawy, M. A. Dahab, A. M. Metwaly, S. S. Elhady, E. B. Elkaeed, I. H. Eissa and K. M. Darwish, Front. Chem., 2021, 9, 661230 CrossRef CAS PubMed.
- M. Y. Behairy, M. A. Soltan, M. A. Eldeen, J. A. Abdulhakim, M. M. Alnoman, M. M. Abdel-Daim, H. Otifi, S. M. Al-Qahtani, M. S. A. Zaki and G. Alsharif, Front. Immunol., 2022, 13, 1008463 CrossRef CAS PubMed.
- N. C. Benson and V. Daggett, J. Phys. Chem. B, 2012, 116, 8722–8731 CrossRef CAS PubMed.
- W. Singh, T. G. Karabencheva-Christova, G. W. Black, J. Ainsley, L. Dover and C. Z. Christov, Sci. Rep., 2016, 6, 20107 CrossRef CAS PubMed.
- J. F. Fatriansyah, R. K. Rizqillah, M. Y. Yandi and M. Sahlan, J. King Saud Univ., Sci., 2022, 34, 101707 CrossRef PubMed.
- A. S. de Souza, B. D. Pacheco, S. Pinheiro, E. M. Muri, L. R. Dias, C. H. Lima, R. Garrett, M. B. de Moraes, B. E. de Souza and L. Puzer, Bioorg. Med. Chem. Lett., 2019, 29, 1094–1098 CrossRef CAS PubMed.
- M. F. Browner, X-ray crystal structure of human cyclooxygenase-2, in New Targets in Inflammation, ed. N. Bazan, J. Botting and J. Vane, Springer, Dordrecht, 1996, vol. 16, pp. 71–74 Search PubMed.
- M. Miciaccia, B. D. Belviso, M. Iaselli, G. Cingolani, S. Ferorelli, M. Cappellari, P. Loguercio Polosa, M. G. Perrone, R. Caliandro and A. Scilimati, Sci. Rep., 2021, 11, 4312 CrossRef CAS PubMed.
- R. Kumari, R. Kumar, O. S. D. D. Consortium and A. Lynn, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef CAS PubMed.
- C. N. Cavasotto, Quantum mechanics in drug discovery, 2020, pp. 257–268 Search PubMed.
- A. Biela, M. Khayat, H. Tan, J. Kong, A. Heine, D. Hangauer and G. Klebe, J. Mol. Biol., 2012, 418, 350–366 CrossRef CAS PubMed.
- J. b. Preikša, V. Petrikaitė, V. Petrauskas and D. Matulis, J. ACS Omega, 2023, 8, 44571–44577 CrossRef PubMed.
- A. V. Makarenkov, S. S. Kiselev, E. G. Kononova, F. M. Dolgushin, A. S. Peregudov, Y. A. Borisov and V. A. Ol'shevskaya, Molecules, 2022, 27, 7484 CrossRef CAS PubMed.
- C.-X. Wei, M. Bian and G.-H. Gong, Molecules, 2015, 20, 5528–5553 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04496g |
| ‡ Both authors contributed equally to this work and share first authorship. |
| This journal is © The Royal Society of Chemistry 2024 |