
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biomass-derived polyol esters as sustainable phase change materials for renewable energy storage†
Magdalena
Gwóźdź
 a,
Marta
Markiewicz
b,
Stefan
Stolte
b,
Anna
Chrobok
a,
David R.
Turner
c,
Karolina
Matuszek
*c and
Alina
Brzęczek-Szafran
*a
a,
Marta
Markiewicz
b,
Stefan
Stolte
b,
Anna
Chrobok
a,
David R.
Turner
c,
Karolina
Matuszek
*c and
Alina
Brzęczek-Szafran
*a
aSilesian University of Technology, Faculty of Chemistry, Gliwice, Poland. E-mail: alina.brzeczek-szafran@polsl.pl
bInstitute of Water Chemistry, Dresden University of Technology, Bergstrasse 66, D-01069 Dresden, Germany
cMonash University, School of Chemistry, Melbourne, Australia. E-mail: karolina.matuszek@monash.edu
First published on 11th October 2024
Abstract
Innovative thermal battery technology has the capability to revolutionize the renewable energy storage market. Its cost-effectiveness, scalability, contribution to CO2 reduction, and lack of reliance on rare earth metals set it apart. Nevertheless, the overall efficiency and sustainability of this technology hinge on crucial factors such as the sources, performance, and cost of the associated phase-change material (PCM). Fatty acid esters with biorenewable origins meet the sustainability criteria yet are limited to low-temperature applications (mostly <70 °C). In this study, we explored a new strategy to fine-tune the operating temperature of esters by adding hydroxyl groups, which are capable of forming H-bonds, positively affecting crystal packing and boosting their thermal properties. OH-group-rich, and biorenewable tartaric and mucic acids were employed as the core of fatty acid esters. Combinations of tartaric acid and fatty alcohols gave sustainable PCMs (confirmed by green chemistry metrics) with high melting enthalpies up to 221 J g−1, improved melting temperatures up to 94 °C, and high stability demonstrated over more than 500 cycles. With the aid of Fourier-transform infrared spectroscopy (FTIR), synchrotron single-crystal X-ray diffraction, and Hirshfeld surface analyses, we obtained insights into the molecular interactions dictating the extraordinary thermal properties of sugar acid-derived esters, which could be feasible as PCMs for sustainable and inexpensive energy storage.
Introduction
Thermal energy storage (TES) is a valuable solution for mitigating the energy crisis by efficiently storing surplus thermal energy and thus easing strain on the power grid.1 Taking into consideration the intensified actions undertaken globally to achieve carbon neutrality, which the EU has committed to achieve by 2050,2 TES has the potential to play a dual role in shaping a more sustainable future:3–6 firstly, by contributing to energy supply and, secondly, by reducing CO2 emissions through minimizing the reliance on fossil fuel-based power generation.7 To fully address the sustainability of these inexpensive, easily scalable technologies, the materials involved cannot be neglected, i.e., their source, performance, price, and utilization/circularity should be considered.8 Phase-change materials (PCMs) have significant potential in TES applications. PCMs harness latent heat during a phase transition, typically solid–liquid, with the operating temperature defined by the melting temperature (Tm) and the energy storage capacity determined by the enthalpy of fusion (ΔHf).9 PCMs should possess a high TES density, undergo reversible crystallization/melting within narrow temperature ranges, and maintain high thermal stability.1 To be fully sustainable, they should also be environmentally acceptable, i.e., show high biodegradability and low toxicity. Current research focuses on organic PCMs, which, in comparison with inorganic PCMs, are less corrosive, exhibit lower supercooling, and do not suffer from phase separation. The operating temperature range of organic PCMs is suitable for applications in TES,10–12 utilized in, e.g., thermo-regulated textiles,13 solar air heaters,14 solar stills,15 solar hot water systems,16 or the heating and cooling of buildings.17–19As organic materials for low/medium temperature applications, paraffins have been recognized as having superior properties, such as high heat capacity, chemical inertness, little supercooling, little volume change during melting, and, additionally, low cost. Unfortunately, their production from crude oil contributes to CO2 emissions and fossil fuel depletion.12,20 Greener alternatives are fatty acids and alcohols, both of which show high enthalpies of phase transitions (ranging from 45 to approximately 250 J g−1), high thermal stability, and Tm ranging from −5 °C to 71 °C, and additionally show no significant supercooling.21–23 They can be obtained from sources such as plant oils, ensuring availability, material circularity, and low cost.24 Modifying the fatty acid/alcohol chain allows the thermal properties to be modified, e.g., longer alkyl chains offer greater degrees of freedom in the liquid phase, leading to increased melting entropy (ΔSf).3 Additionally, they tend to form stronger intermolecular bonds and tighter crystalline structures, necessitating higher temperatures for bond-breaking and ultimately influencing the phase transition temperature and the latent heat of fatty acids.7,21,24,25 Nevertheless, the drawbacks of fatty acids are corrosiveness, unpleasant odor, sublimation during heating, and flammability. On the other hand, a solid–solid transition prior to melting has been identified for fatty alcohols. This transition has the potential to mitigate the released thermal energy under specific circumstances.26,27
A promising solution to overcome these limitations is the derivatization of fatty acids with alcohols to give esters. Fatty acid esters lack the bad odor, exhibit high thermal stability, higher Tm relative to paraffins and fatty acids, and no corrosiveness.28–30 Moreover, this group of PCMs is present in the portfolio of companies offering PCMs,31 manifesting their commercial potential. Nevertheless, their Tm values, which are mostly <70 °C, make them desirable PCMs only for low/medium temperatures.
In initial studies, fatty acid esters obtained by the esterification of stearic and palmitic acids with alcohols (methanol, propanol, or butanol) were studied.32 The resulting esters showed Tm in the range of 17–34 °C, with methyl stearate demonstrating the highest ΔHf of 199 J g−1. With an increase in the alcohol's carbon chain length, ΔHf decreased, while Tm showed an opposite trend. Floros and Narine20 proposed a solvent-free synthesis of diesters derived from fatty acid esters (methyl stearate, methyl palmitate, methyl myristate, and methyl laurate) and a linear diol (ethylene glycol-1,10-decanediol). These fatty esters exhibited Tm ranging from 39 to 77 °C and possessed a remarkably high heat capacity of up to 260 J g−1 for ethane-1,2-diyl dipalmitate, which is the highest known enthalpy among all diesters. Aydin et al.,33–35 Sari et al.,36 Stamatiou et al.,37 Özlem Kaplan et al.,38 and Ravotti et al.28,39 explored a wide series of linear esters synthesized by reacting fatty acids with diols, diacids with fatty alcohols, or glycerin with fatty acids. These materials exhibited high ΔHf (150–226 J g−1) and a wide range of Tm (10–60 °C). Furthermore, abundant, benign, inexpensive sugar alcohols that show high melting enthalpy (even up to 330 J g−1 for galactitol)40–43 have been used as precursors for the synthesis of esters, which were examined as PCMs. Sari et al. esterified glycerol,44 erythritol,45 xylitol,46 mannitol,47 and galactitol48 with fatty acids (stearic, palmitic, myristic, and lauric acid), resulting in a series of tri-, tetra-, penta-, and hexa-esters that exhibited low Tm values (22–65 °C) and high melting enthalpies (145–251 J g−1), with the highest reported being for galactitol hexastearate. Even though the sugar alcohols used as starting materials are rich in hydroxyl groups (which are capable of forming strong H-bonds), the strategy applied was focused on their quantitative esterification with fatty acids, leading to no free groups being left.
The high enthalpies of fusion of esters resulted from the intramolecular van der Waals interactions between their –CH2– units.49 Not surprisingly, initial research was primarily focused on the impact of the hydrocarbon chain length and overall molecular arrangement on the changes in ΔHf and Tm. However, as some authors of this study have shown, H-bonding interactions can also fine-tune the thermal properties of organic PCMs, leading to increased ΔHf values6,50,53 observed also by other groups.51,52,54
Existing PCMs for applications above 70 °C, such as sterilization, are primarily fossil fuel-derived paraffins and amides, while sustainable fatty acids, fatty alcohols, or esters mostly melt at lower temperatures. The state of the art for existing organic PCMs in this temperature range is summarized in Table 6, ESI.†
Driven by the need for sustainability and material circularity, inexpensive, biomass-derived tartaric acid (TA) or mucic acid (MA) were employed as the cores of fatty acid esters, which were capable of forming H-bonds. By varying the number of hydroxyl groups in the core of fatty acid esters and the length of their alkyl chains, structure–property relationships, which were studied by temperature-dependent FT-IR analysis, crystallographic analysis, and Hirshfeld surface analysis, were elucidated, aiming to uncover a way to design highly efficient, sustainable PCMs with tunable operating temperature ranges.39 Additionally, the biodegradability of these materials was studied as a key property influencing their environmental impact.
Experimental
The ESI† section provides a detailed account of the synthesis of tartaric acid diesters, including comprehensive characterization data and a thorough list of the materials used.Materials
The long-chain sugar acid esters were synthesized using L-(+)-tartaric acid (>99.5%), mucic acid (>99%), 1-dodecanol (>98%), 1-hexadecanol (>99%), 1-octadecanol (>99%), and 1-docosanol (>99%) obtained from Sigma-Aldrich. The reaction also used Amberlyst™-15 or p-toluenesulfonic acid as catalysts (Merck). Ethanol (>99%) was used for crystallization to purify the final products. All reagents were used without purification.Characterization
Results and discussion
Synthesis
Two groups of diesters were synthesized by using L-(+)-tartaric acid or mucic acid as precursors with a different number of hydroxyl groups (two or four, respectively). The esters of L-(+)-tartaric acid were synthesized through a straightforward procedure by esterification with fatty alcohols (1-dodecanol, 1-hexadecanol, 1-octadecanol, or 1-docosanol) (Fig. 1). In turn, the high polarity of mucic acid and the resulting difficulty in contacting non-polar fatty alcohols hindered direct esterification. Therefore, mucic acid was first transformed into mucic acid dibutyl ester, which was further reacted with fatty alcohols. | ||
| Fig. 1 Scheme of synthesis of long-chain tartaric acid diesters (C12_TA_C12, C16_TA_C16, C18_TA_C18, and C22_TA_C22) and mucic acid diesters (C12_MA_C12 and C18_MA_C18). | ||
The chosen hydroxyl-group-rich precursors are readily available, fully biobased, and sustainable. Tartaric acid is a naturally occurring organic acid that can be found in various fruits, including grapes, apples, and tamarind.62 It can be naturally sourced through the acid hydrolysis of calcium tartrate, which is derived from potassium tartrate obtained as a byproduct in winemaking62,63 or synthesized from maleic anhydride.64 Likewise, mucic acid is sourced from fruit, as well as from sugar beet.65 It can be produced by oxidation of galactose,66 electrolytic oxidation of D-galacturonate,67 or microbial conversion of D-galacturonate.66,68,69 Valorization of wastes from wine production or the dairy industry not only promotes sustainable resource management and the circular economy, but also does not deplete food resources.70 On the other hand, fatty alcohols are crucial compounds sourced from natural fats and oils from plants, animals, and microorganisms.71 They can be derived from fatty acids found in coconut oil, obtained through the hydrogenation72 of other vegetable oils or produced by the conversion of petroleum feedstock.71
In addition to the source of the precursors, the synthetic procedure applied for diesters was evaluated and analyzed according to green chemistry principles using J. Clark's Green Chemistry metrics toolkit (Appendix 1, ESI†)73 to fully assess its sustainability. All key reaction conditions (reaction temperature, time, catalysts, and purification method) were carefully selected to best adhere to green chemistry parameters as discussed below for a representative tartaric acid derivative (C18_TA_C18), which was used due to its high potential as a sustainable PCM, as discussed in further sections.
The precursor (TA) was transformed into the desired product with high conversion (>99%) and a selectivity of 70% toward diesters (marked with an amber flag), giving a yield of 70% (marked with an amber flag). Nevertheless, stearyl alcohol was used in excess, contributing to a reaction mass efficiency (RME) of 29%. The synthesis was performed solventless (green flag), bolstering the PMI, which could be marked with a green flag. The esterification reaction was conducted with a heterogeneous, reusable catalyst (Amberlyst-15), which could be assigned a green flag; at 70 °C, this translates into a green flag with regard to the energy consumption parameter. The synthesis was conducted in batch mode (amber flag), but potentially it could be performed under continuous flow conditions with a heterogeneous catalyst. The product was purified by crystallization from ethanol, which means that the work-up procedure could be assigned a green flag. Finally, the synthesis included no critical elements (green flag) and no chemicals with H-codes regarding their health and safety parameters. Although full optimization of the synthetic method is beyond the scope of this study (future work should focus on optimization of the reactant ratio and related selectivity toward diesters), the synthesis of tartaric acid esters has full sustainable potential with little environmental impact.
Thermal properties
Thermophysical properties play a significant role in assessing a material's capacity to efficiently store and release thermal energy, thus defining its potential applications. Diesters of tartaric and mucic acids were characterized in terms of their melting temperature (Tm), enthalpy of fusion (ΔHf), crystallization temperature (Tc), enthalpy of crystallization. (ΔHc), and supercooling temperature (ΔT = Tm − Tc) using DSC. Thermal stability was investigated using TGA analysis, and the entropy of fusion (ΔSf) was calculated using the Gibbs equation based on the Tm and ΔHf data. The results are presented in Table 1. Both TA and MA decompose upon melting at 172 °C and 205 °C, respectively, which excludes them from TES applications in their pristine form. However, the esterification of TA with fatty alcohols gave products with Tm values ranging from 67 to 94 °C ± 2 °C and ΔHf values ranging from 133 to 221 J g−1 ± 5%. These values of Tm are comparable to those exhibited by monoamides74 and triglycerides.75 In the series of tartaric acid esters, the increase in the alkyl chain length increased the Tm, reaching 94 °C ± 2 °C for C22_TA_C22 (Fig. 2). This trend can be attributed to stronger intermolecular interactions formed by longer carbon chains.4 As the carbon chain length increases in a series of compounds (e.g. alkanes or fatty acids),76 an increase in the melting temperature and enthalpy of fusion is observed due to stronger van der Waals interactions between molecules and stronger instantaneous dipol-induced dipol interactions. Longer chains can align and pack more efficiently in the solid state, creating a more ordered structure. The greater order in the solid state means a larger entropy increase upon melting, contributing to a higher enthalpy of melting. Therefore, with the increase in the alkyl chain length from C12_TA_C12 to C18_TA_C18, ΔHf values increased, reaching 221 J g−1 ± 5% for C18_TA_C18. | ||
| Fig. 2 DSC traces showing the second heating cycle for the tartaric acid diesters (C12_TA_C12, C16_TA_C16, C18_TA_C18, and C22_TA_C22) and the first heating cycle of the mucic acid diesters (C12_MA_C12 and C18_MA_C18), at a ramp rate of 10 °C min−1. | ||
| Compound | T m [°C] ± 2 °C | ΔHf [J g−1] ± 5% | T c [°C] ± 2 °C | ΔHc [J g−1] ± 5% | ΔSf [J mol−1 K−1] | T d [°C] |
|---|---|---|---|---|---|---|
| a ΔHf taken from the 1st heating cycle due to the decomposition in the second cycle. D – decomposition. | ||||||
| TA | D | — | — | — | — | 172 |
| C12_TA_C12 | 67 | 133 | 51 | 132 | 190 | 288 |
| C16_TA_C16 | 77 | 162 | 60 | 169 | 277 | 286 |
| C18_TA_C18 | 82 | 221 | 71 | 218 | 408 | 297 |
| C22_TA_C22 | 94 | 203 | 80 | 201 | 424 | 289 |
| MA | D | — | — | — | — | 205 |
| C12_MA_C12 | 137 | 205a | — | — | — | 150 |
| C18_MA_C18 | 124 | 194a | — | — | — | 169 |
In the series of tartaric acid diesters, the ΔHf is lower than that for the pure paraffins (C12: 215 J g−1; C16: 235 J g−1; C18: 237 J g−1; and C22: 249 J g−1). Meanwhile, in the literature, it is observed that an increasing number of hydroxyl groups leads to higher melting temperatures and enthalpies, due to more hydrogen bonding that creates a more rigid and ordered structure in the solid state.77 In this case, the two factors counteract each other: (i) longer chains increase hydrophobicity, which can reduce the effectiveness of hydrogen bonding in some cases; (ii) the placement of hydroxyl groups along the carbon chain can affect how easily the molecules can pack in the solid state.
This can also explain why the elongation of the alkyl chain to C22 decreased the ΔHf to 203 J g−1 ± 5%, which can be most probably ascribed to the weakening of the hydrogen bonds,74 as well as van der Waals interactions, simultaneously contributing to lower ΔHf values.
Moreover, the melting peak of C22_TA_C22 displayed heterogeneity in the thermogram registered at 10 °C min−1 (Fig. 2), suggesting possible polymorphism, common for systems with lengthy alkyl chains.75 Nevertheless, when heated at a rate of 1 °C min−1, only one melting transition was observed (Fig. S23†). The melting peak became noticeably narrower, leading to a slight reduction in Tm, which is a typical outcome observed at lower heating rates.78
Comparing Tm and ΔHf values for the investigated hydroxyl group-rich diester of stearyl alcohol and tartaric acid (ΔHf of 221 J g−1 ± 5% and Tm = 82 °C ± 2 °C) and a similar diester without hydroxyl groups (the ester of stearyl alcohol and succinic acid; ΔHf of 194 J g−1 ± 5% and Tm = 64 °C ± 2 °C), a significant role of hydrogen bonds in shifting Tm and ΔHf toward increased values was observed.
To further investigate the influence of the hydroxyl groups on the thermal properties of fatty esters, the tartaric acid core was replaced with mucic acid.79 Increasing the number of hydroxyl groups in the system and, therefore, possibly obtaining more robust hydrogen bonding interactions led to higher Tm values of the resulting esters (from 124 to 137 °C ± 2 °C) than those reported for the series based on tartaric acid (from 67 to 94 °C ± 2 °C). Intriguingly, in contrast to tartaric acid esters, the Tm and ΔHf values decreased with the increase in the alkyl chain length. A similar observation was reported for the ΔHf values of behenic acid monoesters.80 It was found that a larger number of hydroxyl groups could disturb the intermolecular packing of the alkyl chains and thus weaken the van der Waals interactions, resulting in decreased ΔHf, and this topic is further discussed in the Crystallography section.
The high thermal stability of the investigated tartaric acid esters, ranging from 286 °C for C16_TA_C16 to 297 °C for C18_TA_C18, which is well above their maximum working temperature (defined as Tm + 10°),9 indicates their high potential for practical applications (Fig. 3a). The elongation of the alkyl chain in both the TA- and MA-derived series from C12 to C18 led to an increase in the decomposition temperature from 288 °C for C12_TA_C12 to 297 °C for C18_TA_C18 and from 150 °C for C12_MA_C12 to 169 °C for C18_MA_C18. On the other hand, the incorporation of two additional hydroxyl groups into the system led to a marked reduction in thermal stability from 288 °C for C12_TA_C12 to 150 °C for C12_MA_C12. This finding underscores the importance of considering the relationship between possible van der Waals interactions and hydrogen bonding (whose effects can counteract each other) in the future design of sustainable PCMs. Most importantly, despite MA derivatives melting >120 °C with remarkable values of ΔHf, they showed no repetitive phase transitions in a subsequent heating–cooling experiment, which was performed with DSC (Fig. S22†), manifesting their degradation and hindering their application in TES. In contrast, tartaric acid esters, with their finely tuned operating temperature ranging from 67 to 94 °C ± 2 °C and high storage capacity reaching up to 221 J g−1 ± 5% for C18_TA_C18 (which melts at 82 °C ± 2 °C), showed no significant changes after being initially evaluated over three heating–cooling cycles (Fig. S21†).
 | ||
| Fig. 3 (a) TGA curves of tartaric acid (C12_TA_C12, C16_TA_C16, C18_TA_C18, and C22_TA_C22), mucic acid diester (C12_MA_C12 and C18_MA_C18) and their precursors (TA and MA) at a 10 °C min−1 heating rate, and (b) latent heat values of C18_TA_C18 after different thermal cycles, at a ramp rate of 10 °C min−1. | ||
To further confirm the stability of the C18_TA_C18 diester under repetitive heating and cooling conditions, long-term cycling tests were performed for a 5.1 mg sample using DSC (Fig. 3b). C18_TA_C18 was heated and cooled 500 times at 10 °C min−1, in a temperature range of 60–95 °C. The excellent stability of the material was confirmed by no changes in the enthalpy of melting and little supercooling (11 °C). After 500 cycles, the melting peak maintained its original shape, and the Tm showed no deviation. Additional FT-IR and 1H NMR spectra were recorded for the sample after 500 cycles (Fig. S11 and Fig. S26†) and evidenced no changes compared to spectra recorded before cycling, highlighting the potential of tartaric acid esters as promising PCMs with excellent stability during cycling.
Understanding the influence of hydrogen bonds on phase change behavior
The impact of hydrogen bonds on the phase transition behavior and heat storage in PCMs is a key aspect of their design. Hydrogen bonding interactions can either enhance or substantially degrade the thermal properties of PCMs, as discussed in a recent review by Y. Liu et al.81 Interestingly, in the two series of compounds investigated in the present paper, both trends can be observed. To better understand the nature of these interactions and their influence on the thermal properties of the series of diesters, the next section presents detailed characterization of the molecular interactions occurring in both series, with a particular emphasis on hydrogen bonds.FT-IR
Each atom or group of atoms in a molecule contributes to the total enthalpy of melting, depending on its size, dipole moment, ability to form hydrogen bonds, and the surrounding environment. The sum of these interactions is reflected in the Tm and enthalpy.82 To better understand the influence of additional hydroxyl groups in the ester core on a material's thermal properties, we used FT-IR analysis, which offers a precise interpretation of the structural changes found during the phase transitions of the investigated diesters. The FT-IR spectra of the representative C18_TA_C18 and the corresponding C12_MA_C12 in the liquid and solid states are presented in Fig. 4. | ||
| Fig. 4 The FTIR spectra of (a) C18_TA_C18 and (b) C12_MA_C12 collected at room temperature and after melting. | ||
Two peaks at 3484 cm−1 and 3290 cm−1 observed in the solid state of the tartaric diester confirmed the presence of hydrogen bonds formed by the hydroxyl groups. Typically, unbonded hydroxyl groups absorb infrared light at a higher wavenumber than that observed for the investigated esters (>3500 cm−1).83 After melting, only one peak at 3543 cm−1 was observed, indicating the disruption of the hydrogen bonds and the presence of free hydroxyl groups. This observation was confirmed by the redshifted bands corresponding to symmetric and antisymmetric –CH2 stretching at 2852 cm−1 and 2919 cm−1 (shifted by 4–5 cm−1 and 5–7 cm−1, respectively) observed after melting. These shifts are related to the disruption of the diester crystal packing after melting, indicating increased disorder and mobility of the hydrocarbon chain.80,84 Furthermore, the carbonyl band, which is sensitive to the formation of hydrogen bonds, was analyzed.85 In the solid state, symmetric and asymmetric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrational bands at 1724 cm−1 and 1758 cm−1, respectively, were noticeable. As the temperature increased to the phase transition temperature, the hydrogen bonds broke, leading to a new vibrational band at 1749 cm−1. The extension of the alkyl chains in tartaric acid diesters from C18_TA_C18 to C22_TA_C22 resulted in a weakening of the hydrogen bonds, as shown by a shift in the hydroxyl bands (from 3484 cm−1 and 3290 cm−1 to 3487 cm−1 and 3306 cm−1)86 (Fig. 4a and Fig. S24†). These changes may explain the decrease in enthalpy with increasing chain length. Moreover, when the alkyl chain was extended to 22 carbon atoms, the largest increase in Tm in the series was observed.
O vibrational bands at 1724 cm−1 and 1758 cm−1, respectively, were noticeable. As the temperature increased to the phase transition temperature, the hydrogen bonds broke, leading to a new vibrational band at 1749 cm−1. The extension of the alkyl chains in tartaric acid diesters from C18_TA_C18 to C22_TA_C22 resulted in a weakening of the hydrogen bonds, as shown by a shift in the hydroxyl bands (from 3484 cm−1 and 3290 cm−1 to 3487 cm−1 and 3306 cm−1)86 (Fig. 4a and Fig. S24†). These changes may explain the decrease in enthalpy with increasing chain length. Moreover, when the alkyl chain was extended to 22 carbon atoms, the largest increase in Tm in the series was observed.
For mucic acid diesters with four hydroxyl groups, the phase change occurred at a higher temperature, suggesting a robust network of hydrogen bonds that needed more energy to break. Similarly to the tartaric acid diester, the peaks corresponding to the hydroxyl groups at 3325 cm−1 and 3400 cm−1 disappeared during melting. Nevertheless, this time, they were replaced by a broad peak at 3437 cm−1 (Fig. 4b). Interestingly, in the spectrum of the solid C12_MA_C12, only one peak within the CO bond region, at 1743 cm−1, was observed. After melting, this peak remained mostly unchanged, but a new peak appeared at 1798 cm−1, at a different position to the new peak found with the tartaric acid diester. It is worth noting that further analysis of the C12_MA_C12 diester after recrystallization did not reveal the characteristic peaks of the original ester. Instead, the spectrum reflected that of the molten diester (Fig. S25†). This finding suggests decomposition/evaporation of C12_MA_C12 after melting, even though the TGA showed no mass loss up to 220 °C (Fig. S17 and S18†). The peak formed at 1798 cm−1 and the broad band at 3437 cm−1 suggest that a mixture of diester and lactone was formed after melting. The 1H NMR spectrum recorded after keeping the sample melted and stirred for 3 days further confirms the absence of –OH groups, as evidenced by the disappearance of their characteristic peaks (δ 4.82–4.63 and δ 3.75 (Fig. S12†)). Additionally, a slightly shifted peak at 4.5 ppm suggests the formation of a lactone. This is presumably caused by acid catalysis induced by water, as previously reported for other polyhydroxylated compounds. Some water is essentially always present in such hygroscopic polyhydroxylated compounds, which during prolonged high-temperature treatment can catalyze dehydration and cyclization reactions, shifting the equilibrium of H2O towards H3O+ at 100 °C.87 The degradation reaction is only observed during melting of mucic acid derivatives, which melt at higher temperature (124 and 137 °C) compared to the esters of tartaric acid (67–94 °C).
Crystallography
To gain further insight into the role of hydrogen bonding in the hydroxyl group-rich diesters, we investigated the crystal structures of the representative C12_TA_C12 and C18_TA_C18 obtained through single-crystal X-ray diffraction using synchrotron radiation on the best quality crystals available. It should be noted that determining the crystal structure of long-chain organic compounds from powder samples is often a labor-intensive process and not always feasible.51 This was a problem we faced in this study, which limited the analysis to two materials and required synchrotron radiation. It was not possible to collect full data for C18_TA_C18 but enough to unambiguously resolve the structure. C12_TA_C12 crystallized in the monoclinic C2 space group with four molecules in a unit cell. Taking into consideration the hydroxyl groups’ orientation, two out of the four molecules were directed toward each other with carbon chains arranged alternately in homologous, diagonal layers, parallel to the bc crystallographic plane (Fig. 5).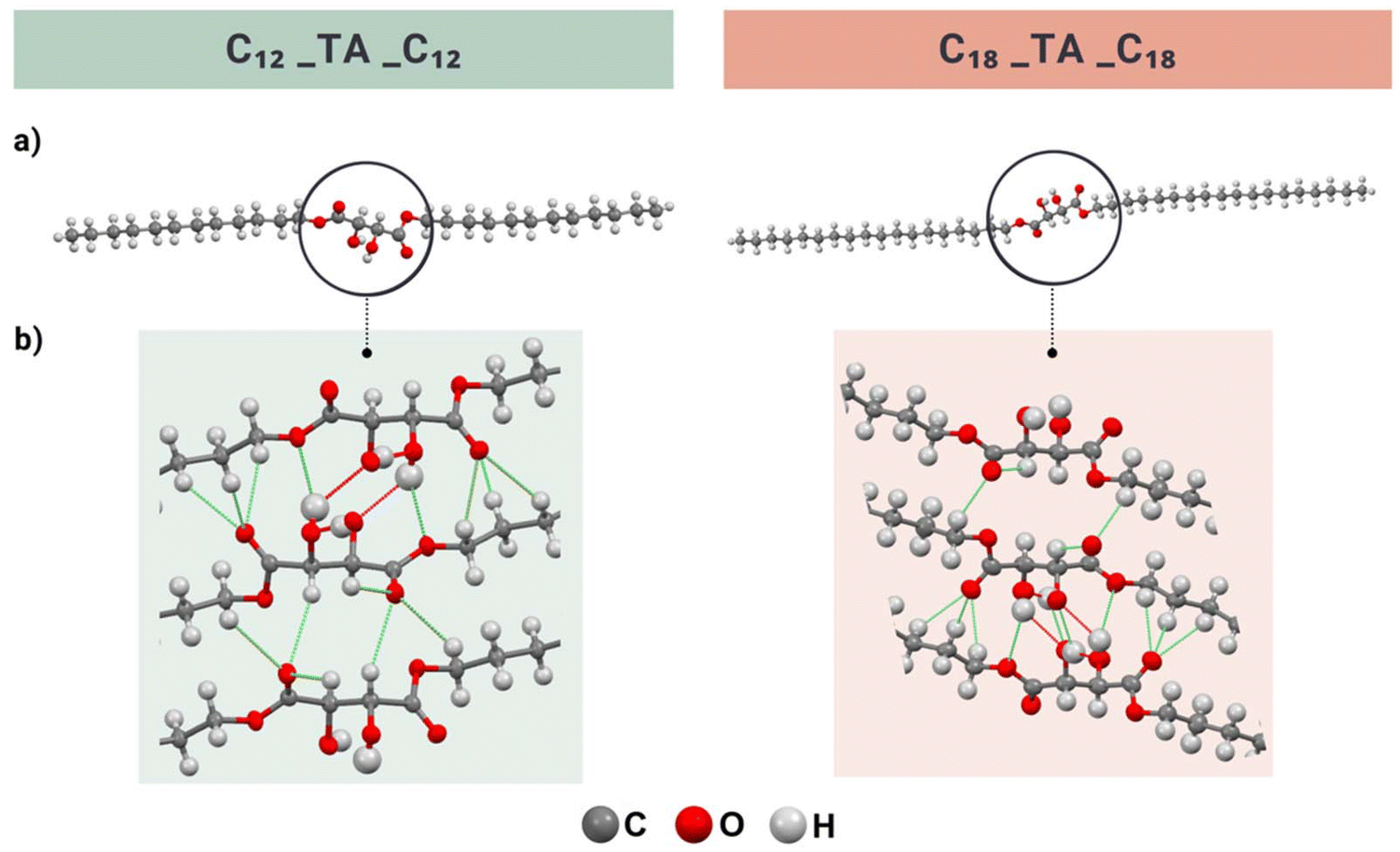 | ||
| Fig. 5 (a) Lettice structure of C12_TA_C12 and C18_TA_C18 and (b) hydrogen bonding and core group interactions (red represents strong hydrogen bonds and green represents non-classical hydrogen bonds) in C12_TA_C12 and C18_TA_C18, viewed down the b axis. | ||
Adjacent chains from adjacent sheets were offset face-to-face, with an interplanar distance of 4.253 Å (C1–C1′ distance) (Fig. S29†). A similar molecular arrangement is observed in the structure of C18_TA_C18, which also crystallizes in the monoclinic C2 space group, with four molecules in the unit cell. Here, the interplanar distance is 4.010 Å (C40–C40′ distance), indicating denser packing in the C18_TA_C18 crystal (Fig. S30†).
In the studied esters, hydrogen bonds typically vary in length from 2.6–3.0 Å (O–H⋯O); within this range, stronger hydrogen bonds tend to be shorter and have closer to linear angles.88 In contrast to conventional hydrogen bonds, non-classical hydrogen bonds exhibit weaker strength and a longer distance between the donor and acceptor, extending up to ∼3.5 Å.89,90 These weak C–H⋯O bonds play a crucial role in stabilizing molecules, particularly in proteins, sugars, DNA, and crystal packing, having a significant impact on the thermal properties of materials.91
In the TA crystal structure, different types of hydrogen bonds were present, including shorter bonds (2.6–2.7 Å), formed by carboxylic acid hydrogens, and slightly longer bonds (2.8–2.9 Å), formed between the hydroxyl groups.92 Both types of hydrogen bonds had O–H⋯O angles ≥165°, indicating their strong character. In turn, the C12_TA_C12 molecule participated in four strong, conventional intermolecular hydrogen bonding interactions and several weaker, non-classical hydrogen bonds (Fig. 5, Tables S2 and S4†). Each hydroxyl group in the core acted as both a hydrogen bond donor and acceptor, forming connections with hydroxyl groups from two neighboring molecules. This configuration created an extensive network of hydrogen bonds formed by the tartaric acid-derived core of the adjacent molecules. In C12_TA_C12, the hydrogen bonds had similar lengths (O–H⋯O), ranging from 2.64 to 2.83 Å, but varied significantly in their angles. When the hydroxyl group acted as a donor, the bond angle was 174°, whereas when it acted as an acceptor, the angle was 126°. The notable presence of a considerable number of C–H⋯O bonds was particularly interesting. In the C12_TA_C12 crystal, these bonds also formed between the oxygens of the carbonyl groups and the hydrogens of the alkyl groups nearest to the core or the hydrogen atoms of the hydroxyl group. They exhibited a length range of 3.07–3.55 Å, and the angles between the donor and acceptor ranged from 116–155°. Given the considerable length of the C–H⋯O bonds, the acceptor can be connected to two or three H atoms, resulting in bifurcated and trifurcated bonds.90 In the C18_TA_C18 molecule, a similar hydrogen bonding pattern is observed. Specifically, there are four strong hydrogen bonds with lengths ranging from 2.63 to 2.67 Å and angles from 123° to 168° (Fig. 5, Tables S3 and S4†). It was also observed that the hydrogen atom from the hydroxyl group forms both intramolecular and intermolecular hydrogen bonds, with a length of 3.07 Å and an angle of 114°.
The desired PCM should have enough strong hydrogen bonds in the crystalline phase to require a significant amount of energy to break them, resulting in efficient PCM loading. On the other hand, the H-bonds need to be capable of rearranging during the reverse process.6 In the series of hydroxyl-group rich fatty esters, long symmetrical carbon chains and ester groups contribute to the flexibility of the molecule, facilitating the rearrangement of the crystal lattice upon crystallization.3 Thus, the interplay between the hydrogen bonds and van der Waals interactions is crucial for ensuring both a high enthalpy of fusion and the stability of the PCM.
Hirshfeld surface analysis
The intermolecular interactions and their influence on crystal packing were further assessed using Hirshfeld surface analysis with CrystalExplorer software.93 The analysis helps to visualize and quantify the interactions within a molecular crystal. The colors indicate the intensity and distance of contacts, where the normalized contact distance is based on the distance between the nuclei on the surface and the nearest nuclei outside the surface. The contacts at the van der Waals radius distance are shown in white, while those at shorter or longer distances from the van der Waals radii are displayed in red and blue, respectively. In contrast to tartaric acid molecules,92 with numerous intensely red spots, distributed throughout the surface, which confirmed the presence of an extensive hydrogen bond network (Fig. 6), on the surface of C12_TA_C12 and C18_TA_C18, a significant number of white regions were observed, corresponding to van der Waals interactions. In unmodified tartaric acid, both hydroxyl and carboxyl groups formed strong H-bonds, as indicated by the intense red spots. In turn, in the tartaric acid-derived esters, the red spots present on the hydroxyl groups were much more intense than those located on the carbonyl groups. This observation supports the much weaker character of the H-bonds formed by the CO group, resulting from their much longer distances, as discussed in the previous section. This plot can be decomposed into atoms involved in specific interactions and their relative percentage contributions (Fig. 6).
 | ||
| Fig. 6 (a) 3D Hirshfeld surface area and (b) 2D Hirshfeld fingerprint plots of TA, C12_TA_C12 and C18_TA_C18. | ||
To calculate the percentage contribution of specific interactions, a two-dimensional fingerprint plot was generated. In C12_TA_C12, the hydrogen bonds (depicted as long, slender spikes) made up 15% of the total Hirshfeld surface (Fig. S32†). The great majority (83%) constituted H⋯H interactions, observed as a characteristic area between the spikes. In the case of C18_TA_C18, hydrogen bonds make up 11% of the interactions, while H⋯H interactions account for almost 88% (Fig. S33†). This is not surprising considering the longer alkyl chains present in the structure. Additionally, the highlighted red area in the middle of the fingerprint plot is more intense in the case of the ester with a longer alkyl chain, indicating that a substantial fraction of points on the Hirshfeld surfaces consisted of closely aligned H⋯H contacts between the long n-alkyl chains (i.e., almost linear orientation between adjacent molecules).
This leads to a significant number of dispersive interactions.94,95 For unmodified tartaric acid, the percentage contribution of specific interactions was the opposite. The H⋯O/O⋯H interactions constituted as much as 71%, while the H⋯H interactions accounted for 15% (Fig. S31†). The lighter blue color of the spikes in the TA histogram compared to those in the ester derivative indicated a more frequent occurrence of hydrogen interactions in the unmodified tartaric acid crystal structure. The examination of both the precursor and one of the diesters facilitated the discernment of distinct patterns. Through an analysis of enthalpy changes within the compound series, we speculate that further chain extension is unlikely to exert a pronounced impact on the distribution of hydrogen bonds, although it may influence their intensity.
Biodegradability
The ultimate biodegradability (i.e., complete mineralization to CO2, H2O, and microbial biomass) was tested at concentrations of approximately 15 mg L−1. C12_TA_C12 (78%) and C16_TA_C16 (38%) showed appreciable degradation (Fig. 7a). None of these compounds could be considered readily biodegradable (at least 60% degradation within a 10-day window, starting when the degradation reaches 10%),61 yet C12_TA_C12 was close to the threshold (a degradability window of 12.2 days). All the other compounds showed degradability within the error of the measurement. Considering structure–biodegradability relationships, the tested diesters do not possess fragments that would make them resistant to degradation (e.g., branching and presence of halogen atoms); in contrast, the straight alkyl chains found in their structure are usually readily degradable.96 Nevertheless, if such alkyl chains are too long, they can exhibit considerable microbial toxicity.97 It is, therefore, possible that some of the diesters were not degraded, not because they were recalcitrant but because the concentrations used in the test were too high and inhibited the activity of microbial inoculum. Concentrations in the mg L−1 range are unlikely to occur in the environment (organic trace pollutants are present in surface water in concentrations of ng L−1 to μg L−1); consequently, toxic inhibition of microorganisms is also unlikely.98 Unfortunately, the respirometric method does not allow for testing lower concentrations; however, we could examine if toxic inhibition indeed occurred in our test or not. To this end, we conducted another test in which easily degradable sodium benzoate was added to all the samples containing diesters and the degradation of sodium benzoate was followed for 5 days (since within this time, sodium benzoate reached over 60% degradation, but none of diesters did so). | ||
| Fig. 7 (a) Ultimate biodegradation of tartaric and mucic acid diesters. SB: sodium benzoate (positive control), (b) toxicity of tartaric acid diesters (C12_TA_C12, C16_TA_C16, C18_TA_C18, and C22_TA_C22) and mucic acid diesters (C12_MA_C12 and C18_MA_C18) to microbial inoculum. Solid lines represent the biodegradation of SB in the presence of diesters at concentrations of approximately 15 mg L−1, while dashed lines show tests conducted at diester concentrations of approximately 4 mg L−1. | ||
Indeed, all the diesters inhibited the activity of the microbial inoculum, as shown by the biodegradation of sodium benzoate tested alone being higher than its biodegradation in the presence of diesters (Fig. 7). As expected, both longer diester alkyl chains and higher diester concentrations gave a greater degree of inhibition. Interestingly, the didodecyl substituted ester of mucic acid (C12_MA_C12) was more toxic toward the inoculum than the ester of tartaric acid having the same substituents (C12_TA_C12, Fig. 7a) when tested at concentrations of 15 mg L−1, which might explain why the latter was highly degraded, but the former was not (Fig. 7b). Considering that homologs having short alkyl chains are degradable to a great extent (despite showing some degree of inoculum inhibition) and that long chain homologs showed considerable inhibition of the inoculum (at 15 mg L−1), it can be expected that the latter are also degradable if tested at lower, non-inhibitory concentrations. However, more detailed tests at lower concentrations would be required to confirm this hypothesis (e.g., OECD 309 biodegradation simulation test).99
Conclusions
A new group of sustainable materials with physical properties suitable for application as PCMs have been developed using renewable, inexpensive precursors. Sugar acid-derived esters showed very high enthalpies of fusion, from 133 up to 221 J g−1 ± 5%, and Tm values ranging from 67 to 137 °C ± 2 °C, suitable for low-cost TES. Increasing the number of hydroxyl groups in the molecule improved thermal properties but reduced stability, except for a tartaric acid derivative that remained stable over 500 heating/cooling cycles. This hypothesis, however, would need to be tested on samples with more OH groups.In addition to their sustainability, confirmed by green chemistry metrics, the materials are not expected to be persistent in the environment. Tartaric acid esterified with dodecyl chains showed high biodegradability (slightly below the readily biodegradable criterion), while the hexadecyl homolog showed lower biodegradability. Higher homologs were not degraded, likely due to the high test concentrations inhibiting microbial activity. However, at environmentally realistic concentrations (at least three orders of magnitude lower), toxic inhibition is not expected, and the biodegradability of larger homologs may be higher. These bioderived PCMs show promise for sustainable thermal energy storage applications, balancing hydrogen bonding and van der Waals interactions to tune physical properties.
Author contributions
Conceptualization: M. G., K. M., and A. B.-S., data curation: M. G., formal analysis: M. G. and A. B.-S., funding acquisition: M. G. and A. B.-S., investigation: M. G. and M. M., methodology: M. G., M. M., K. M., and A. B.-S., visualization: M. G., writing – original draft: M. G., A. B.-S., and S. S. and writing – review & editing: A. C., D. T., and K. M.Data availability
The data supporting this article have been included as part of the ESI. Crystallographic data for C12_TA_C12 and C18_TA_C18 have been deposited at the Cambridge Structural Database, such as CCDC 2369962 and 2369963.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. G., A. B.-S., and K. M. acknowledge the Polish National Agency for Academic Exchange under the Strategic Partnerships programme (BPI/PST/2021/1/00039)., A. B.-S. acknowledges the Excellence Initiative – Research University Programme under Grant No. 04/050/SDU/10-21-01, Rector's Pro-quality Grant, Silesian University of Technology 04/050/RGJ23/0168 and the habilitation grant 04/050/RGH22/0162. M. G. acknowledges the Silesian University of Technology (04/050/BKM24/0196). The contributions of Mr Szymon Ruczka and Dr Anna Kolanowska are appreciated.This research was undertaken using the MX2 beamline at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector.
References
- A. A. Mehrizi, H. Karimi-Maleh, M. Naddafi and F. Karimi, J. Energy Storage, 2023, 61, 106859 CrossRef.
- I. Energy Agency, Net Zero by 2050 - A Roadmap for the Global Energy Sector, 2050 Search PubMed.
- N. Soodoo, K. D. Poopalam, L. Bouzidi and S. S. Narine, J. Energy Storage, 2022, 51, 104355 CrossRef.
- A. Stamatiou, M. Obermeyer, L. J. Fischer, P. Schuetz and J. Worlitschek, Renewable Energy, 2017, 108, 401–409 CrossRef CAS.
- S. L. Piper, C. M. Forsyth, M. Kar, C. Gassner, R. Vijayaraghavan, S. Mahadevan, K. Matuszek, J. M. Pringle and D. R. MacFarlane, RSC Sustainability, 2023, 207890 Search PubMed.
- K. Matuszek, R. Vijayaraghavan, M. Kar and D. R. Macfarlane, Cryst. Growth Des., 2020, 20, 1285–1291 CrossRef CAS.
- Y. Yuan, N. Zhang, W. Tao, X. Cao and Y. He, Renewable Sustainable Energy Rev., 2014, 29, 482–498 CrossRef CAS.
- K. Matuszek, S. L. Piper, A. Brzęczek-Szafran, B. Roy, S. Saher, J. M. Pringle and D. R. Macfarlane, Adv. Mater., 2024, 36, 23 CrossRef PubMed.
- S. L. Piper, C. M. Forsyth, M. Kar, C. Gassner, R. Vijayaraghavan, S. Mahadevan, K. Matuszek, J. M. Pringle and D. R. MacFarlane, RSC Sustainability, 2023, 1, 470–480 RSC.
- K. Du, J. Calautit, Z. Wang, Y. Wu and H. Liu, Appl. Energy, 2018, 220, 242–273 CrossRef CAS.
- J. F. Hinojosa, S. F. Moreno and V. M. Maytorena, Energies, 2023, 16(7), 3078 CrossRef CAS.
- D. Cabaleiro, F. Agresti, S. Barison, M. A. Marcos, J. I. Prado, S. Rossi, S. Bobbo and L. Fedele, Appl. Therm. Eng., 2019, 159, 113868 CrossRef CAS.
- D. Patel, W. Wei, H. Singh, K. Xu, C. Beck, M. Wildy, J. Schossig, X. Hu, D. C Hyun, W. Chen and P. Lu, ACS Sustainable Chem. Eng., 2023, 11, 11570–11579 CrossRef CAS PubMed.
- A. E. Kabeel, A. Khalil, S. M. Shalaby and M. E. Zayed, Energy Convers. Manage., 2016, 113, 264–272 CrossRef CAS.
- M. Faegh and M. B. Shafii, Desalination, 2017, 409, 128–135 CrossRef CAS.
- R. Padovan and M. Manzan, Sol. Energy, 2014, 103, 563–573 CrossRef.
- Q. Al-Yasiri and M. Szabó, J. Build. Eng., 2021, 36, 102122 CrossRef.
- F. Souayfane, F. Fardoun and P. H. Biwole, Energy Build., 2016, 129, 396–431 CrossRef.
- X. Wang, W. Li, Z. Luo, K. Wang and S. P. Shah, Energy Build., 2022, 260, 111923 CrossRef.
- M. C. Floros and S. S. Narine, Energy, 2016, 115, 924–930 CrossRef CAS.
- H. Mehling and M. A. White, Chem. Phys. Impact., 2023, 6, 100222 CrossRef.
- N. R. Jankowski and F. P. McCluskey, Appl. Energy, 2014, 113, 1525–1561 CrossRef.
- Q. Zhang, H. Yan, Z. Zhang, J. Luo, N. Yin, Z. Tan and Q. Shi, Mater. Today Sustain., 2021, 11–12, 100064 Search PubMed.
- S. Kahwaji and M. A. White, Molecules, 2021, 26, 21 CrossRef PubMed.
- D. Rozanna, T. G. Chuah, A. Salmiah, T. S. Y. Choong and M. Sa'ari, Int. J. Green Energy, 2005, 1, 495–513 CrossRef.
- J. C. Van Miltenburg, H. A. J. Oonk and L. Ventola, J. Chem. Eng. Data, 2001, 46, 90–97 CrossRef CAS.
- W. Acree and J. S. Chickos, J. Phys. Chem. Ref. Data, 2017, 46, 1 CrossRef.
- R. Ravotti, O. Fellmann, N. Lardon, L. J. Fischer, A. Stamatiou and J. Worlitschek, Appl. Sci., 2019, 9, 2 Search PubMed.
- H. Fatahi, J. Claverie and S. Poncet, Appl. Sci., 2022, 12(23), 12019 CrossRef CAS.
- L. Raghunanan, M. C. Floros and S. S. Narine, Thermochim. Acta, 2016, 644, 61–68 CrossRef CAS.
- H. Nazir, M. Batool, F. J. Bolivar Osorio, M. Isaza-Ruiz, X. Xu, K. Vignarooban, P. Phelan, Inamuddin and A. M. Kannan, Int. J. Heat Mass Transfer, 2019, 129, 491–523 CrossRef CAS.
- D. Feldman, D. Banu and D. Hawes, Sol. Energy Mater. Sol. Cells, 1995, 36, 311–322 CrossRef CAS.
- A. A Aydin, Sol. Energy Mater. Sol. Cells, 2013, 113, 44–51 CrossRef.
- A. A. Aydin and H. Okutan, Sol. Energy Mater. Sol. Cells, 2011, 95, 2417–2423 CrossRef CAS.
- A. A. Aydin and A. Aydin, Sol. Energy Mater. Sol. Cells, 2012, 96, 93–100 CrossRef CAS.
- A. Sari, A. Biçer and A. Karaipekli, Mater. Lett., 2009, 63, 1213–1216 CrossRef CAS.
- A. Stamatiou, M. Obermeyer, L. J. Fischer, P. Schuetz and J. Worlitschek, Renewable Energy, 2017, 108, 401–409 CrossRef CAS.
- Ö. Kaplan, N. G Tosun, İ. Gökçe and C. Alkan, Energy Sources, Part A, 2023, 45, 608–622 CrossRef.
- R. Ravotti, O. Fellmann, N. Lardon, L. J. Fischer, A. Stamatiou and J. Worlitschek, Appl. Sci., 2018, 8, 7 Search PubMed.
- E. P. del Barrio, A. Godin, M. Duquesne, J. Daranlot, J. Jolly, W. Alshaer, T. Kouadio and A. Sommier, Sol. Energy Mater. Sol. Cells, 2017, 159, 560–569 CrossRef CAS.
- A. Solé, H. Neumann, S. Niedermaier, I. Martorell, P. Schossig and L. F. Cabeza, Sol. Energy Mater. Sol. Cells, 2014, 126, 125–134 CrossRef.
- C. Barreneche, A. Gil, F. Sheth, A. I Fernández and L. F. Cabeza, Sol. Energy, 2013, 94, 344–351 CrossRef CAS.
- A. Kaizawa, N. Maruoka, A. Kawai, H. Kamano, T. Jozuka, T. Senda and T. Akiyama, Heat Mass Transfer, 2008, 44, 763–769 CrossRef CAS.
- A. Sari, A. Biçer and A. Karaipekli, Mater. Lett., 2009, 63, 1213–1216 CrossRef CAS.
- A. Sari, R. Eroglu, A. Biçer and A. Karaipekli, Chem. Eng. Technol., 2011, 34, 87–92 CrossRef CAS.
- A. Biçer and A. Sar, Sol. Energy Mater. Sol. Cells, 2012, 102, 125–130 CrossRef.
- A. Sari, Energy Convers. Manage., 2012, 64, 68–78 CrossRef CAS.
- A. Sari and A. Biçer, Sol. Energy Mater. Sol. Cells, 2012, 101, 114–122 CrossRef CAS.
- S. H. Yalkowsky and D. Alantary, J. Pharm. Sci., 2018, 107, 1211–1227 CrossRef CAS PubMed.
- S. L. Piper, C. M. Forsyth, M. Kar, D. R. MacFarlane, K. Matuszek and J. M. Pringle, Mater. Adv., 2021, 2, 7650–7661 RSC.
- K. D. Poopalam, L. Raghunanan, L. Bouzidi, S. K. Yeong and S. S. Narine, Sol. Energy Mater. Sol. Cells, 2019, 201 DOI:10.1002/er.4711.
- T. Inagaki and T. Ishida, J. Phys. Chem. C, 2016, 120, 7903–7915 CrossRef CAS.
- B. Gaida, J. Kondratowicz, S. L. Piper, C. M. Forsyth, A. Chrobok, D. R. Macfarlane, K. Matuszek and A. Brzeczek-Szafran, ACS Sustainable Chem. Eng., 2024, 12, 623–632 CrossRef CAS.
- N. Biedermann, J. Schnizer, D. Lager, M. Schnürch and C. Stanetty, J. Org. Chem., 2024, 89(8), 5573–5588 CrossRef CAS PubMed.
- D. Aragão, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson and T. T. Caradoc-Davies, J. Synchrotron Radiat., 2018, 25, 885–891 CrossRef PubMed.
- T. M. McPhillips, S. E. McPhillips, H. J. Chiu, A. E. Cohen, A. M. Deacon, P. J. Ellis, E. Garman, A. Gonzalez, N. K. Sauter, R. P. Phizackerley, S. M. Soltis and P. Kuhn, J. Synchrotron Radiat., 2002, 9, 401–406 CrossRef CAS PubMed.
- W. Kabsch, J. Appl. Crystallogr., 1993, 26, 795–800 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- OECD, Test No. 301: Ready Biodegradability, OECD Publishing, 1992, 301 Search PubMed.
- K. Izawa, Y. Amino, M. Kohmura, Y. Ueda and M. Kuroda, Comprehensive Natural Products II: Chemistry and Biology, 2010, vol. 4, pp. 631–671 Search PubMed.
- Y. Ukaji and T. Soeta, Compr. Chirality, 2012, 3, 176–201 CAS.
- I. Goldberg and J. S. Rokem, Encyclopedia of Microbiology, 3rd edn, 2009, pp. 421–442 Search PubMed.
- E. Anet and T. Reynolds, Nature, 1954, 174, 930 CrossRef CAS.
- R. Sakuta and N. Nakamura, Int. J. Mol. Sci., 2019, 20 Search PubMed.
- D. Barth and M. G. Wiebe, Appl. Microbiol. Biotechnol., 2017, 101, 4033–4040 CrossRef CAS PubMed.
- D. Mojzita, M. Wiebe, S. Hilditch, H. Boer, M. Penttilä and P. Richard, Appl. Environ. Microbiol., 2010, 76, 169–175 CrossRef CAS PubMed.
- D. Concha, K. Rodríguez-Núñez, L. Castillo, R. Martínez and C. Bernal, Biochem. Eng. J., 2022, 187, 108646 CrossRef CAS.
- O. Okogeri and V. N. Stathopoulos, Int. J. Thermofluids, 2021, 10, 100081 CrossRef CAS.
- P. Munkajohnpong, C. Kesornpun, S. Buttranon, J. Jaroensuk, N. Weeranoppanant and P. Chaiyen, Biofuels, Bioprod. Biorefin., 2020, 14, 986–1009 CrossRef CAS.
- A. Krishnan, B. McNeil and D. Stuart, Front. Bioeng. Biotechnol., 2020, 8, 610936 CrossRef PubMed.
- C. R. Mcelroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111 RSC.
- K. D. Poopalam, L. Raghunanan, L. Bouzidi, K. Y. Shoot and S. Narine, Int. J. Energy Res., 2019, 43, 13 Search PubMed.
- R. Ravotti, J. Worlitschek, C. R. Pulham and A. Stamatiou, Molecules, 2020, 25(23), 5572 CrossRef CAS PubMed.
- K. Matuszek, M. Kar, J. M. Pringle and D. R. Macfarlane, Chem Rev., 2023, 123(1), 491–514 CrossRef CAS PubMed.
- T. Inagaki and T. Ishida, J. Am. Chem. Soc., 2016, 138, 11810–11819 CrossRef CAS PubMed.
- S. Cabus, K. Bogaerts, J. Van Mechelen, M. Smet and B. Goderis, Cryst. Growth Des., 2013, 13, 3438–3446 CrossRef CAS.
- S. Petit, R. Ralainirina, S. Favre and R. D. Baynast, WO. Pat., 1993002039A1, 1993.
- W. Wei, F. Feng, B. Perez, M. Dong and Z. Guo, Green Chem., 2015, 17, 3475–3489 RSC.
- Y. Liu, Y. Deng, J. Zheng, F. Wu, J. Lu, S. Sun, D. Wu and T. Wu, Sol. Energy Mater. Sol. Cells, 2022, 248 Search PubMed.
- S. J. Grabowski, W. A. Sokalski and J. Leszczynski, Chem. Phys., 2007, 337, 68–76 CrossRef CAS.
- T. Kondo and C. Sawatari, Polymer, 1996, 37, 393–399 CrossRef CAS.
- R. N. A. H. Lewis and R. N. McElhaney, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 2347–2358 CrossRef CAS PubMed.
- L. Hunger, L. Al-Sheakh, D. H. Zaitsau, S. P. Verevkin, A. Appelhagen, A. Villinger and R. Ludwig, Chem. – Eur. J., 2022, 28, 46 Search PubMed.
- M. S. Cintrón and D. J. Hinchliffe, Fibers, 2015, 3, 30–40 CrossRef.
- A. Yamaguchi, N. Hiyoshi, O. Sato and M. Shirai, Green Chem., 2011, 13, 873–881 RSC.
- T. K. Harris and A. S. Mildvan, Proteines – Structure, Function, Bioinformation, 1999, 35, 275–282 CrossRef CAS.
- L. Jiang and L. Lai, J. Biol. Chem., 2002, 277, 37732–37740 CrossRef CAS PubMed.
- T. Steiner, Crystallogr. Rev., 2003, 9, 177–228 CrossRef CAS.
- Y. Liu, Y. Deng, J. Zheng, F. Wu, J. Lu, S. Sun, D. Wu and T. Wu, Sol. Energy Mater. Sol. Cells, 2022, 248, 112031 CrossRef CAS.
- D. Mroz, R. Wang, U. Englert and R. Dronskowski, CrystEngComm, 2021, 23, 1052–1058 RSC.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- S. J. Prathapa, C. Slabbert, M. A. Fernandes and A. Lemmerer, CrystEngComm, 2019, 21, 41–52 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- R. S. Boethling, E. Sommer and D. DiFiore, Chem. Rev., 2007, 107, 2167–2820 CrossRef PubMed.
- M. Markiewicz, M. Piszora, N. Caicedo, C. Jungnickel and S. Stolte, Water Res., 2013, 47(9), 2921–2928 CrossRef CAS PubMed.
- J. C. G. Sousa, A. R. Ribeiro, M. O. Barbosa, M. F. R. Pereira and A. M. T. Silva, J. Hazard. Mater., 2018, 344, 146–162 CrossRef CAS PubMed.
- OECD, Guidelines for testing of chemicals 309: Aerobic mineralisation in serface water - Simulation biodegradation test, 2004 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2369962 and 2369963. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4gc03460k |
| This journal is © The Royal Society of Chemistry 2024 |