
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering lignin-derivable diacrylate networks with tunable architecture and mechanics†
Yu-Tai
Wong
 a and
LaShanda T. J.
Korley
*ab
a and
LaShanda T. J.
Korley
*ab
aDepartment of Chemical and Biomolecular Engineering, University of Delaware, Newark, Delaware 19716, USA. E-mail: lkorley@udel.edu
bDepartment of Materials Science and Engineering, University of Delaware, Newark, Delaware 19716, USA
First published on 3rd July 2024
Abstract
Network engineering strategies offer a promising pathway toward tunable thermomechanical properties of bio-derivable, aromatic (meth)acrylate thermosets to expand their application library. In this work, a series of acrylate thermosets, synthesized from lignin-derivable vanillyl alcohol/bisguaiacol F diacrylates (VDA/BGFDA) and a bio-based n-butyl acrylate (BA), were designed as a sustainable platform to explore structure-architecture-property relationships. Using this approach, we examined a series of processable, fully bio-derivable acrylates. Increasing the diacrylate content across all networks improved storage moduli at 25 °C (E′25) by up to 2 GPa (1.1 GPa for VDA/BA-25/75 vs. 3.1 GPa for VDA/BA-75/25, and 1.5 GPa for BGFDA/BA-25/75 vs. 1.7 GPa for BGFDA/BA-50/50), and led to a more inhomogeneous network as evidenced by lower acrylate group conversion and a broader tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ peak, indicating heterogeneous relaxation modes. Modifying the aromatic content of the starting diacrylate impacted the final inhomogeneity of the network, with increasing inhomogeneity observed for the bis-aromatic BGFDA relative to the mono-aromatic VDA. Similarly, combining the mono- and bis-aromatic diacrylates generated a network with a biphasic-like thermal relaxation mode. By correlating network architecture and material performance, the increasing architectural complexity suggested a more convoluted thermal relaxation mode while the enhancement of thermomechanical properties could still be achieved for potential application as damping materials. Overall, we presented a design strategy utilizing bio-derivable acrylates to expand the suite of renewable material platforms from a network engineering perspective.
δ peak, indicating heterogeneous relaxation modes. Modifying the aromatic content of the starting diacrylate impacted the final inhomogeneity of the network, with increasing inhomogeneity observed for the bis-aromatic BGFDA relative to the mono-aromatic VDA. Similarly, combining the mono- and bis-aromatic diacrylates generated a network with a biphasic-like thermal relaxation mode. By correlating network architecture and material performance, the increasing architectural complexity suggested a more convoluted thermal relaxation mode while the enhancement of thermomechanical properties could still be achieved for potential application as damping materials. Overall, we presented a design strategy utilizing bio-derivable acrylates to expand the suite of renewable material platforms from a network engineering perspective.
1 Introduction
Aromatic-based, thermosets are generally utilized in high-performance applications that require high mechanical strength, high glass transition temperatures (Tgs), and superior thermal stability due to their structural rigidity and dimensional stability.1–6 Among various classes of thermosets, aromatic-based (meth)acrylates (i.e., methacrylates and acrylates) are some of the most common polymer components in coatings and composites due to their high toughness and low cost.1,7,8 Additionally, the relatively fast curing kinetics of (meth)acrylates paired with an appropriate formulation allows diverse processing techniques to be used, such as liquid molding and additive manufacturing.7–9 However, current commercial precursors for aromatic (meth)acrylates are primarily petroleum-based with negative impacts on both human health and the environment. For example, bisphenol F (BPF) and bisphenol A (BPA), two widely used aromatic precursors for dental materials and commercial composites, are petroleum-derived and are known as human endocrine disruptors.8,10–12 Moreover, (meth)acrylate networks usually require a high concentration of reactive diluent (>40 wt%) to lower viscosities and increase the gel points for improved processability during curing. One of the most applied diluents, styrene, is sourced from petroleum resources and is a suspected volatile carcinogen.7,8,13–16 Given these sustainability and safety hurdles, there is a pressing need to identify alternatives to petroleum-derived precursors with reduced toxicity and environmental impact.To address these challenges in the development of high-performance polymeric materials, lignin serves as a promising replacement for petroleum feedstocks due to its aromatic structure, abundance in nature, and potentially lower toxicity.1,17 Lignin exists in the cell walls of plants as a crosslinked biopolymer and is mostly underutilized as a by-product from the industrial pulping process.17–20 More than 98% of this pulp waste is burned for energy recovery with a relatively low rate of valorization due to its complex and recalcitrant structure.20 Although this unmodified lignin has been used in concrete fillers and composites, associated issues regarding unpleasant odors and dark colors limit the expansion of its applications.21–24 Alternatively, lignin fractionation via cleaving β-O-4 linkages unravels numerous pathways to yield substituted phenols (i.e., p-hydroxyphenyl, guaiacyl, and syringyl units), which can be further upgraded into various (bis)phenols through chemical modifications.1,17,20,25 These (bis)phenols possess structural similarity to petroleum-derived counterparts with additional methoxy groups and other functional handles on the aromatic rings that can be leveraged to tune thermomechanical properties of the resultant polymers.1,2,17,18,26 From a toxicity perspective, lignin-derivable bisphenols (i.e., bisguaiacols and bissyringols) exhibit lower estrogenic activity compared to petroleum-derived analogues, which is attributed to the additional methoxy groups that lower their binding affinity to estrogen receptors.27,28 In addition to reduced health risks, lignin-derivable bisguaiacols have been reported to exhibit enhanced mechanical properties.29,30 For example, introducing additional hydrogen bonding in a thermoset non-isocyanate polyurethane (NIPU) through inherent methoxy groups of bisguaiacols increased both elongation-at-break and toughness values of the final network.29 The bio-derivable, crosslinked NIPUs also showcased similar network architecture to petroleum-derived systems even with the presence of additional methoxy groups as evidenced by comparable moduli.29 Altogether, lignin-derivable (bis)phenolics provide a material platform for high-performance thermosets with improved renewability, lower health impacts, and comparable or even superior thermal and mechanical performance to their petroleum-derived counterparts.7,29,31–38
Over the past decade, lignin-derivable (meth)acrylates have gained increasing attention as viable replacements for petroleum-derived counterparts to achieve high-performance and promote sustainability.1,7,13–15,39–41 Methacrylated lignin model compounds (e.g., methacrylated vanillin, guaiacol, and eugenol) have been studied as substitutes for styrene as reactive diluents, reducing carcinogenic hazards and lowering volatilities due to the presence of the larger methacrylate group.7,13–15,39 Resins incorporated with lignin-derivable methacrylates further exhibited comparable thermomechanical properties (e.g., Tg and storage moduli) to those with styrene due to their structural similarity.7,13–15,39 However, these bio-derivable methacrylates were observed to have higher viscosities, requiring additional reactive diluents to facilitate curing and thus hindering the direct replacement of traditional diluents in additive manufacturing and composites applications.7,41 To expand their application beyond diluents, Bassett et al. explored the substitution of petroleum-derived building blocks with lignin-derivable analogues to assess their potential to impart structural rigidity in performance-driven methacrylates.40 Despite the promising Tgs and moduli of these lignin-derivable dimethacrylates for high-performance applications, this study was limited to the use of petroleum-based reactive diluents (styrene) and only compared resins from different feedstocks.40 To this end, this work aims to explore the design of processable acrylates utilizing lignin-derivable diacrylates with varying aromatic content and an aliphatic, bio-derivable acrylate as the reactive diluent for fully bio-derivable network formation.
Herein, the thermomechanical properties of a series of acrylate networks—photopolymerized through the reaction of lignin-derivable (bis)phenols (vanillyl alcohol or bisguaiacol F) diacrylates with a bio-derivable n-butyl acrylate (BA)—are investigated (Scheme 1). The chemistry and architecture of the final polymer are controlled using network engineering strategies by varying the diacrylate composition and the aromatic content of diacrylates to study the resulting structure–architecture–property relationships. Specifically, the effect of chemical composition on storage modulus (E′), tanδ, Young's modulus (Et), and elongation-at-break (εb) are examined. The resulting network architectures and subsequent inhomogeneities are associated with (1) the final acrylate conversion, and (2) thermal transition modes characterized by infrared spectra and the deconvolution study of tanδ curves, respectively. Through this design, we demonstrate a fully-renewable acrylate platform that utilizes lignin-derivable precursors to achieve high storage moduli and incorporates a bio-derivable aliphatic acrylate to enhance the resin processability. This research highlights the role of network engineering toward tunable thermomechanical properties to expand the versatility of renewable (meth)acrylates for high-performance applications, ranging from coatings to composites.
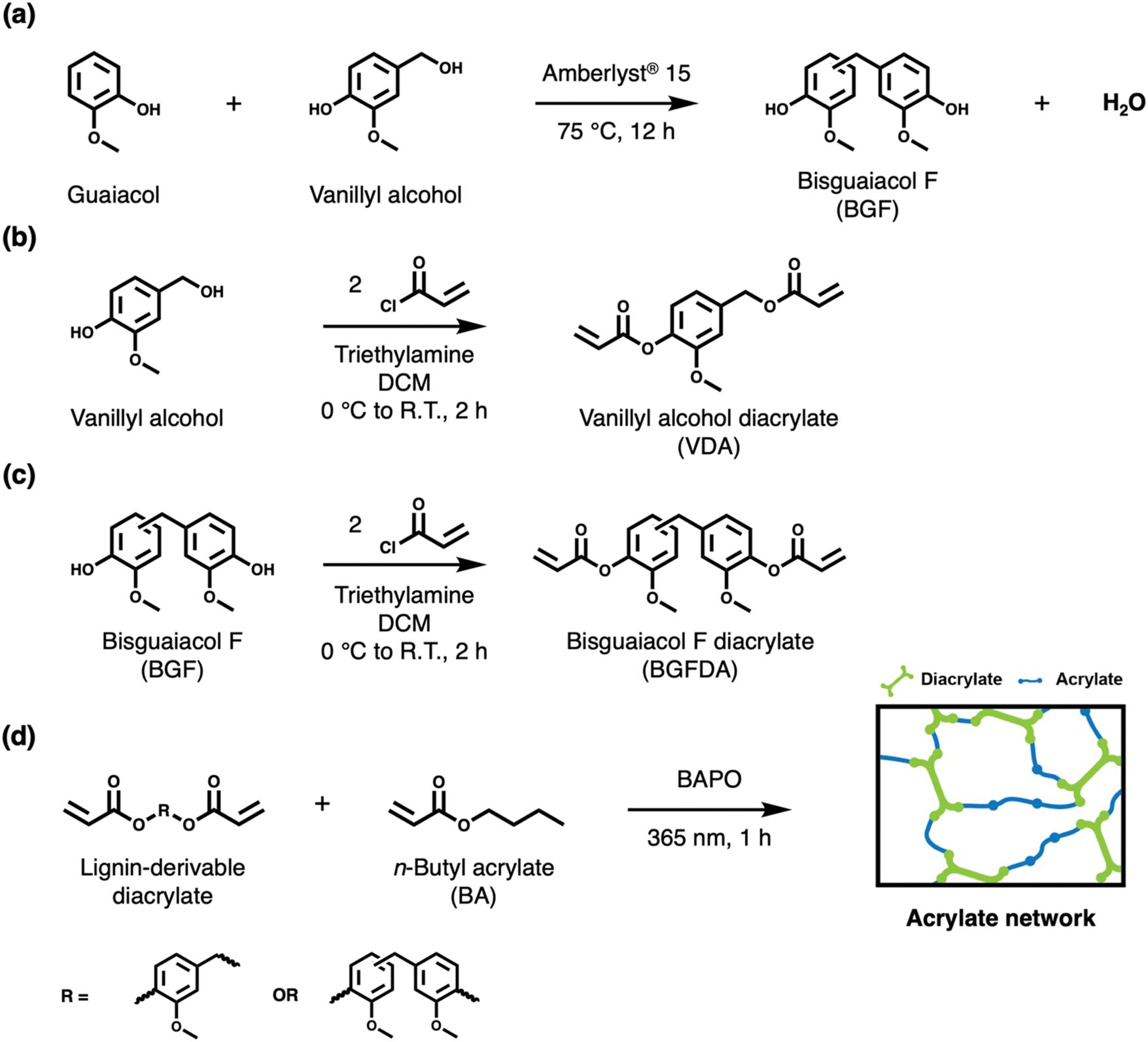 | ||
| Scheme 1 Synthetic scheme of lignin-derivable diacrylates and acrylate networks. (a) Reaction of guaiacol and vanillyl alcohol to form bisguaiacol F (BGF). (b) Acrylation of vanillyl alcohol to yield vanillyl alcohol diacrylate (VDA). (c) Acrylation of BGF to yield bisguaiacol F diacrylate (BGFDA). (d) Photopolymerization to form acrylate networks with n-butyl acrylate (BA), where the diacrylate is either VDA, BGFDA, or a mixture of both. | ||
2 Experimental
2.1. Materials
Dichloromethane (DCM; ≥99.5%), sodium bicarbonate (≥99.7%), sodium chloride (≥99.5%), hexanes (99%), ethyl acetate (≥99.5%), chloroform (>99.8%), and hydrochloric acid solution (36.5 to 38.0 wt%) were purchased from Fisher Scientific. Vanillyl alcohol (≥98%), guaiacol (98%), Amberlyst® 15 hydrogen form (dry; MQ200), triethylamine (≥99%), acryloyl chloride (≥97%), deuterated chloroform (CDCl3; ≥99.9%) and phenylbis(2,4,6-trimethylbenzoyl)phosphine oxide (BAPO; 97%) were purchased from Sigma-Aldrich. n-Butylacrylate (BA; 99+%) was purchased from Acros Organics. BA was destabilized by passing through a basic alumina column before use. All chemicals except for BA were used as received.2.2. Synthesis of bisguaiacol F (BGF)
BGF was synthesized as described in the literature (Scheme 1a).29,31 Vanillyl alcohol (0.03 mol, 5 g) and guaiacol (0.13 mol, 16 g) were added into a single-neck round-bottom flask equipped with a magnetic stir bar. The reaction mixture was stirred and heated at 75 °C with an Ar sparge for 40 min. The solid catalyst, Amberlyst® 15 hydrogen form (1.5 g, 30% w/w relative to vanillyl alcohol), was subsequently added to the reaction mixture. The reaction proceeded for 12 h. The reaction mixture was then cooled to ∼20 °C and dissolved in ∼50 mL of DCM. The solid catalyst was removed by filtration. The filtrate was washed with ∼50 mL of deionized (DI) water three times in a separatory funnel. The organic phase was collected, and excess DCM was removed by rotary evaporation. The residue was further purified by automated flash column chromatography (Biotage® Selekt, Sfär Silica Column—60 μm particle size, 100 Å pore size, 100 g silica gel) with a step gradient of ethyl acetate (33% v/v) and hexanes (67% v/v) as the mobile phase. The collected product was concentrated by rotary evaporation, and further dried in a vacuum oven at 25 °C for 12 h to yield BGF as a white solid (purity: >99%, yield: ∼54 mol%). Proton (1H) Nuclear Magnetic Resonance (NMR) (600 MHz, CDCl3, 16 scans, 25 °C) δ 6.85–6.64 (m, 6H), 5.48 (s, 2H), 3.85–3.83 (m, 8H). Fourier Transform Mass Spectrometry (FTMS) (Electrospray ionization [ESI], m/z): calculated for C15H16O4 260.1049; found 260.1041. The 1H NMR spectrum with peak assignments is shown in Fig. S1 (ESI†).2.3. General procedure for the synthesis of (di)acrylates
In a typical diacrylate synthesis, 1 eq. of alcohol (vanillyl alcohol or BGF), 4 eq. of triethylamine, and 0.3 M (relative to alcohol) of DCM were added into a single-neck round-bottom flask equipped with a magnetic stir bar. The reaction mixture was stirred and sparged with Ar for 40 min in an ice bath. Under Ar flow, 3 eq. of acryloyl chloride was added dropwise into the reaction mixture, and the reaction was warmed back to ∼20 °C followed by proceeding for 2 h. Next, the solution was concentrated by rotary evaporation to remove DCM, and the residue was dissolved in ethyl acetate. The solution mixture was subsequently washed sequentially in: (1) 1 m HCl solution, (2) saturated sodium bicarbonate solution, and (3) saturated brine in a separatory funnel. The organic phase was collected, and excess ethyl acetate was removed by rotary evaporation. Both diacrylates were purified by automated flash column chromatography (Biotage® Selekt, Sfär Silica Column—60 μm particle size, 100 Å pore size, 100 g silica gel) with a step gradient of ethyl acetate (33% v/v) and hexanes (67% v/v) as the mobile phase. The diacrylates were recovered and dried at ∼20 °C under vacuum to afford white solids.2.4. Preparation of acrylate networks
All diacrylate networks were prepared via radical photopolymerization by reacting the diacrylate and acrylate in different compositions with BAPO as the photoinitiator (1% w/w relative to the monomer mixture) under ambient conditions. Due to the solid form of both VDA and BGFDA at room temperature and their varying solubility in the BA, VDA/BA networks were cured in bulk with 25, 50, and 75 mol% of diacrylate, while BGFDA/BA networks were cured with 25 and 50 mol% of diacrylate. Herein, materials formed can be denoted as follows: A/B(/C)-X/Y(/Z), where A, B, and C refer to the monomer used, and X, Y, and Z refer to the mol% of the monomer on a molecule basis. It also was noted that the two acrylate groups of VDA can differ in reactivity depending on whether the ester oxygen is connected to a benzyl or a phenyl group, which will be discussed in a later section.The fabrication procedure for VDA/BA-75/25 is detailed as an example. To a scintillation vial, VDA (0.86 g, 3.28 mmol), BA (0.14 g, 1.09 mmol), and BAPO (10 mg, 2.4 × 10−2 mmol) were dissolved in 5 mL of DCM, and mixed by shaking on a shaker plate at 600 rpm for 10 min, with the vial covered in aluminum foil to prevent early initiation until complete solubilization. The solution was then poured into a PTFE evaporating dish having dimensions of 12 mm × 65 mm (height × diameter), and covered to prevent early termination of the photoinitiator. After the complete evaporation of DCM for 24 h, the film was cured in an ultraviolet (UV) chamber (SPDI UV, λ = 365 nm) at an intensity ∼13 mW cm−2 for 1 h and followed by a thermal post-curing at 150 °C for additional 24 h to ensure maximum conversion (Fig. S5 and S6, ESI†). The temperature for thermal post-curing after photopolymerization was determined by the exothermic peaks in differential scanning calorimetry (DSC) (Fig. S7, ESI†). Rectangular films were then cut from the bulk film after thermal post-curing to provide specimens having dimensions of 20 mm × 3 mm × 0.2 mm (length × width × thickness) for further characterization.
2.5. Soxhlet extraction
After network formation, the gel fraction was probed via Soxhlet extraction. Samples were weighed to obtain the initial mass (m0) before the extraction. The extraction was then completed for 24 h with a single cycle duration ∼15 min at 90 °C using ∼250 mL of chloroform as the extraction solvent. Samples were subsequently dried in a vacuum oven at ∼20 °C for 24 h and weighed to obtain the dry mass (mdry). The gel fraction was estimated as mdry/m0. Soxhlet extraction was performed three times on each sample, and the reported values are the average gel fraction with the error reported as a standard deviation.2.6. 1H NMR spectroscopy
All NMR samples were prepared in CDCl3 and analyzed using a Bruker AVANCE III 600 MHz (5-mm SMART probe) spectrometer. Processing and analysis of 1H NMR spectra was completed using the MestReNova software package.2.7. Mass spectrometry
The masses of BGF, lignin-derivable diacrylates, and BA were measured by ESI-FTMS using a Q-Exactive Orbitrap (Thermo Fisher Scientific) mass spectrometer. Prior to the analysis, samples were dissolved in DCM at a concentration of 0.1 mg mL−1. Direct syringe injection of the samples into the mass spectrometer was conducted in a positive ion mode for ESI-FTMS.2.8. Attenuated total reflectance- Fourier transform infrared (ATR-FTIR) spectroscopy
ATR-FTIR spectroscopy was performed using a Thermo Nicolet NEXUS 870 FTIR with a deuterated triglycine sulfate/potassium bromide (DTGS/KBr) detector. All acrylate network films were scanned (128 scans) at a resolution of 4 cm−1 in the wavenumber range of 400–4000 cm−1. Network conversion was assessed by the examination of the acrylate double bond stretching band at ∼1635 cm−1 and carbonyl stretching band at ∼1724 cm−1 as an internal reference (Fig. S8, ESI†) by eqn (1):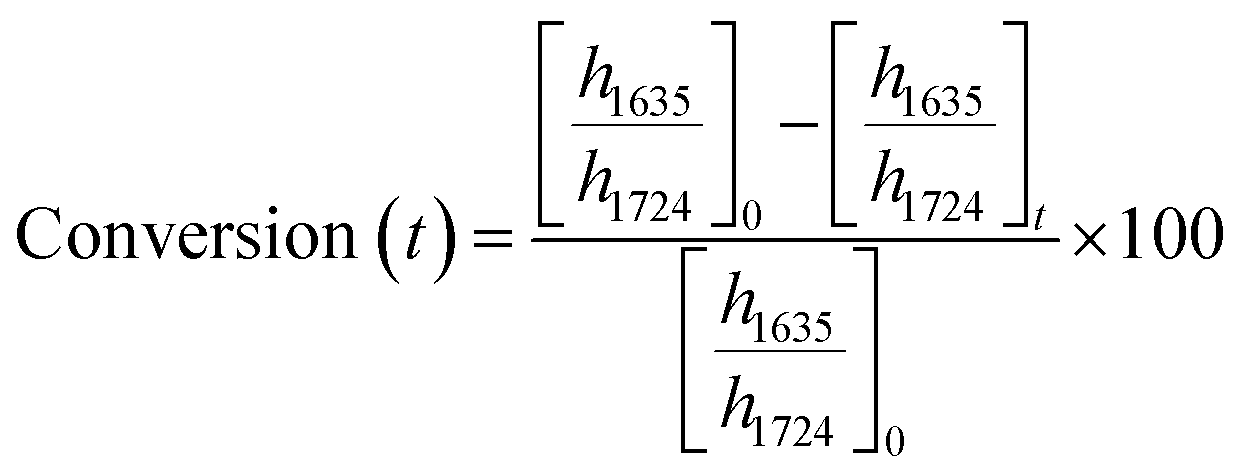 | (1) |
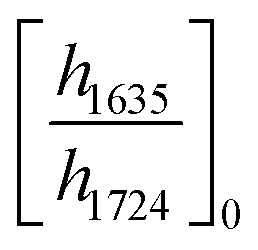 and
and 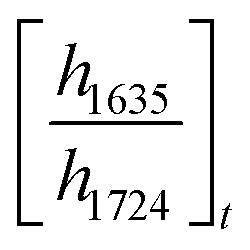 are the ratios before and after UV exposure for time t, respectively, between the height of the acrylate double bond stretching band (∼1635 cm−1) and the internal reference (∼1724 cm−1). Reported conversions are an average of triplicate measurements on different samples with errors representing standard deviations.
are the ratios before and after UV exposure for time t, respectively, between the height of the acrylate double bond stretching band (∼1635 cm−1) and the internal reference (∼1724 cm−1). Reported conversions are an average of triplicate measurements on different samples with errors representing standard deviations.
2.9 Differential scanning calorimetry (DSC)
DSC (Discovery DSC, TA Instruments) was employed to examine the additional curing peaks after UV curing of acrylate networks. Each sample was encapsulated in a Tzero aluminum pan sealed with the corresponding hermetic lid. Two heating/cooling cycles were performed between 0 and 250 °C under a continuous N2 flow (50 mL min−1) at a rate of 10 °C min−1. The temperature corresponding to the maximum of the exothermic peak was identified as an additional curing temperature. DSC curves are presented in Fig. S7 (ESI†).2.10 Thermogravimetric analysis (TGA)
TGA (Discovery TGA, TA Instruments) was used to study the thermal stability of acrylate networks. Each sample was placed in a 100 μL platinum pan. All samples were heated to 700 °C from ∼20 °C at a heating rate of 10 °C min−1 under a continuous N2 flow (50 mL min−1). The temperature at 5 wt% loss (Td,5%) was used to assess the thermal stability of crosslinked acrylate networks. TGA curves are presented in Fig. S9 and summarized in Table S1 (ESI†).2.11. Dynamic mechanical analysis (DMA)
An RSA-G2 Solids Analyzer (TA Instruments) was used to determine the storage modulus (E′), loss modulus (E′′), and tanδ (E′′/E′) of each specimen using oscillatory tension mode with a strain amplitude of 0.075%, frequency of 1 Hz, and an axial force of 0.4 N. Rectangular samples (20 mm × 3 mm × 0.2 mm [length × width × thickness]) were first heated to 250 °C, and then cooled to −100 °C followed by a heating cycle to 250 °C at a rate of 3 °C min−1. Data from the final heating trace were used for analysis. Reported moduli and tanδ are an average of individually triplicate measurements with errors representing standard deviations.
2.12. Tensile testing
Tensile properties of the acrylate networks were measured on a Zwick/Roell tensile tester with a load cell of 100 N and a clamp force of 20 N at ∼20 °C. Rectangular samples (20 mm × 3 mm × 0.2 mm [length × width × thickness]) were subjected to an extension rate of 10% min−1 until film breakage. The ultimate tensile strength (σm), elongation-at-break (εb), Young's modulus (Et), and toughness results are reported as an average of triplicate measurements with errors representing standard deviations.2.13. Small-angle X-ray scattering
SAXS data were collected using a Xenocs Xeuss 2.0. X-rays were generated at 50 kV/0.6 mA at a beam wavelength of 1.542 Å (Cu Ka radiation) and a sample-to-detector distance of 1200 mm. The scattered beam was recorded on a CCD detector with a pixel resolution of 172 × 172 mm. 2D patterns were azimuthally integrated to obtain the scattering intensity as a function of the absolute value of the scattering vector, Q = |![[Q with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0051_20d1.gif) | = 4πλ−1sinθ, where λ and θ denote the wavelength of the X-ray beam and the half of the scattering angle, respectively.
| = 4πλ−1sinθ, where λ and θ denote the wavelength of the X-ray beam and the half of the scattering angle, respectively.
3. Results and discussion
The aromatic nature of lignin-derivable (bis)phenols diacrylates suggests their potential to impart rigidity to high-performance acrylates networks designed from renewable feedstocks. Meanwhile, the incorporation of a bio-derivable, aliphatic acrylate with low viscosity can further facilitate resin processability and serve as a precursor for network engineering. The combination of a rigid crosslinker (e.g., VDA, BGFDA) and a flexible spacer (i.e., BA) allowed modulation of the network chemistry and architecture by controlling diacrylate compositions and the aromatic content of the diacrylate (monophenol vs. bisphenol) toward tunable thermomechanical properties. To examine this network engineering strategy, two lignin-derivable (bis)phenols, vanillyl alcohol and BGF, were acrylated through nucleophilic reaction with acryloyl chloride, respectively, to form diacrylates (VDA and BGFDA, Schemes 1b and c), while BGF was synthesized via the acid-catalyzed electrophilic aromatic substitution between guaiacol and vanillyl alcohol as reported (Scheme 1a).29,31 Bio-derivable BA was chosen as the reactive diluent to control the distance between junction points due to its low viscosity and aliphatic structure, providing additional flexibility to the networks. All networks were subsequently prepared via bulk free-radical photopolymerization using either VDA or BGFDA as the crosslinker and BA as the reactive diluent. Compositions of diacrylate ranging from 25 mol% to 75 mol% depending on the solubility of diacrylate in BA were utilized to investigate the connection between network chemistry, architectural features, and thermomechanical properties. In addition to two-component networks, we also investigated a mixed diacrylate system using an equal amount of VDA and BGFDA (i.e., VDA/BGFDA/BA-25/25/50) to compare the effect of the aromatic, diacrylate content within two-component networks (VDA/BA-50/50 and BGFDA/BA-50/50).Preparation of acrylate networks
Network formation was monitored by ATR-FTIR for the disappearance of the acrylate alkene stretching band at ∼1635 cm−1, and subsequently conversions were quantified using eqn (1). Unreacted groups were present in all cases, indicated by the incomplete conversion (final conversion <100%, Table 1) due to the decreased chain mobility after gelation, which inhibits pendent acrylate groups from reacting.42 Increasing the content of aromatic diacrylate led to earlier local gelation, which hindered the mobility of reactive groups to diffuse and react with each other as noted by the drop in final conversions from 92.4% to 61.6% in VDA/BA networks, and 86.6% to 78.7% in BGFDA/BA networks. However, the final conversions of BGFDA/BA networks were slightly lower than VDA/BA networks (∼4–6% lower) when a similar amount of diacrylate was used. This lower conversion is possibly caused by the higher observed viscosity of the BGFDA/BA resins during formulation preparation, subsequently reducing the mobility and propagation of reactive groups as reported in earlier studies.42 Notably, the final conversions of networks differing in diacrylate type (VDA/BA-50/50, BGFDA/BA-50/50, and VDA/BGFDA/BA-25/25/50) showed no significant difference after post-curing. The network with mixed diacrylates (VDA/BGFDA/BA-25/25/50) exhibited the lowest average conversion (∼73.1%) compared to the single diacrylate networks (82.3% of VDA/BA-50/50 and 78.7% of BGFDA/BA-50/50), but all of the thermosets were within the range of error. The slight variation between these networks was potentially driven by qualitative viscosity differences of the resin solutions. The gel fractions of all networks were over 95% in all cases (Table 1), demonstrating that the networks are highly crosslinked. To highlight the evolution of network formation using lignin-derivable diacrylates, a conversion profile under different curing times for the VDA/BA networks is detailed in Fig. S10 (ESI†). Conversions were obtained at cure times of 15, 30, 45, and 60 min, and after thermal post-curing. All VDA/BA networks approached their maximum extent of curing within 30 min, highlighting the fast curing kinetics and also the inevitable formation of pendant groups in highly-crosslinked networks.| Sample | Final conversion (%)a | Gel fraction (%)b |
|---|---|---|
a Determined by ATR-FTIR using the disappearance of the acrylate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C peak (∼1635 cm−1) after thermal post-curing at 150 °C.
b Determined by Soxhlet extraction with chloroform as the extraction solvent. C peak (∼1635 cm−1) after thermal post-curing at 150 °C.
b Determined by Soxhlet extraction with chloroform as the extraction solvent.
|
||
| VDA/BA-25/75 | 92.4 ± 0.4 | 95.7 ± 2.3 |
| VDA/BA-50/50 | 82.3 ± 7.4 | 99.9 ± 0.1 |
| VDA/BA-75/25 | 61.6 ± 11.8 | 99.9 ± 0.1 |
| BGFDA/BA-25/75 | 86.6 ± 4.8 | 98.2 ± 2.1 |
| BGFDA/BA-50/50 | 78.7 ± 10.4 | 97.7 ± 4.0 |
| VDA/BGFDA/BA-25/75 | 73.1 ± 2.8 | 99.9 ± 0.1 |
Thermomechanical properties of acrylate networks
The thermomechanical properties of acrylate networks were examined by DMA and are summarized in Table 2. The storage moduli at 25 °C (E′25) and 200 °C (E′200) were used to approximate the moduli in the glassy and rubbery states, respectively. However, it also should be noted that all networks showed no distinct glassy and rubbery state reflected in the shape of E′ curves (Fig. 1), which is an indication of the inhomogeneity that will be discussed in the next section.| Sample | E′25 (GPa)ab | E′200 (GPa)ab | E t (MPa)cd | ε b (%)d |
|---|---|---|---|---|
| a Reported values are the average of triplicate measurements with errors representing standard deviations. b Determined from DMA experiments in oscillatory film tension mode. c Young's modulus was determined from the slope of the initial elastic region in the engineering tensile stress–strain curves. d All data are based on tensile stress–strain curves obtained from the uniaxial tensile testing. Each measurement is triplicated with errors representing standard deviations. | ||||
| VDA/BA-25/75 | 1.1 ± 0.4 | 0.1 ± 0.1 | 1.3 ± 0.1 | 4.3 ± 1.6 |
| VDA/BA-50/50 | 2.6 ± 0.3 | 0.4 ± 0.1 | 2.0 ± 0.4 | 1.6 ± 0.3 |
| VDA/BA-75/25 | 3.1 ± 0.2 | 0.9 ± 0.1 | 2.0 ± 0.8 | 1.9 ± 0.4 |
| BGFDA/BA-25/75 | 1.5 ± 0.3 | 0.2 ± 0.1 | 0.8 ± 0.2 | 4.4 ± 0.2 |
| BGFDA/BA-50/50 | 1.7 ± 0.4 | 0.2 ± 0.1 | 1.5 ± 0.1 | 1.8 ± 0.3 |
| VDA/BGFDA/BA-25/25/50 | 2.2 ± 0.1 | 0.7 ± 0.1 | 1.4 ± 0.1 | 2.3 ± 0.3 |
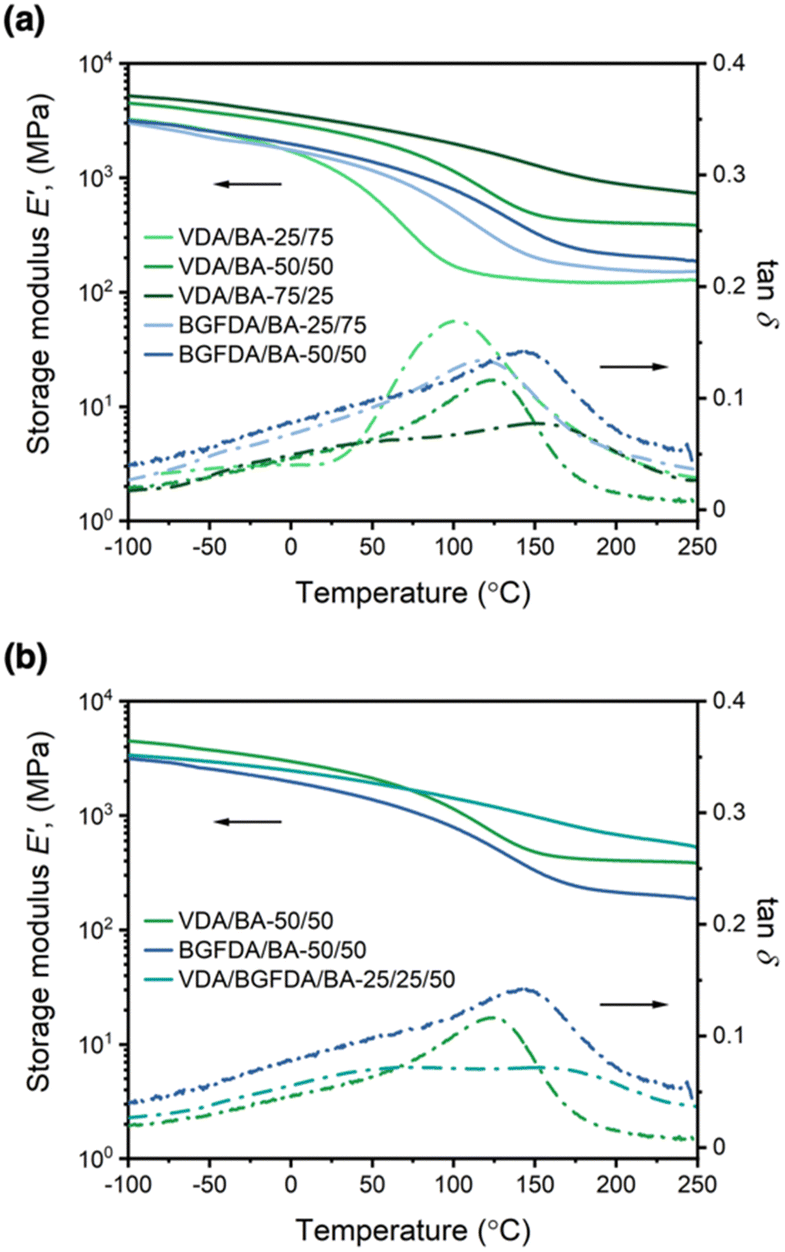 | ||
Fig. 1 Storage modulus (solid line, —) and tanδ (dashed line, ![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) as a function of temperature for (a) varying diacrylate content and (b) varying diacrylate type with a total diacrylate content at 50 mol%. ) as a function of temperature for (a) varying diacrylate content and (b) varying diacrylate type with a total diacrylate content at 50 mol%. | ||
Overall, the composition of diacrylate in the network dictates the value of E′25 and E′200 as shown in Fig. 1a. For example, E′25 for VDA/BA networks was enhanced from ∼1.1 GPa to 3.1 GPa by increasing the VDA content from 25 mol% to 75 mol%, and E′25 for BGFDA/BA networks shifted slightly from ∼1.5 GPa to 1.7 GPa by increasing the BGFDA amount from 25 mol% to 50 mol%. Similarly, E′200 for VDA/BA was enhanced from ∼0.1 GPa to 0.9 GPa by increasing VDA content from 25 mol% to 75 mol%, and E′200 for BGFDA/BA remained similar at ∼0.2 GPa with BGFDA content increasing from 25 mol% to 50 mol %. This trend in E′ was likely the result of a denser network resulting from the incorporation of a higher diacrylate content. Additionally, the transition of E′ from a glassy to rubbery state becomes less apparent with increasing diacrylate content, suggestive of restricted chain mobility triggered by temperature change, when a denser network is obtained.43,44 Beyond the E′ tunability of VDA/BA and BGFDA/BA networks, these bio-derivable networks also showed comparable E′25 (ranging from 1–3 GPa) to petroleum-derived, commercial (meth)acrylate thermosets with E′25 in the range of 2–3 GPa.1,42
The trend of E′ is more complex when comparing networks with varying diacrylate type while maintaining the total diacrylate content at 50 mol% (Fig. 1b). The BGFDA/BA network exhibited the lowest E′ throughout the temperature range (E′25 ∼1.7 GPa and E′200 ∼0.21 GPa), whereas the VDA/BA network had the highest E′25 (∼2.6 GPa) and an intermediate E′200 (∼0.40 GPa). Interestingly, the network with mixed diacrylates, VDA/BGFDA/BA-25/25/50, showed an intermediate E′25 (∼2.2 GPa) and the highest E′200 (∼0.68 GPa). The lower E′25 and E′200 of BGFDA/BA was likely due to a network with a slightly lower density of elastically effective junctions. However, the response of E′ to temperature change for the VDA/BGFDA/BA-25/25/50 network suggested a network with a higher content of various architectural features, including unreacted groups and clusters of crosslinks, that induce chain motion across a wider range of temperature.45
Unlike conventional thermosets, distinct Tgs were unable to be identified using the tanδ curves for these highly-crosslinked networks due to the broad nature of the transition. While tanδ is defined as E′′/E′ (the ratio between viscosity and elasticity effects), the peak is commonly applied as an indication of glass transition behavior. However, we observed that width of the tanδ peaks increased as the diacrylate content increased (Fig. 1a). The flattening and broadening of tanδ curves with increased diacrylate content suggested a more inhomogeneous network, leading to complex thermal relaxation modes.43,44,46,47
Network inhomogeneity and architecture
Defects, such as loops and unreacted groups, inevitably arise during free-radical crosslinking due to the interplay between reaction kinetics and diffusion limits.46 These molecular-scale defects are further encompassed in a non-uniform spatial distribution of crosslink junctions and subsequently dictate the network architecture and thermomechanical performance.46,48 In this work, we anticipated that unreacted groups and non-uniform spatial distribution of junctions would dominate the inhomogeneous nature of our networks because loops are less likely to occur in concentrated gels, such as a bulk polymerization.49–51 To investigate the occurrence of these architectural features, unreacted groups were characterized by conversion studies, and tanδ curves from DMA that reflect the thermal response of chain motions were examined to semi-quantitatively determine the overall inhomogeneity. Interestingly, these bio-derivable acrylate networks exhibited broad tanδ curves, which are one of the essential features for damping materials.52 The broad tanδ curves are likely a result of the formation of different domains, and prompted the exploration of microscopic network architecture of these bio-derivable acrylate networks via deconvolution of tanδ curves into Gaussian peaks (Fig. 2).53 Each deconvoluted Gaussian peak was treated as a domain with relaxation temperatures corresponding to a distinct chemical composition and architecture embedded within the bulk network. The tanδ curves of these networks can be deconvoluted into 3–4 Gaussian bands, which are summarized in Table 3. Based on the peak positions, we further assigned these relaxation bands to different compositions in the order of increasing temperature to domains of BA, BA-rich, diacrylate-rich, and diacrylate. Notably, the assignments of deconvoluted peaks at the lowest temperature was supported by the reported Tg for poly(n-butyl acrylate) at ∼−50 °C, and peaks at the highest temperature were consistent with estimated Tgs for VDA and BGFDA homopolymers at ∼190 and 220 °C, respectively, as determined by the polymer properties prediction website, Polymer Genome (Fig. S11, ESI†).54–56 These deconvoluted peaks also are quite broad with a maximum peak width spanning over 200 °C, indicating the inhomogeneous nature of each domain themselves. These domains are likely comprised of micro-structured defects, including unreacted groups and junction clusters, which were unobservable by small-angle X-ray scattering (i.e., no apparent peaks were observed, Fig. S12, ESI†).
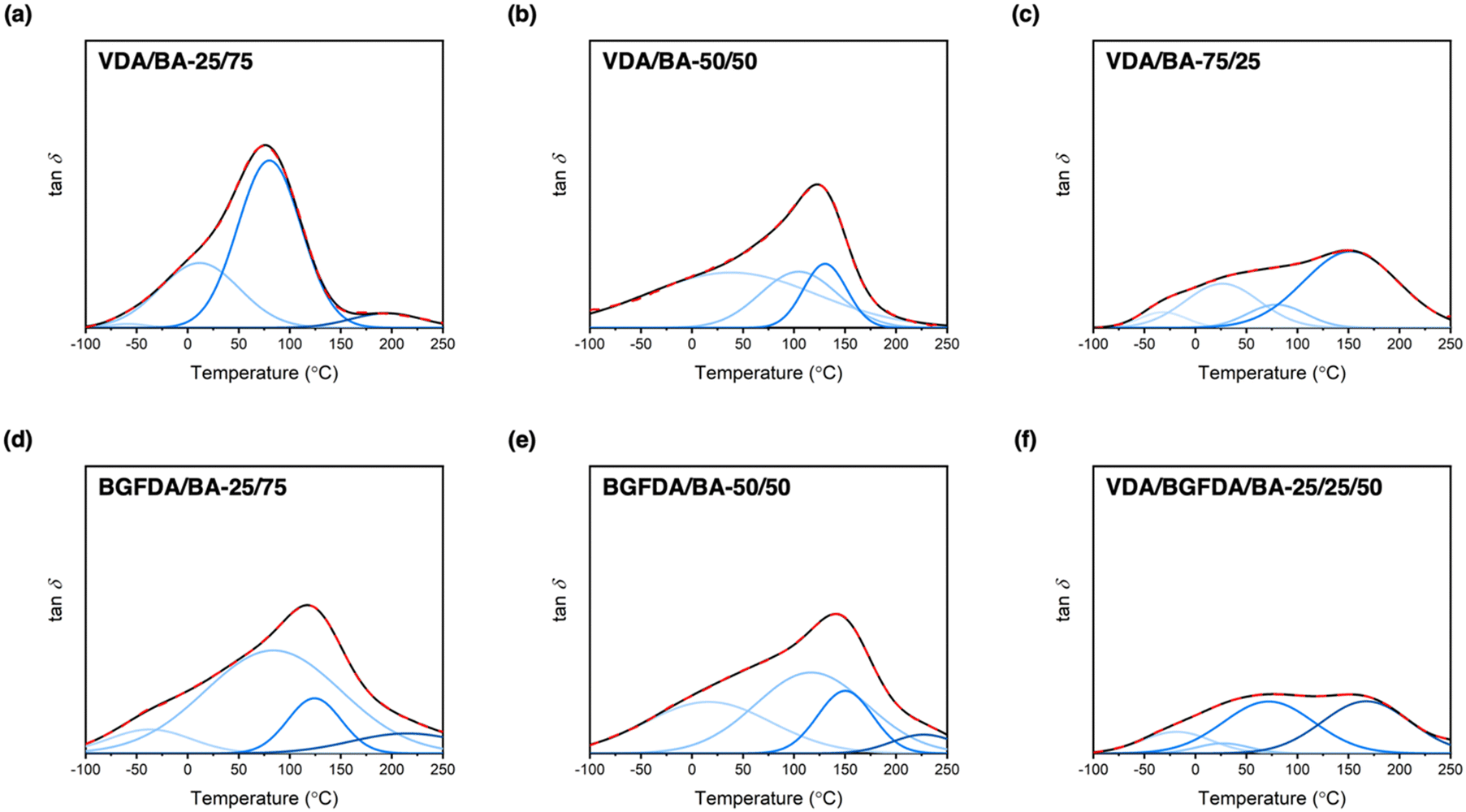 | ||
| Fig. 2 Deconvolution of tanδ curves of acrylate networks. The red dashed line () denotes the original curve, and the black solid line (—) is the integrated result of deconvoluted curves. All y-axes are on the same scale. | ||
| Sample | Max tanδ temperature (°C) |
Phase 1 | Phase 2 | Phase 3 | Phase 4 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Center (°C) | FWHM (°C)a | Center (°C) | FWHM (°C)a | Center (°C) | FWHM (°C)a | Center (°C) | FWHM (°C)a | ||
|
a Determined as the full width at half maximum (FWHM) of the deconvoluted tanδ curve.
|
|||||||||
| VDA/BA-25/75 | 77 | −58 | 44 | 11 | 92 | 80 | 85 | 195 | 81 |
| VDA/BA-50/50 | 123 | — | — | 38 | 197 | 105 | 99 | 130 | 54 |
| VDA/BA-75/25 | 151 | −32 | 54 | 26 | 86 | 80 | 67 | 152 | 109 |
| BGFDA/BA-25/75 | 119 | −37 | 94 | 84 | 157 | 124 | 85 | 215 | N.A. |
| BGFDA/BA-50/50 | 143 | 16 | 144 | 117 | 129 | 150 | 63 | 227 | N.A. |
| VDA/BGFDA/BA-25/25/50 | 78 | −18 | 79 | 27 | 60 | 72 | 104 | 167 | 102 |
| 154 | |||||||||
As detailed in Table 3, no distinct trends in each domain were revealed by the deconvolution studies. Alternatively, the height and position of the maximum tanδ peaks were examined to determine the network structure and overall inhomogeneity. Increasing the diacrylate content resulted in shifts of both the maximum tan delta peak and the dominant deconvoluted peak to higher temperatures, indicating a denser network structure with the incorporation of more crosslinker. For example, the maximum tanδ peak for VDA/BA networks shifted from ∼77 °C to ∼151 °C when increasing the VDA content from 25 mol% to 75 mol%, and the peak for BGFDA/BA networks shifted from ∼119 °C to ∼143 °C when the BGFDA content was increased by 25 mol%. The height of the maximum tanδ peak was reduced when a higher content of diacrylate was incorporated, indicating increased inhomogeneity with domains more evenly relaxing over a wide range of temperatures for both VDA/BA and BGFDA/BA networks.
Varying the diacrylate type (i.e., VDA, BGFDA, or a mix of VDA and BGFDA) had a more complex connection to the thermomechanical behavior because the size of the diacrylate unit and reaction kinetics need to be considered simultaneously. VDA/BA-50/50 exhibited a slightly higher maximum tanδ peak temperature than BGFDA/BA-50/50 (123 °C vs. 119 °C), which is potentially a result of the slightly lower density of elastically effective junctions for BGFDA/BA network. However, BGFDA/BA-50/50 showed a higher extent of inhomogeneity with a more pronounced shoulder peak at the lower temperature range (0–50 °C), agreeing with the trend of more unreacted groups due to a higher resin viscosity. The higher extent of inhomogeneity exhibited by BGFDA/BA networks indirectly suggests that the reactivity of the two acrylate groups of VDA shows no significant difference. In contrast, a notable difference in the reactivity would likely result in a more inhomogeneous network characterized by the presence of non-uniformly distributed junction clusters. Surprisingly, the network with mixed diacrylates (VDA/BGFDA/BA-25/25/50) showed a biphasic-like thermal transition with two prominent domains at 72 °C and 167 °C. The VDA/BGFDA/BA network displayed a more inhomogeneous network compared with VDA/BA and BGFDA/BA networks despite having an intermediate aromatic content. This result is likely due to the different reactivities between VDA, BGFDA, and BA that impart more complexity to the reaction kinetics, which require future investigation.
Overall, increased inhomogeneity was observed with increasing diacrylate content or by changing the diacrylate from monophenol (VDA) to bisphenol (BGFDA), agreeing with the expected influence of unreacted groups. A schematic representation of the impact of diacrylate content and aromatic content of diacrylate is summarized in Fig. 3. The increase in E’ and the broad tanδ curves can be further leveraged for damping applications despite the network inhomogeneity.
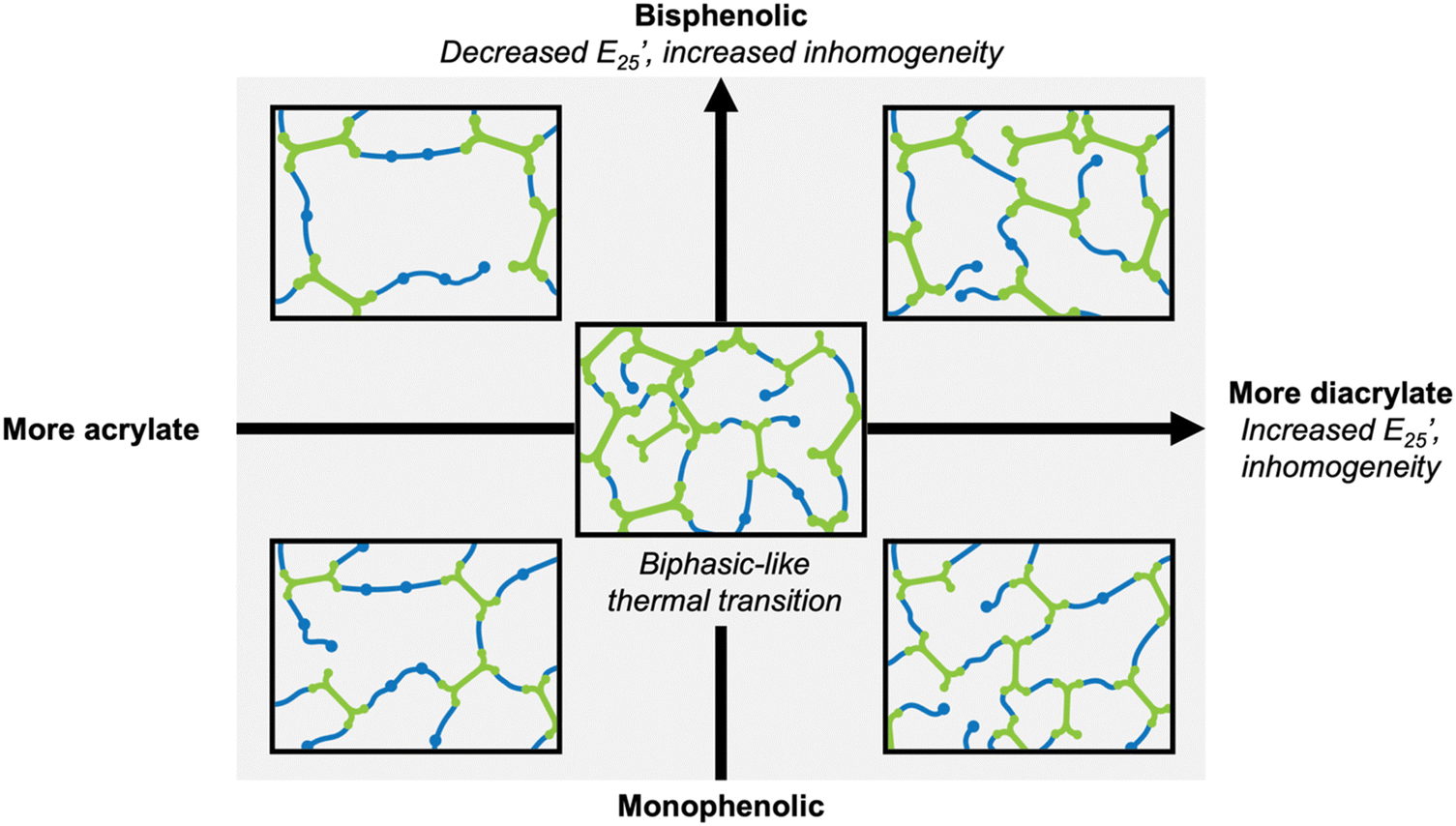 | ||
| Fig. 3 Schematic representation of the effect of chemical structure on network architecture and thermomechanical properties. | ||
Tensile properties of acrylate networks
The mechanical behavior of each network was examined through uniaxial tensile testing to probe the influence of network architectures on Et and εb (Fig. 4). Similar to the trend of E′, increasing the diacrylate content enhanced Et as a result of a denser network (Fig. 4a), where the Et of VDA/BA-50/50 was ∼0.7 GPa higher than VDA/BA-25/75 (2.0 GPa vs. 1.3 GPa, Table 2), and Et of BGFDA/BA-50/50 was ∼0.7 GPa higher than BGFDA/BA-25/75 (1.5 GPa vs. 0.8 GPa). However, the Et values of VDA/BA-75/25 (2.0 ± 0.8 GPa) and VDA/BA-50/50 (2.0 ± 0.4 GPa) were not significantly different, indicating that the elastically ineffective junctions start to dictate the tensile properties with higher diacrylate content. Furthermore, VDA/BA-50/50 showed the highest Et value among the networks when varying diacrylate types due to the smaller diacrylate unit leading to a denser structure, while BGFDA/BA and VDA/BGFDA/BA have similar Et within error (1.5 ± 0.1 GPa vs. 1.4 ± 0.1 GPa, respectively).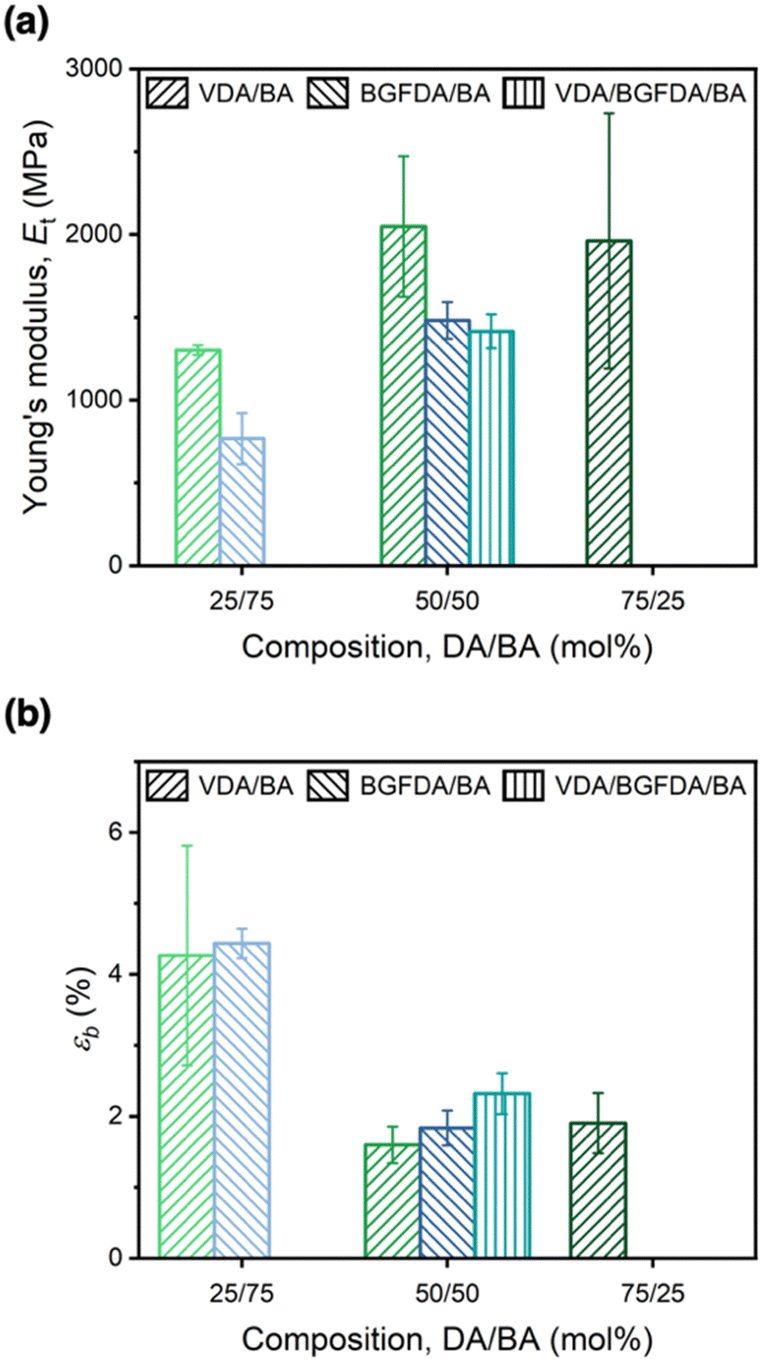 | ||
| Fig. 4 (a) Young's modulus (Et) and (b) elongation at break (εb) of acrylate networks. Error bars represent the standard deviations from triplicate measurements. | ||
Unsurprisingly, the acrylate networks were mechanically brittle (εb < 5% and toughness < 1.1 MJ m−3, Table S2, ESI†) due to their highly crosslinked structure. In contrast to the trend of Et, increasing the diacrylate content led to a reduction in εb (Fig. 4b) with a denser network limiting network extensibility. As with Et, εb values of VDA/BA-75/25 and VDA/BA-50/50 were statistically the same (1.6 ± 0.3% vs. 1.9 ± 0.4%), which can similarly be attributed to the pronounced presence of elastically ineffective junctions due to inhomogeneities at high diacrylate content. Interestingly, networks using 50 mol% of diacrylate showed εb ∼2%, while VDA/BGFDA/BA has a slightly higher εb. This finding agreed with a previous study that concluded that brittle networks show no significant impact on εb as the extent of inhomogeneity increases; εb is mainly dictated by the diacrylate content.57
4. Conclusions
This work examined engineering strategies utilizing the composition and aromatic content of lignin-derivable diacrylates as tunable handles to investigate thermomechanical properties of a bio-derivable and processable acrylate platform. We demonstrated that the content of diacrylate incorporated in the network is the driving factor for tailoring both thermomechanical properties and network architecture. Specifically, increasing the amount of diacrylate led to an enhancement in E′25 due to a denser structure and higher extent of inhomogeneity attributed to the presence of more unreacted groups and non-uniformly distributed junctions that result in multiple thermal relaxation modes. Additionally, the shift from monophenolic to bisphenolic diacrylate in binary component networks reduced the E′25 due to the lower density of elastically effective junctions. Notably, a ternary system with mixed diacrylate types generated a network with a biphasic-like thermal relaxation mode and the highest extent of network inhomogeneity due to the formation of two distinct domains. A deconvolution study of tanδ curves probed the highly inhomogeneous nature of a densely crosslinked system composed of bulky aromatic precursors. While all networks were highly inhomogeneous, these renewable acrylate networks showed tunable and comparable E′25 (∼1–3 GPa) to petroleum-based acrylates, displaying potential for use in traditional composites, as well as broad tanδ curves harnessed from defects, further expanding their application as damping materials. Overall, this work showcased a network engineering strategy to enable processable, renewable (meth)acrylates, exploiting the rigidity of lignin-derivable (bis)phenols and the low viscosity of a bio-derivable, aliphatic oil to tailor thermomechanical performance.
Data availaibliity
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by a National Science Foundation grant (NSF GCR CMMI 1934887). The authors thank the University of Delaware (UD) Advanced Materials Characterization Laboratory for the use of the ATR-FTIR, DSC, TGA, and SAXS instruments, and the UD NMR facility and UD Mass Spectrometry facility for the use of NMR and Mass Spectrometers, respectively. The authors thank the kind help from Dr. Zachary Hinton, Dr. Zoé Schyns, and Dr. Daseul Jang for data analysis discussions. This research was also supported as part of the Center for Plastics Innovation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under award DE-SC0021166, for the use of the RSA-G2 instrument.References
- J. S. Mahajan, R. M. O’Dea, J. B. Norris, L. T. J. Korley and T. H. Epps, ACS Sustainable Chem. Eng., 2020, 8, 15072–15096 CrossRef CAS.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Macromol. Rapid Commun., 2016, 37, 9–28 CrossRef CAS PubMed.
- V. Schimpf, A. Asmacher, A. Fuchs, K. Stoll, B. Bruchmann and R. Mülhaupt, Macromol. Mater. Eng., 2020, 305, 2000210 CrossRef CAS.
- E. Feghali, K. M. Torr, D. J. van de Pas, P. Ortiz, K. Vanbroekhoven, W. Eevers and R. Vendamme, Top. Curr. Chem., 2018, 376, 32 CrossRef PubMed.
- R. M. O’Dea, P. A. Pranda, Y. Luo, A. Amitrano, E. O. Ebikade, E. R. Gottlieb, O. Ajao, M. Benali, D. G. Vlachos, M. Ierapetritou and T. H. Epps, Sci. Adv., 2022, 8, eabj7523 CrossRef PubMed.
- S. Zhao, X. Huang, A. J. Whelton and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2018, 6, 7600–7608 CrossRef CAS.
- J. F. Stanzione, J. M. Sadler, J. J. La Scala and R. P. Wool, ChemSusChem, 2012, 5, 1291–1297 CrossRef CAS PubMed.
- S. K. Yadav, K. M. Schmalbach, E. Kinaci, J. F. Stanzione and G. R. Palmese, Eur. Polym. J., 2018, 98, 199–215 CrossRef CAS.
- M. K. Porwal, M. M. Hausladen, C. J. Ellison and T. M. Reineke, Green Chem., 2023, 25, 1488–1502 RSC.
- A. V. Krishnan, P. Stathis, S. F. Permuth, L. Tokes and D. Feldman, Endocrinology, 1993, 132, 2279–2286 CrossRef CAS PubMed.
- N. Olea, R. Pulgar, P. Pérez, F. Olea-Serrano, A. Rivas, A. Novillo-Fertrell, V. Pedraza, A. M. Soto and C. Sonnenschein, Environ. Health Perspect., 1996, 104, 298–305 CrossRef CAS PubMed.
- S. Takayanagi, T. Tokunaga, X. Liu, H. Okada, A. Matsushima and Y. Shimohigashi, Toxicol. Lett., 2006, 167, 95–105 CrossRef CAS PubMed.
- J. F. Stanzione III, J. M. Sadler, J. J. La Scala, K. H. Reno and R. P. Wool, Green Chem., 2012, 14, 2346 RSC.
- J. F. Stanzione, P. A. Giangiulio, J. M. Sadler, J. J. La Scala and R. P. Wool, ACS Sustainable Chem. Eng., 2013, 1, 419–426 CrossRef CAS.
- Y. Zhang, Y. Li, L. Wang, Z. Gao and M. R. Kessler, ACS Sustainable Chem. Eng., 2017, 5, 8876–8883 CrossRef CAS.
- B. T. White, V. Meenakshisundaram, K. D. Feller, C. B. Williams and T. E. Long, Polymer, 2021, 223, 123727 CrossRef CAS.
- R. M. O’Dea, J. A. Willie and T. H. Epps, ACS Macro Lett., 2020, 9, 476–493 CrossRef PubMed.
- P. J. De Wild, W. J. J. Huijgen and R. J. A. Gosselink, Biofuels, Bioprod. Biorefin., 2014, 8, 645–657 CrossRef CAS.
- A. Tolbert, H. Akinosho, R. Khunsupat, A. K. Naskar and A. J. Ragauskas, Biofuels, Bioprod. Biorefin., 2014, 8, 836–856 CrossRef CAS.
- W. Schutyser, T. Renders, S. Van den Bosch, S.-F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- A. Vishtal and A. Kraslawski, BioResources, 2011, 6, 3547–3568 Search PubMed.
- V. K. Thakur, M. K. Thakur, P. Raghavan and M. R. Kessler, ACS Sustainable Chem. Eng., 2014, 2, 1072–1092 CrossRef CAS.
- G. F. Bass and T. H. Epps, Polym. Chem., 2021, 12, 4130–4158 RSC.
- X. Shi, S. Gao, C. Jin, D. Zhang, C. Lai, C. Wang, F. Chu, A. J. Ragauskas and M. Li, Green Chem., 2023, 25, 5907–5915 RSC.
- S. Gao, Z. Cheng, X. Zhou, Y. Liu, J. Wang, C. Wang, F. Chu, F. Xu and D. Zhang, Chem. Eng. J., 2020, 394, 124896 CrossRef CAS.
- G. Yang, B. J. Rohde, H. Tesefay and M. L. Robertson, ACS Sustainable Chem. Eng., 2016, 4, 6524–6533 CrossRef CAS.
- A. Amitrano, J. S. Mahajan, L. T. J. Korley and T. H. Epps, RSC Adv., 2021, 11, 22149–22158 RSC.
- Y. Peng, K. H. Nicastro, T. H. Epps, III and C. Wu, Food Chem., 2021, 338, 127656 CrossRef CAS PubMed.
- S. V. Mhatre, J. S. Mahajan, T. H. Epps and L. T. J. Korley, Mater. Adv., 2023, 4, 110–121 RSC.
- J. S. Mahajan, Z. R. Hinton, E. Nombera Bueno, T. H. Epps Iii and L. T. J. Korley, Mater. Adv., 2024, 5, 3950–3964 RSC.
- K. H. Nicastro, C. J. Kloxin and T. H. Epps, ACS Sustainable Chem. Eng., 2018, 6, 14812–14819 CrossRef CAS.
- M. Fache, B. Boutevin and S. Caillol, Eur. Polym. J., 2015, 68, 488–502 CrossRef CAS.
- A. L. Holmberg, N. A. Nguyen, M. G. Karavolias, K. H. Reno, R. P. Wool and T. H. Epps, Macromolecules, 2016, 49, 1286–1295 CrossRef CAS.
- A. L. Holmberg, K. H. Reno, N. A. Nguyen, R. P. Wool and T. H. Epps, ACS Macro Lett., 2016, 5, 574–578 CrossRef CAS PubMed.
- S. Cousinet, A. Ghadban, E. Fleury, F. Lortie, J.-P. Pascault and D. Portinha, Eur. Polym. J., 2015, 67, 539–550 CrossRef CAS.
- M. Fache, E. Darroman, V. Besse, R. Auvergne, S. Caillol and B. Boutevin, Green Chem., 2014, 16, 1987–1998 RSC.
- C. Veith, F. Diot-Néant, S. A. Miller and F. Allais, Polym. Chem., 2020, 11, 7452–7470 RSC.
- E. D. Hernandez, A. W. Bassett, J. M. Sadler, J. J. La Scala and J. F. Stanzione, ACS Sustainable Chem. Eng., 2016, 4, 4328–4339 CrossRef CAS.
- C. Zhang, S. A. Madbouly and M. R. Kessler, Macromol. Chem. Phys., 2015, 216, 1816–1822 CrossRef CAS.
- A. W. Bassett, C. M. Breyta, A. E. Honnig, J. H. Reilly, K. R. Sweet, J. J. La Scala and J. F. Stanzione, Eur. Polym. J., 2019, 111, 95–103 CrossRef CAS.
- A. W. Bassett, A. E. Honnig, C. M. Breyta, I. C. Dunn, J. J. La Scala and J. F. Stanzione, ACS Sustainable Chem. Eng., 2020, 8, 5626–5635 CrossRef CAS.
- R. Anastasio, R. Cardinaels, G. W. M. Peters and L. C. A. Breemen, J. Appl. Polym. Sci., 2020, 137, 48498 CrossRef CAS.
- M. Podgórski, J. Appl. Polym. Sci., 2009, 112, 2942–2952 CrossRef.
- A. M. Ortega, S. E. Kasprzak, C. M. Yakacki, J. Diani, A. R. Greenberg and K. Gall, J. Appl. Polym. Sci., 2008, 110, 1559–1572 CrossRef CAS.
- L. Rey, J. Duchet, J. Galy, H. Sautereau, D. Vouagner and L. Carrion, Polymer, 2002, 43, 4375–4384 CrossRef CAS.
- S. Seiffert, Polym. Chem., 2017, 8, 4472–4487 RSC.
- A. Jangizehi, F. Schmid, P. Besenius, K. Kremer and S. Seiffert, Soft Matter, 2020, 16, 10809–10859 RSC.
- Y. Gu, J. Zhao and J. A. Johnson, Angew. Chem., Int. Ed., 2020, 59, 5022–5049 CrossRef CAS PubMed.
- H. Zhou, J. Woo, A. M. Cok, M. Wang, B. D. Olsen and J. A. Johnson, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19119–19124 CrossRef CAS PubMed.
- M. Zhong, R. Wang, K. Kawamoto, B. D. Olsen and J. A. Johnson, Science, 2016, 353, 1264–1268 CrossRef CAS PubMed.
- N. J. Rebello, H. K. Beech and B. D. Olsen, ACS Macro Lett., 2021, 10, 531–537 CrossRef CAS PubMed.
- C.-L. Qin, W.-M. Cai, J. Cai, D.-Y. Tang, J.-S. Zhang and M. Qin, Mater. Chem. Phys., 2004, 85, 402–409 CrossRef CAS.
- D. Ratna, V. G. Dalvi, S. Billa, S. K. Sharma, S. K. Rath, K. Sudarshan and P. K. Pujari, ACS Appl. Polym. Mater., 2021, 3, 5073–5086 CrossRef CAS.
- E. Penzel, N. Ballard and J. M. Asua, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2018, pp. 1–20 Search PubMed.
- C. Kim, A. Chandrasekaran, T. D. Huan, D. Das and R. Ramprasad, J. Phys. Chem. C, 2018, 122, 17575–17585 CrossRef CAS.
- C. Kuenneth, W. Schertzer and R. Ramprasad, Macromolecules, 2021, 54, 5957–5961 CrossRef CAS.
- A. Shundo, M. Aoki, P. Wang, T. Hoshino, S. Yamamoto, S. Yamada and K. Tanaka, Macromolecules, 2023, 56, 3884–3890 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00159a |
| This journal is © The Royal Society of Chemistry 2024 |