
Advancement of modification engineering in lean methane combustion catalysts based on defect chemistry
Ruishan
Qiu
 a,
Wei
Wang
a,
Zhe
Wang
a and
Haiwang
Wang
*b
a,
Wei
Wang
a,
Zhe
Wang
a and
Haiwang
Wang
*b
aSchool of Materials Science and Engineering, Northeastern University, Shenyang, Liaoning 110819, P.R. China
bHebei Provincial Laboratory for Dielectric and Electrolyte Materials, School of Resources and Materials, Northeastern University at Qinhuangdao, Qinhuangdao, Hebei 066004, P.R. China. E-mail: whwdbdx@126.com
First published on 6th March 2023
Abstract
The direct emission of lean methane (0.1–1.0 vol%) into the atmosphere causes a serious greenhouse effect and energy waste. Catalytic methane combustion technology has great potential in reducing the emission of lean methane. However, the properties of catalysts affect the complete oxidation efficiency of lean methane at low temperatures. In this paper, the critical applications of defect engineering in the modification of methane combustion catalysts are mainly reviewed. Specifically, this study first introduces the characteristics and mechanisms of catalytic methane combustion reactions. Then, the types of defects and the influence of defects on catalytic performance are discussed. On this basis, the preparation strategies for defects in methane combustion catalysts are highlighted and complemented by research advances in machine learning prediction and exploration. Finally, the research challenges and opportunities for defect engineering in methane combustion catalysts are summarized. It is hoped that this review will be instructive for researchers in the reasonable and controllable construction of methane combustion catalysts with suitable defect structures for the promotion of catalytic performance.
1. Introduction
With the development of global industrial and the transformation of the energy pattern, the impact on the earth's ecological environment is becoming increasingly prominent due to the large number of greenhouse gases emitted from human production activities. Methane, the other commonly emitted greenhouse gas in addition to CO2, has about 25 times the greenhouse effect of CO2.1–3 The emissions of lean methane lead to energy wastage and environmental pollution, and its main sources include natural gas vehicles, ships, as well as coal mine, ventilation air methane (VAM),4 and oil-gas fields.5,6 The catalytic methane combustion (CMC) technology avoids the direct emission of lean methane into the atmosphere, by way of improving the combustion efficiency of lean methane at low temperatures.7 It also reduces the production of toxic NOx (nitrogen and oxygen are thermodynamically more likely to produce nitrogen oxides at temperatures generally in the range 1773–2773 K), CO, and unsaturated hydrocarbons at low concentrations of methane during high-temperature combustion.8–10 As a result, methane combustion catalysts have received increasing attention as a hot research issue, as evidenced by the significant increase in the number of papers published on CMC over the last 20 years or so (Fig. 1A).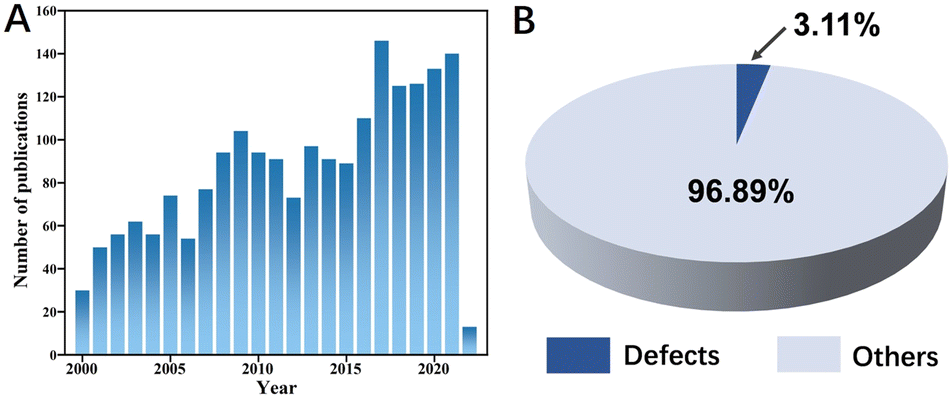 | ||
| Fig. 1 (A) Number of publications on methane combustion. (B) The percentage of defects and other papers in the total methane combustion-related literature (source: Web of Science; keyword: methane-combustion; date: 29 March 2022). | ||
A large number of methane combustion catalysts have been investigated by researchers, among which the excellent catalytic performance of noble metal-based catalysts such as Pd, Pt, Au, Ag, and Ir has been verified by numerous research results.11–18 However, noble metal-based catalysts are prone to poisoning (deactivation by SO2, H2O, etc.),19–21 poor thermal stability, carbon build-up, and high cost in practical applications.22–24 Researchers also have devoted more interest to the design of non-noble metal catalysts,25 for example, single metal oxides such as CuO,26,27 Co3O4,28–30 MnOx,31 Fe2O3,32 and multimetallic oxides with structures such as perovskite,33–35 spinel,36–38 hydrotalcite,39,40 and hexaaluminate.41,42 Such metal oxides have good redox ability, thermal stability, and oxygen mobility, which would facilitate the catalytic oxidation of methane.43–46 Moreover, the thermal catalytic potential of high-entropy oxides (HEO) has been demonstrated.47–49 Hu et al.50 prepared high-entropy oxide nanoparticles containing 10 metal elements, which successfully solved the problem of the “deactivation” of methane combustion catalysts. In recent years, researchers have developed a series of strategies to further improve the catalytic performance of catalysts as materials synthesis techniques and equipment have evolved. Common modification strategies include morphology and crystal plane modulation, composite loading, grain size modulation, and interfacial modulation.
The researchers have found that defects are the key to the highly efficient catalytic oxidation of metal oxide catalysts. For example, as an anionic defect that can effectively activate surface oxygen molecules,51,52 researchers have constructed oxygen vacancies in catalysts through various ways, which can enhance the adsorption and mobility of oxygen on the material surface.53 We know that these are important factors affecting the catalytic activity of CMC. As of March 29, 2022, there were approximately 2187 scientific papers published on methane catalytic combustion (Fig. 1A), of which approximately 3.11% of the total were related to defect construction (Fig. 1B), indicating the rise of the defect construction strategies in the field of methane combustion catalyst modification. Defect engineering, as a novel and versatile strategy, has extraordinary tunability concerning the physical and chemical properties of materials, allowing the construction of defect-enriched (functionalized) materials by using physical or chemical methods. Given the complexity of the building defect responses. It is therefore extremely important to gain insight into how the material defects are constructed and to regulate their concentration and spatial distribution. Also, the influence of defects on the catalytic activity of methane combustion at low temperatures and its mechanism deserves further exploration.
As a universal and effective modification approach, defect engineering has been widely used in the fields of photocatalysis,54,55 electrocatalysis,56–58 energy storage,59,60 and so on. However, there is currently no detailed review of the defect engineering of methane combustion catalysts. In this paper, we review the progress of defect engineering in catalytic methane combustion and explain how defects can be used wisely to improve catalytic performance. Typically, we clarify the basic reaction mechanisms involved in CMC. Secondly, based on the classification of catalyst defects, we discuss the role of defect engineering in enhancing the catalytic activity from the perspective of fundamental reaction mechanisms, and the progress of research on some representative defective catalytic materials in relevant applications. Strategies for the construction of defects in catalysts are then introduced in the context of catalytic methane combustion. Finally, the current challenges and future perspectives are presented. For all we know, this is the first general review of defect design in methane combustion catalysts.
2. Methane combustion catalytic reaction mechanism
The methane molecule, being the simplest organic compound, is extremely stable due to its tetrahedral symmetric structure (Fig. 3A) and has a bond energy of 439.3 KJ mol−1 for C–H.61 Therefore, the activation and conversion of methane are very difficult. The complete oxidation of CH4 proceeds through the following reaction:| CH4(g) + 2O2(g) = CO2(g) + 2H2O(g) |
| ΔH0r(298 K) = −803 kJ mol−1 |
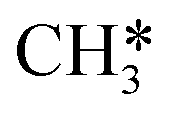 is the most difficult step.62 With the development of characterization techniques and in-depth studies, a clearer understanding of the reaction mechanisms involved in the reaction of CMC has been gained.63,64 In general, the catalytic reaction mechanism is determined by the catalyst material type (such as noble metal, non-noble metal, single metal oxide, and multimetallic oxide), structure, reaction conditions, and other factors. The four reaction models of CMC currently proposed by researchers are shown in Fig. 2.32 They are mainly divided into the Langmuir–Hinshelwood (L–H) and the Eley–Rideal (E–R) mechanism dominated by surface-adsorbed oxygen, the Mars–van Krevelen (M–vK) mechanism dominated by lattice oxygen, and the Two–Term (T–T) mechanism co-participated by surface adsorbed oxygen and lattice oxygen.64
is the most difficult step.62 With the development of characterization techniques and in-depth studies, a clearer understanding of the reaction mechanisms involved in the reaction of CMC has been gained.63,64 In general, the catalytic reaction mechanism is determined by the catalyst material type (such as noble metal, non-noble metal, single metal oxide, and multimetallic oxide), structure, reaction conditions, and other factors. The four reaction models of CMC currently proposed by researchers are shown in Fig. 2.32 They are mainly divided into the Langmuir–Hinshelwood (L–H) and the Eley–Rideal (E–R) mechanism dominated by surface-adsorbed oxygen, the Mars–van Krevelen (M–vK) mechanism dominated by lattice oxygen, and the Two–Term (T–T) mechanism co-participated by surface adsorbed oxygen and lattice oxygen.64
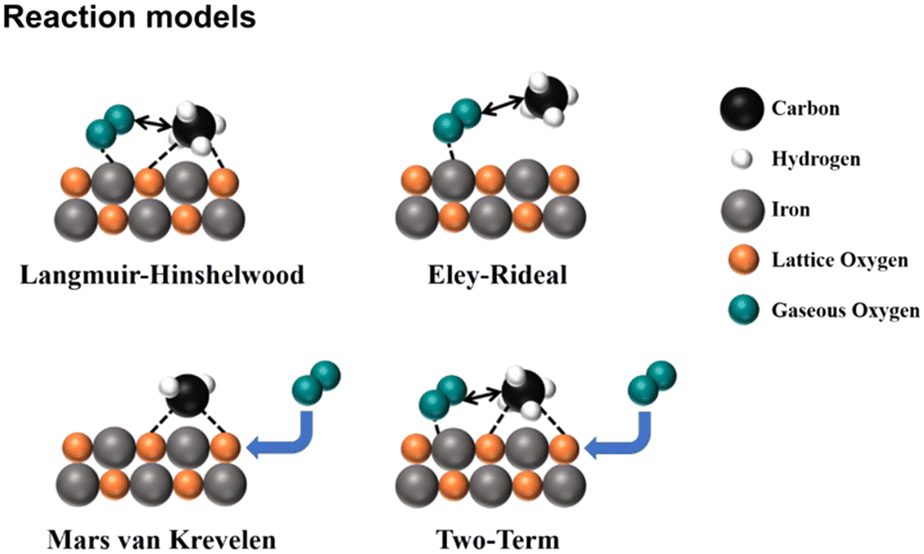 | ||
| Fig. 2 Schematic diagram of the reaction mechanism of catalytic methane combustion. Reproduced from ref. 32 with permission from the American Chemical Society. | ||
In the Mv–K mechanism, CH4 and O2 are first adsorbed onto the catalyst surface. Then, the adsorbed CH4 molecules react with the surface lattice oxygen of the catalysts to generate CO2 and H2O. Meanwhile, the lattice oxygen on the catalyst surface decreases and oxygen vacancies are formed. Afterward, gas-phase oxygen molecules fill the oxygen vacancy and the internal lattice oxygen migrates to the surface. In the L–H mechanism, gas-phase oxygen molecules are adsorbed on the catalyst surface and activated as reactive oxygen species. The adsorbed methane molecules and reactive oxygen species then react. However, the E–R model involves a single active state, in which only one reactant molecule (methane or gas-phase oxygen) is adsorbed on the surface of the catalysts and converted to an active state, and then reacts with another gas-phase molecule. All these reaction mechanisms are widely supported by many experimental results.61,65–68
For example, He et al.32 investigated catalytic methane combustion reaction intermediates and mechanisms on α-Fe2O3 (Fig. 3B). At low temperatures, CH4 molecules are activated by lattice oxygen, instead of by gas-phase oxygen molecules. Yet, the latter is more critical at higher temperatures, where a catalytic cycle is formed in the vacancy-promoted adsorption of molecular dioxygen. Isotope tracing experiments support the M–vK mechanism: (1) CH4 first dissociates with an antiferromagnetically-coupled iron dimer adsorbed onto the lattice oxygen O3c at the centre of the tetraferrous iron to form a methoxy CH3–O species; (2) then, it is converted to an adsorbed formate intermediate; (3) formate intermediates first forms bidentate bridging formate and then transitions to a monodentate conformation. (4) Finally, CO2 and H2O are formed and desorbed, leaving oxygen vacancies on the surface of the catalyst. Neighboring lattice oxygen and oxygen molecules, however, replenish the vacancies and regenerate the lattice oxygen to reconstitute the active center. This reaction model is mainly dependent on the lattice oxygen (Olatt) of the catalysts. Alternatively, a slightly different reaction mechanism to Mv–K has been proposed, where only the catalytic oxidation of methane by adsorbed reactive oxygen species on the catalyst surface is involved.69
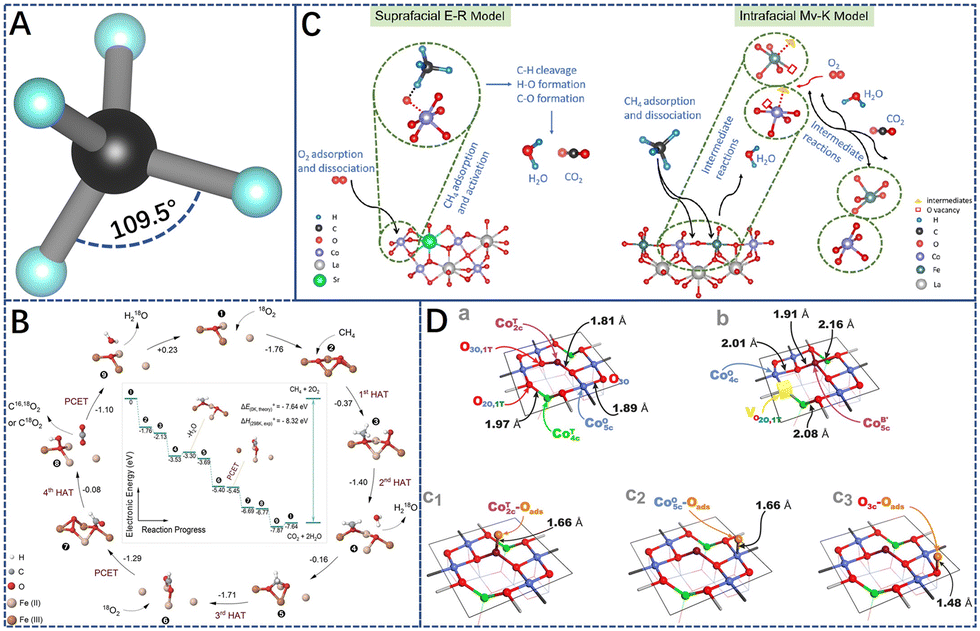 | ||
| Fig. 3 (A) The symmetric structure of methane molecule. (B) Proposed catalytic cycle for the catalytic methane combustion (CMC) reaction at higher temperatures via a molecular-oxygen-assisted pathway with calculated relative electronic energies. Reproduced from ref. 32 with permission from the American Chemical Society. (C) Schematic diagrams of the proposed suprafacial E–R mechanism (left panel) and intrafacial M–vK mechanism (right panel) for LaCoO3-based perovskite oxide catalysts. Reproduced from ref. 45 with permission from Elsevier. (D) Perspective views of various cobalt spinel (100) terminations. The stoichiometric bare (100)-S termination (a) and the oxygen defected (100)-VO surface with an oxygen vacancy in the O2O,1T position (b). The (100) terminations with an oxygen adatom stabilized on the Co2cT (c1), Co5cO (c2), and O3c (c3) active sites. Color coding: Co5cO, blue; Co2cT, purple; Co4cT, green; spinel O, red; oxygen adatom, orange; oxygen vacancy, yellow cube. Reproduced from ref. 70 with permission from the American Chemical Society. | ||
Due to the complexity of the heterogeneous catalytic reactions, in some cases, it is extremely challenging to explain the reaction process over the catalysts by a single mechanism. For example, Wang et al.45 combined the experimental results of methane oxidation catalyzed by LaCoO3 series perovskites and DFT calculations to reveal the interplay of the homogeneous E–R and intrafacial M–vK mechanism of the catalysts at different reaction temperature intervals. At lower reaction temperature intervals, methane oxidation typically follows the E–R mechanism: (1) CH4 is first adsorbed on the catalyst surface; (2) oxygen molecule is adsorbed on the metal sites for rapid conversion to the reactive oxygen species, which reacts with CH4(ad) to form intermediates; (3) finally, intermediates are rapidly converted to CO2(ad) and H2O(ad), which are then desorbed from the catalyst surface. Reactive oxygen, therefore, features prominently in the catalytic combustion of methane and the rate of methane oxidation is affected by the desorption rate of the product molecules (CO2 and H2O). In contrast, in the high-temperature reaction range, methane oxidation follows the M–vK mechanism and the reaction is mainly dependent on the lattice oxygen (Olatt) in the oxide (Fig. 3C).
In addition, Zasada et al.70 investigated the process of catalytic methane combustion on the (100) crystalline surface of cobalt spinel nanocubes through steady-state isotopic transient kinetic analyses (SSITKA), and temperature-programmed surface reactions (TPSR) catalytic studies, Density Functional Theory (DFT) calculations (Fig. 3D). The results showed that methane was activated by single-atom oxygen O–CoT species (ΔEa = 0.83 eV) from 300 °C to 450 °C, operating in the Langmuir–Hinshelwood (L–H) mechanism. At 450 °C < T < 650 °C, the O3O intrafacial sites (two 3-fold oxygen anions linked to CoO exclusively, Fig. 3D) acted in concert with Co5cO (four singly truncated octahedral ions, Fig. 3D) to gradually participate in methane oxidation. The slight reduction of the (100) surface is attributed to the formed oxygen vacancies by in-plane dehydroxylation and decarboxylation of the catalysts, and the vacancies actually filled with oxygen molecules are evidence that the L–H and Mv–K mechanisms may be operating simultaneously. Above 650 °C, the generation of oxygen vacancies in most of the cobalt spinel and the high involvement of lattice oxygen suggests that the Mv–K reaction model is dominant. Following this, Zasada et al.71 further investigated the molecular characterization of the reaction mechanisms involved in the conversion of methane to H2O and CO2 on the (100) surface of cobalt spinel nanocubes. The results of DFT modeling and catalytic isotopic studies indicated that complete oxidation of methane occurs along suprafacial and interfacial pathways on oxygen-covered and oxygen-depleted/bare (100) surfaces of cobalt-spinel nanocubes. These different methane activation mechanisms evolve with differences in the properties and accessibility of the surface and lattice oxygen species, but the activation of the first C–H bond in the methane molecule remains a kinetically relevant step. Under high-temperature or oxygen-poor conditions, where surface oxygen species are scarce, critical reaction steps (CH4 activation, CO2 and H2O product release) may occur under an interfacial mechanism involving lattice oxygen. The labile peroxo O22− species participate in intermediate reaction steps leading to the final formation of H2O and CO2 products.
Popescu et al.72 researched the correlation between the semiconductor and redox properties of LaCo1−yO3 (y = 0 and 0.2) and LaCo1−xFexO3 (x = 0.6 and 1) perovskites and the catalytic oxidation behavior of methane in the 300–800 °C temperature range by in situ electrical conductivity measurements. The results showed that the reaction of LaCo1−yO3 perovskite was always in a metal conductive state, and no reduction occurred in the methane–air mixing reaction and pure methane reaction. The reaction only involves adsorbed oxygen species, indicating that the reaction mechanism is the suprafacial mechanism. The LaCo1−xFexO3 (x = 0.6 and 1) perovskite behaves as a P-type semiconductor with a partial reduction in both air and methane–air mixing reactions, and its main carriers are positive holes. The transition of LaCo0.4Fe0.6O3 from the semiconductor state to metal conductive state occurred in the range of the reaction temperature, which proved that the suprafacial mechanism and redox mechanism occurred simultaneously on the LaCo0.4Fe0.6O3 perovskite. However, the catalytic reaction on the LaFeO3 perovskite only involved the surface lattice oxygen O− species, indicating that the reaction mechanism is the Mars–van Krevelen mechanism. Similarly, Mihai et al.73 investigated the conductivity of spinel MnCo2O4, NiCo2O4, and CuCo2O4 to study variations in their catalytic behavior. Their activities in the complete methane oxidation in terms of both T10 and the natural reaction rates are as follows: Cu-cobaltite > Ni-cobaltite > Mn-cobaltite. The results show that Ni-cobaltite is in a metallic conductivity state and cannot be reduced under reaction conditions, which indicates that it acts through the suprafacial mechanism. Cu-cobaltite acts as a P-type semiconductor but is not reduced in the reaction mixture at the reaction temperature of 350 °C, suggesting that at least under these conditions it also works by a suprafacial mechanism. Mn-cobaltite has n-type semiconductor properties and is reduced under reaction conditions, indicating that heterogeneous redox mechanism is involved.
3. Understanding of defects in catalytic methane combustion
Specifically, the CMC reaction consists of four processes: the adsorption and activation of CH4 and O2, the conversion of the reactant species, and the desorption of products. From these reaction processes and the reaction mechanism described in section 2, it can be seen that the catalytic efficiency of methane combustion is closely related to the adsorption and activation of CH4 and O2 molecules, and the oxygen mobility on the catalyst surface and in the bulk. The formation of defects can impart some unique properties to the catalysts that are beneficial for specific applications.74 In the methane combustion catalyst, precise modulation of defects can enhance the adsorption and activation of reactant molecules, and improve the electron transfer and oxygen mobility on the catalyst surface. Through the precise combination of these features, the catalytic methane conversion efficiency can be finely tuned and optimized. To better understand the role of defects in CMC, we indicate the types of defects and the advantages of catalytic methane conversion efficiency in this section.3.1 Defect types in catalyst materials
Crystals have a regular structure of atomic arrangements, but defects are common in crystals. Crystal defects or defect sites exhibit high reactivity due to a specific arrangement of atoms in the defect site that differs from the crystal surface. In general, defect structures are created during the synthesis and modification of materials. There are several types of material defects such as vacancy, surface step/kink, impurity doping, twin/grain boundary, and lattice distortion/disorder defects (Fig. 4). Among them, lattice distortion and vacancy defects are widely utilized in the modification of methane combustion catalysts.75,76 In addition to lattice distortion and vacancy defects, the influence of other defects have also been investigated in CMC reactions.77–80 As shown in Fig. 5A and C, twin boundary defects and lattice defects can be observed in metal oxides prepared by ball milling.81,82 Furthermore, Fig. 5B shows the surface step defects formed by the quenching strategy.83 These defects have a remarkable contribution to the catalytic performance of catalysts.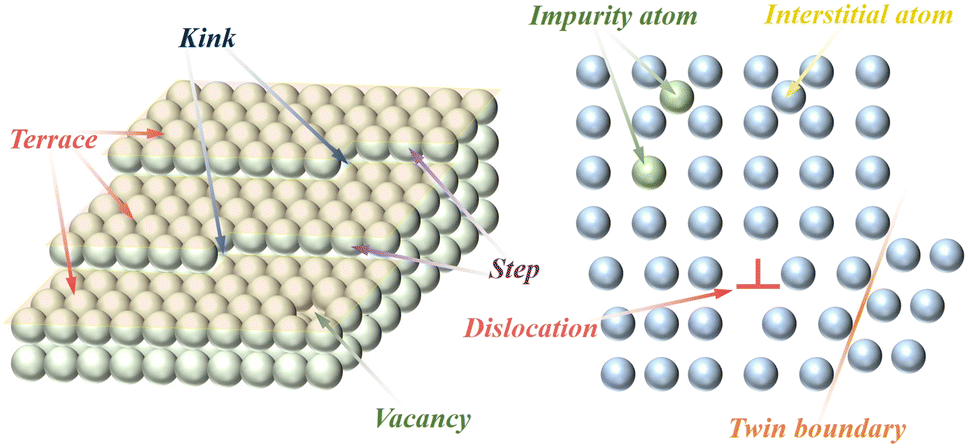 | ||
| Fig. 4 Schematic illustrating the different types of defects in catalyst materials. | ||
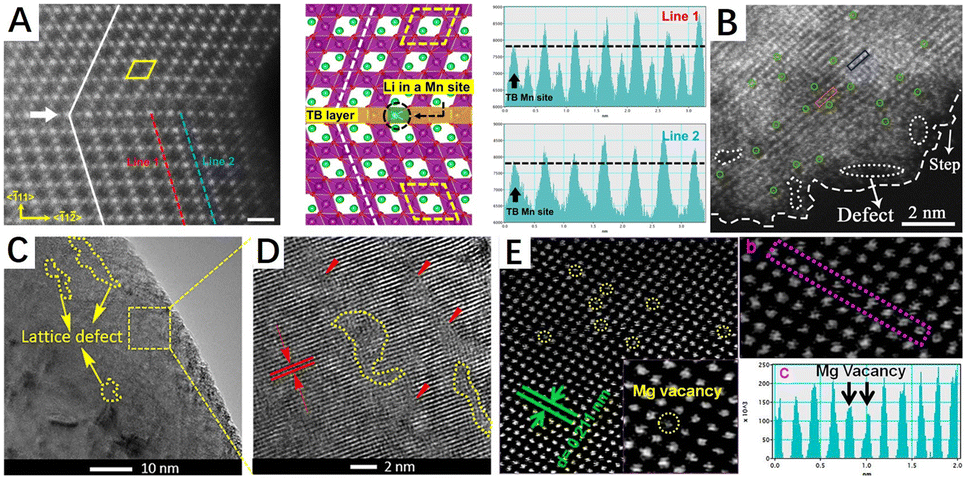 | ||
| Fig. 5 Different types of defects in some materials (A) HAADF-STEM images and structural schematic of the grain boundary. Reproduced from ref. 81 with permission from Springer Nature. (B) HAADF-STEM image of surface defects and step. Reproduced from ref. 83 with permission from Elsevier. (C and D) Lattice defect in A-TiO2 materials. Reproduced from ref. 82 with permission from the American Chemical Society. (E) HAADF-STEM image of the carbon-doped MgO sample: (a) magnesium vacancies (yellow circles), (b) line profile showing the image intensity versus position in the purple area, and (c) magnesium vacancies in the purple line profile in panel b. Reproduced from ref. 87 with permission from the American Chemical Society. | ||
In principle, vacancies and impurity doping are the two main types of point defects, which can be formed by the absence of an atom or the irregular placement of atoms in a regular crystal structure. Vacancies are created by the removal of a lattice atom from the crystal structure and are classified as anionic or cationic vacancies that alter the physical/chemical properties of the crystalline materials, the most common anionic vacancy being an oxygen vacancy. In metal oxides, surface oxygen vacancies (Ovs) tend to adsorb and activate gaseous oxygen to form reactive oxygen species (ROS) such as O22−, O2− and O− necessary for catalytic oxidation reactions,51,84 while bulk vacancies improve the mobility and activity of the lattice oxygen species through their transmission effect.85,86 Meanwhile, metal vacancies as cation vacancies also exert an influential impact on the catalytic activity of methane combustion. As shown in Fig. 5E, Mg atomic vacancies in the carbon-doped MgO catalyst can be clearly observed in the HAADF-STEM diagram.87 Since metal vacancies in the metal oxide catalysts can alter the metal–O bonding energy and oxygen coordination environment, enhancing their surface lattice oxygen mobility.88 Moreover, it can enable the formation of electron-rich sites or even lower valence states on metal oxides, thus enhancing the chemical adsorption of reactants by metal oxides. For example, Co, Fe, and O vacancies can be formed in CoFe LDHs materials by nitric acid etching and have a significant impact on their catalytic properties.89 In contrast to vacancy defects, impurity doping can change the physical and chemical properties of materials by introducing a dopant into the lattice of the substrate material, which is classified as an anion or cation. At the same time, impurity doping can also be divided into substituting-site doping and interstitial-site doping according to the position of the dopant in the crystal lattice (depending on their size relative to the host atom/ion). As shown in Fig. 4, there are interstitial defects (self-interstitial, heteroatomic interstitial) and substituting-site doping, where the self-interstitial defect is distinguished from the other two. Doping defects also have been widely used in the modification of methane combustion catalysts.90–92
The types of defects mentioned above have a profound effect on the physical/chemical properties of the materials,93–97 and therefore defect engineering has a good potential for research applications as an effective modification route for methane combustion catalysts. However, the defect structure–activity enhancement response in the catalyst materials is complex, in terms of defect structure, type, and distribution concentration. Therefore, the effects of defects on the catalytic activity of methane combustion and the controlled construction of defects in the catalyst materials need to be investigated in more depth to explore more effective modification pathways.
3.2 Defects as active sites
Due to the high energy of the defect sites, the defect structures in catalysts have high reactivity with reactant molecules.97,98 The defects can also optimize the electronic structure and density distribution of the material and induce localized strain. Therefore, the defect sites have the advantage of directly serving as the reactive sites.99 For example, the atomic arrangement at the grain boundary transitions from one orientation to the other and is therefore in a transition state.100 Huang et al.77 found that high-temperature steam pretreatment of Pd/Al2O3 catalyst can significantly improve the methane combustion reaction activity of Pd (Fig. 6B). In the experiment, it was found that the reasons leading to the improvement of catalytic activity were twin boundaries (TBs) and grain boundaries (GBs) in the palladium catalysts (Fig. 6A). DFT calculations showed that the increase of catalytic activity was attributed to the surface strain present in the immediate vicinity of GBs, which verified that the twin/grain boundary is an excellent catalytic active site. In other studies, Polo-Garzon et al.101 used SrTiO3 (STO) as a model perovskite and regulated the Sr content on its surface by different preparation and post-processing methods (chemical etching, immersion, and roasting). Electron microscopy results showed that the different Sr-enriched STOHT catalysts had much richer step sites on their surfaces (Fig. 6C). The experimental results showed that the number of such stepped structures was directly related to the methane combustion reaction rate. DFT calculations and in situ infrared experimental results showed that methane had the lowest C–H activation energy (0.32 eV) on the Sr-enriched surface with stepped structures. This work demonstrates that the surface step structure is the active center for CH4 combustion.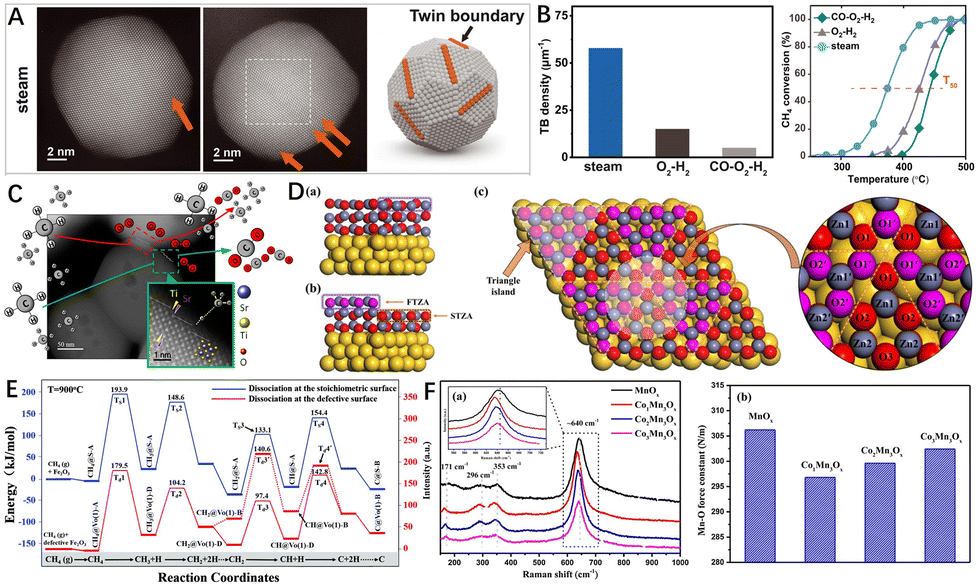 | ||
| Fig. 6 (A) HAADF-STEM images of Pd/Al2O3 preprocessed by steam (600 °C); (B) TB density statistical histogram and light-off curves of the Pd/Al2O3 catalyst after different pretreatments. Reproduced from ref. 77 with permission from the American Association for the Advancement of Science. (C) HAADF-STEM images of STO particles (after calcination at 750 °C for 5 h) recorded along [001] zone axis. Reproduced from ref. 101 with permission from the American Chemical Society. (D) Side view of the (a) ZA and (b) TZA with the indication of FTZA and STZA. (c) Top view of the (2 × 2) TZA with the labeled atoms. The yellow, red, and grey represent Au, O, and Zn, respectively. The pink represents O in FTZA in order to distinguish it from the O in STZA. Reproduced from ref. 120 with permission from Elsevier. (E) Energy profile for CH4 sequential dissociation on the stoichiometric and defective α-Fe2O3(001) surface (T = 900 °C). Ts denotes the transition state on the stoichiometric α-Fe2O3 (001) surface and Td denotes the transition state on the defective surface. Reproduced from ref. 121 with permission from the Royal Society of Chemistry. (F) (a) Ex situ Raman spectra and (b) Mn–O force constant of the prepared catalysts. Reproduced from ref. 132 with permission from Elsevier. | ||
In addition, surface defect sites can also anchor single atoms, atomic clusters, or groups to form active sites.102,103 In heterogeneous catalysis, the Strong Metals–Support Interaction (SMSI) can significantly enhance the adsorption of CH4 and the activity of lattice oxygen of the supports,104–106 and strongly modify the dispersion and morphology of the metal particles.107–109 For example, Wu et al.110 prepared a highly dispersed palladium catalyst supported on defective Al2O3–CeO2 (RAl2O3–CeO2: ceria was immobilized on the coordinatively unsaturated Al3+penta sites of γ-alumina activated by pre-reduction to fabricate hybrid-oxide support) for methane combustion. The RAl2O3–CeO2 interface as such can be effective in enhancing the catalytic activity because such an interface increases the dispersion of the deposited Pd species. The strong electronic interactions increase the concentration of reactive oxygen species.
3.3 Promoting the adsorption of reactant molecules on the catalysts
Adsorption is an important initial step in all heterogeneous catalysis processes, and the adsorption of reactant molecules on the catalyst surface can initiate many chemical reactions.111 Defects can improve the electronic state structure of the catalyst surface to change the adsorption energy of the reactant molecules,112–114 which is beneficial to the adsorption of reactant molecules. It has been proposed through experiments and theoretical calculations that reactants are preferentially adsorbed at defect sites (such as oxygen vacancies, and steps) in heterogeneous catalysis.115 For example, Berdiyorov et al.116 used DFT calculations to investigate the influence of vacancy defects (Ca ions and charge-neutral CO3) on the adsorption properties of CH4 molecules on the calcite surface. Both types of vacancy defects lead to the strong adsorption of methane molecules compared to defect-free samples. Wang et al.46 successfully synthesized a unique La–Co–C–O hybrid (La2O2CO3/LaCoO3/Co3O4 phase composition), and the chaotic microstructure leads to the increase of crystal defects and the formation of multi-level pore size distribution, which is beneficial to the reaction gas adsorption and diffusion. In addition, the appropriate number and strength of Lewis/Brønsted acid sites are more beneficial to the adsorption performance of the catalysts,117,118 and the acid sites are responsible for the desorption of CO2 from the catalyst surface.1193.4 Promoting the activation of reactant molecules on the catalysts
The dissociation of the first C–H bond of the CH4 molecule is the rate-controlling step in the CMC. An appropriate concentration of defects can effectively improve the ability of the catalyst surface to activate reactant molecules (methane and oxygen molecular). For example, vacancy defects have unsaturated coordination, which is more advantageous to the adsorption of methane and the activation of C–H.87 Liu et al.120 researched the activation mechanism for the dissociation of methane adsorbed on the triangular defect surface of ZnO@Au by DFT calculations (surface model is shown in Fig. 6D, simulating the triangular defects reported in the literature). The coordinatively unsaturated O and Zn sites induced by the triangular defects on ZnO@Au were found to be capable not only of stabilizing methane but also polarizing the C–H bond by dynamically frustrated Lewis pairs. The defective ZnO@Au could activate methane through a precursor-mediated mechanism, and its activation energy (0.21 eV) was lower than the absolute value of adsorption energy (0.48 eV). Similarly, in the presence of oxygen vacancies in α-Fe2O3, oxygen vacancies reduced the dissociation barriers of CH3, CH2, and CH radicals, significantly promoting the activation of CH4 (Fig. 5E).121Further, another key issue for the CMC is the activation efficiency of the catalyst surface oxygen (oxygen molecules, lattice oxygen), as it determines the number and conversion frequency of reactive oxygen species available for the methane combustion reaction. Defects such as vacancies play a very important role in the activation of oxygen molecules and the generation of oxygen species at the active interface,122–125 and promote the dehydrogenation mechanism of the catalyst.126 For example, Li et al.127 prepared a series of Ce1−xFexO2−δ (0 ≤ x ≤ 0.4) catalysts, Fe-doped CeO2 formed Fe–Ce–O solid solution, and the catalytic performance was mainly affected by the concentration of oxygen vacancies. The gaseous oxygen-replenished oxygen vacancies and created reactive oxygen species. These reactive oxygen species could react directly with the activated CH4 molecules to generate CO2 and H2O.
3.5 Improving the migration of lattice oxygen in the catalysts
In the Mv–K and T–T reaction mechanisms, lattice oxygen acts as an essential factor as its mobility directly affects the activation and oxidation efficiency of methane molecules. The introduction of defects is usually accompanied by the formation of thermodynamically unstable structures, resulting in an increase in the density of the surface coordinated with unsaturated atoms, thus enhancing electron mobility. For example, defects such as oxygen vacancies and metal vacancies can weaken the metal–O bond strength,88,128 promoting the activation of the lattice oxygen and interfacial oxygen molecules and the formation of reactive oxygen species, thereby improving the oxygen mobility on the material surface.129–132 Zhu et al.133 reported surface defect engineering to modulate the surface lattice oxygen species in Co3O4 catalysts by an alkaline earth metal doping etching strategy, and effectively improved their oxygen mobility. Li et al.132 prepared amorphous Co–Mn binary oxides with enhanced defects via a facile self-redox method. The amorphous Co1Mn3Ox catalyst had abundant oxygen vacancy defects, which weakened the Mn–O bond strength (Fig. 6F), promoting the mobility of surface lattice oxygen, and significantly improved the catalytic activity for propane oxidation. Besides, Jeong et al.134 introduced Fe2O3 into the CeO2–ZrO2 lattice to form oxygen vacancies through a charge compensation mechanism, the number of oxygen vacancies increased, and the redox reaction of Fe3+/Fe2+ further enhanced the oxygen release and oxygen storage capacity. The results showed that the activity of 11.2 wt% PdO/16 wt% Ce0.67Zr0.18Fe0.15O2−δ/γ-Al2O3 was the highest, and it reached complete oxidation at 280 °C.4. Creation of defects in methane combustion catalysts
Defects indirectly or directly influence the performance of catalysts and have a significant effect on CMC reactions. The insight into the structural response at the defective sites during catalytic reactions offers a unique possibility to manipulate material defects to improve their catalytic activity.135 The controlled construction of defects in catalyst materials with respect to the performance response is a complex process. Therefore, an in-depth understanding of defect construction methods is extremely important. Currently, researchers have developed several methods to prepare defect-enriched catalysts, divided into two main categories according to the defect material synthesis process: the direct creation of defects during the synthesis of the catalyst (atomic doping, ball milling, etc.), and the introduction of defects in the pre-synthesized catalyst (etching, quenching, etc.).4.1 Direct synthesis of defect-rich catalysts
Materials are inevitably prepared with defects, and catalysts containing different types of defects can be prepared by controlling the synthesis method and the reaction conditions, which involve the crystal growth process and reaction kinetics of the catalyst material. For instance, defects such as vacancies and disordered structures can be created through lattice distortion caused by atomic doping, as well as defects such as grain boundaries can be constructed by the ball milling method.In metal oxide catalysts such as spinels and perovskites, atomic doping by co-precipitation, sol–gel, solution combustion, and hydrothermal methods to form solid solutions that alter the original structure of the material to increase the concentration of crystal defects or oxygen vacancies.90,147,148 The doping of spinel with dopants of different oxidation states results in various changes in structural stability and electronic properties.149 Zheng et al.150 used N-butylamine as a precipitator to prepare Co–In-x oxide nanocomposites (Fig. 7A), in which the CH4 conversion rate of Co–In-0.2 catalyst could reach 99% at 395 °C (Fig. 7C). The doped In3+ enters the spinel structure of Co3O4 and forms the Co–In–O solid solution phase with smaller particle size. The appropriate amount of In3+ mainly replaces the Co3+ position in the octahedral position of the spinel structure, increasing the proportion of active species Co2+, enriching the species of reactive oxygen species, and optimizing the surface acidity. Chen et al.151 prepared Co1−xMxCr2O4 (M = Li, Zr; x = 0–0.2) catalysts by citric acid method. It was found that Li+ or Zr4+ cations, which tend to occupy tetrahedral (A) sites, lead to the decrease or increase of catalytic activity respectively. The catalytic activity of Co0.95Zr0.05Cr2O4 was the best, reaching 90% of methane conversion at 448 °C, which was 66 °C lower than CoCr2O4. The results showed that lithium or zirconium substitution can change the structure and electronic properties of spinel. Zirconium substitution enhances the reducibility of zirconium-doped catalysts and reduces the strength constants of Co–O and Cr–O bands, which may contribute to their catalytic activity for methane combustion. For doped perovskite oxides, it is possible to synthesize multicomponent perovskites by cations that partially replace the A and B positions. The substitution of A or B sites in perovskite can change the oxidation state of the metal ions in the crystal structure and lead to the formation of defects such as oxygen vacancies.152 For example, Tan et al.153 prepared a series of perovskite composite oxide catalysts doped with manganese BaCeO3 (BaCe1−xMnxO3−δ) by the sol–gel method. The prepared perovskite composite oxide catalysts showed good catalytic performance and high-temperature stability. With the increase in the Mn doping amount, the catalytic activity firstly increased and then decreased. When the doping amount was 0.4, the catalytic activity was the best, the ignition temperature of methane was 249.7 °C, and the catalytic conversion temperature T90 was 423.2 °C. The electrical conductivity of the sample was measured from the AC impedance spectrum. The catalytic performance of the catalyst varies with the change in the conductivity.
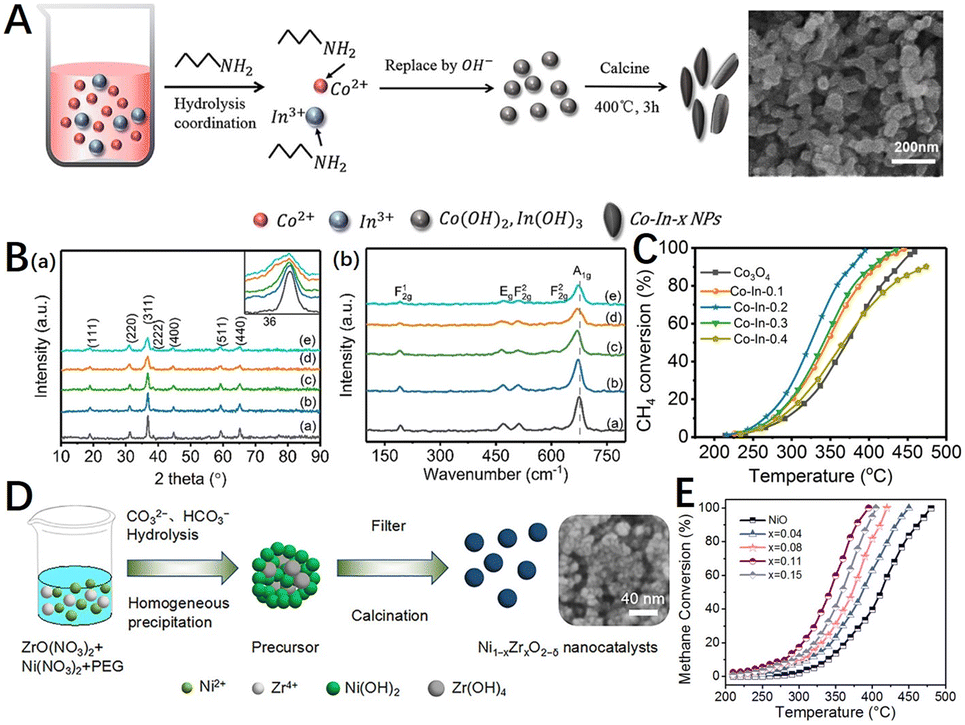 | ||
| Fig. 7 (A) Schematic diagram of the Co–In-x nanoparticles via a modified precipitation method adopting organic base N-butylamine as the precipitator; (B) (a) XRD diffraction patterns and (b) Raman spectra of Co–In-x catalysts: (a) Co3O4, (b) Co–In-0.1, (c) Co–In-0.2, (d) Co–In-0.3, and (e) Co–In-0.4; (C) catalytic activity of catalysts for CH4 combustion. Reproduced from ref. 150 with permission from the American Chemical Society. (D) Synthesis of Zr-doped NiO nanocatalysts (Ni1−xZrxO2−δ) via a designed co-precipitation method; (E) methane conversion as a function of temperature over NiO and Ni1−xZrxO2−δ nanocatalysts. Reproduced from ref. 146 with permission from the American Chemical Society. | ||
In addition, the properties of the monomeric oxides can also be regulated by doping. Wang et al.146 achieved the incorporation of Zr into the NiO lattice by a homogeneous co-precipitation method (Fig. 7D). The results showed that the Ni0.89Zr0.11O2−δ nanocatalyst with moderate Zr content exhibited excellent low-temperature activity with a T90 of 380 °C (Fig. 7E). The optimized incorporation of Zr resulted in the formation of the Ni0.89Zr0.11O2−δ nanocatalyst with a small grain size, large specific surface area, and increased surface acid-basic sites. A large amount of active Ni2+ and surface oxygen were generated during the conversion of Ni3+ to Ni2+, which played a key role in the adsorption and activation of CH4 molecules. Cerium oxide (CeO2) is also widely used in catalytic methane combustion due to its excellent redox capacity, oxygen storage capacity, and tunability. Su et al.154 used the density functional theory to demonstrate that two Pd2+ cations can substitute for one Ce4+, thus forming a very stable structure containing highly coordinated unsaturated Pd cations that can strongly adsorb CH4 and dissociate the first C–H bond with a low energy barrier, leading to the effective activation of methane. In comparison with single dopants, multi-dopants show greater potential to enhance the catalytic performance of methane combustion by combining the advantages of two or more dopants.155–157 Toscani et al.158 prepared nanostructured mixed oxides of CeO2–ZrO2–Sc2O3 by citrate complexation. The effect of the co-substitution of ZrO2 and Sc2O3 in CeO2 was evaluated in terms of the sample's reducibility and its catalytic activity to methane oxidation. Methane catalytic combustion experiments showed that the sample containing Sc2O3 performed better than the sample containing CeO2–ZrO2.
In addition, metal–organic framework materials (MOFs) are organic–inorganic hybrid polymers formed by the coordination polymerization of metal ions and organic ligands.159,160 Using MOF materials as porous precursors, metal element species can be mixed at the atomic level. Due to the pyrolysis of organic ligands during MOF synthesis, the active metals are highly dispersed throughout the framework.161 The good distribution of ions results in high dispersion of the metal ions in binary or hybrid metal oxides, which is an important advantage compared to the traditional methods for synthesizing binary or hybrid metal oxides.162,163 Meanwhile, the porous heterostructure generated by the thermal degradation of MOFs can inhibit the sintering of core metal components. For example, Wu et al.164 prepared 1D Pd@CeO2–BDC catalysts by solvothermal synthesis (Fig. 8A). It was found that the one-dimensional mesoporous Pd@CeO2–BDC catalyst was rich in oxygen vacancies. The EDS analysis (Fig. 8B) showed that the elemental composition in the catalyst was uniformly distributed, and Pd species were uniformly distributed on the CeO2 (110) surface in the form of small PdO clusters and PdxCe1−xO2−σ (Fig. 8D). Owing to the strong interaction of the metal supports in the MOF framework, part of the Pd atoms replaced Ce4+ in the CeO2 lattice, and the heterovalent substitution of Pd and Ce accelerated the formation of the reactive oxygen species. As depicted in Fig. 8E, the 1%Pd@CeO2–BDC bimetallic MOF catalyst showed the superior catalytic properties and stability, achieving 90% methane conversion at 342 °C (WHSV = 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL g−1 h−1). Similarly, hydrotalcite can be used as the precursor of metal oxide catalysts. By controlling its thermal decomposition, mesoporous and thermally stable mixed metal oxide with a high surface area can be obtained, which has controlled redox and acid–base properties, and metal cations are highly dispersed in mixed oxide materials.109,165–167 Zhao et al.168 deposited Ag nanoparticles on layered double hydroxide (LDH) layers (Ag–CoAl-LDHs) through simple autoxidation reduction. By controlling its calcination temperature, the highly homogeneous and thermally stable dispersion of Ag NPs, the efficiency and catalytic stability of Ag–CoAlO-600 °C provide great application potential for methane combustion (Fig. 8G).168
000 mL g−1 h−1). Similarly, hydrotalcite can be used as the precursor of metal oxide catalysts. By controlling its thermal decomposition, mesoporous and thermally stable mixed metal oxide with a high surface area can be obtained, which has controlled redox and acid–base properties, and metal cations are highly dispersed in mixed oxide materials.109,165–167 Zhao et al.168 deposited Ag nanoparticles on layered double hydroxide (LDH) layers (Ag–CoAl-LDHs) through simple autoxidation reduction. By controlling its calcination temperature, the highly homogeneous and thermally stable dispersion of Ag NPs, the efficiency and catalytic stability of Ag–CoAlO-600 °C provide great application potential for methane combustion (Fig. 8G).168
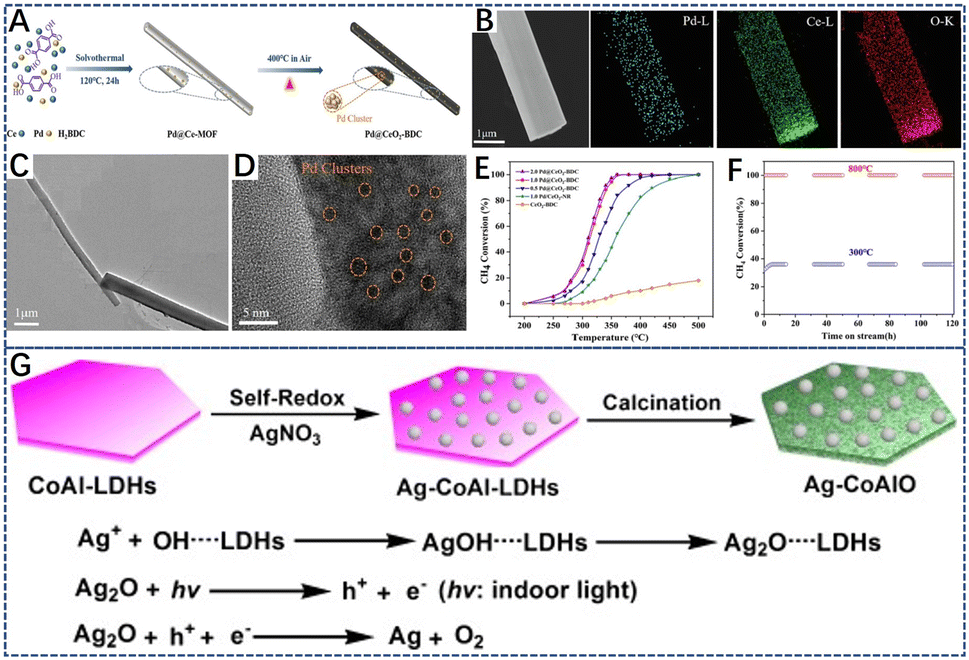 | ||
| Fig. 8 (A) Synthesis scheme of the Pd@CeO2–BDC catalyst; (B) EDX elemental mapping of Pd@CeO2–BDC catalyst; (C and D) TEM and HRTEM images of 1.0Pd/Ce-MOF (the precursor of Pd@CeO2–BDC catalyst); (E) CH4 conversion over the different catalysts; (F) catalytic stability of 1.0 Pd@CeO2–BDC at 300 °C and 800 °C. Reproduced from ref. 164 with permission from Elsevier; (G) the scheme of the preparation of Ag–CoAl-LDHs and Ag–CoAlO. Reproduced from ref. 168 with permission from Wiley-VCH. | ||
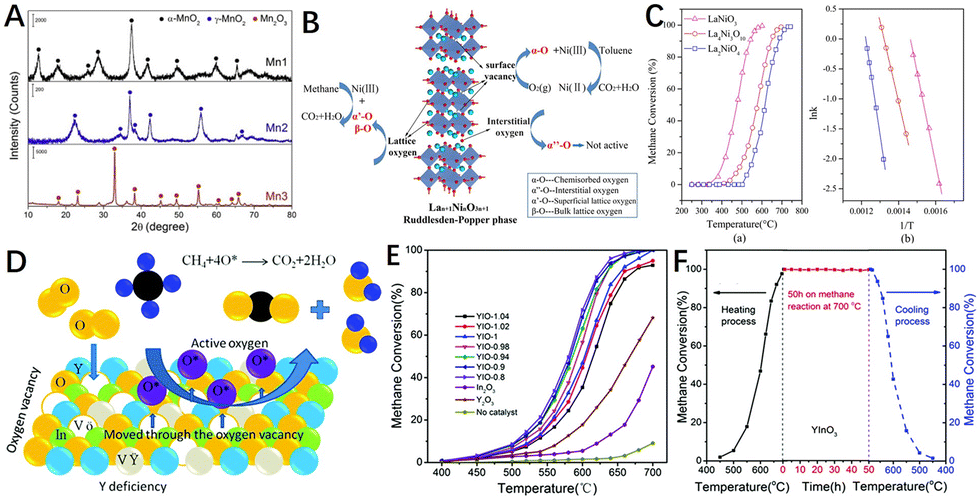 | ||
| Fig. 9 (A) XRD patterns of Mn1, Mn2, and Mn3 catalysts. Reproduced from ref. 170 with permission from Elsevier. (B) Proposed mechanism of oxygen transfer on the Lan+1NinO3n+1 layered perovskites in catalytic oxidation of toluene and methane; (C) conversion rates and Arrhenius plots of La2NiO4 (□), La4Ni3O10 (○), and LaNiO3 (△) in methane oxidation. Condition: methane (1 vol%), O2 (10 vol%), 0.5 g catalyst, GHSV = 19200 mL gcat−1 h−1. Reproduced from ref. 171 with permission from the American Chemical Society. (D) Schematic representation of the catalytic methane oxidation on the YxInO3+δ catalyst; (E) methane conversion as a function of temperature over the YIO-x catalysts; (F) the stability test performed at 700 °C within 50 h under 1% vol. CH4, 10% vol. O2 in nitrogen; GHSV = 30000 mL g−1 h−1. Reproduced from ref. 172 with permission from the Royal Society of Chemistry. | ||
Additionally, layered perovskites have a greater degree of oxygen non-stoichiometric structure than perovskites, which makes them potentially more suitable for catalytic applications. Meng et al.171 prepared Lan+1NinO3n+1 layered perovskites (Fig. 9B) by co-precipitation in a supercritical water reaction environment. The results showed that La4Ni3O10 (n = 3) with higher lattice oxygen mobility had the lowest activation energy for methane oxidation (Fig. 9C, Ea = 111.0 kJ mol−1). Xiao et al.172 synthesized a novel non-stoichiometric YxInO3+δ (YIO-x, 0.8 ≤ x ≤ 1.04) perovskite catalyst by using the glycine self-propagating gel combustion method, which had more oxygen vacancies and a high specific surface area (Fig. 9D). The results showed that the low levels of non-stoichiometry in the A site of YxInO3+δ effectively increased the amount of oxygen desorption by 39–42% when compared to the original (YIO-1) due to Y-deficiency and oxygen vacancies. The non-stoichiometry also resulted in significant changes in Lewis acid sites on the surface of the samples, confirming that it greatly promoted the catalytic combustion of methane (Fig. 9E). After 50 h of stability test, the catalyst maintained high activity, indicating its good catalytic stability (Fig. 9F).
Ball milling is usually used for the mechanochemical construction of defects. The mechanical forces generated by ball milling can disrupt the structure of the material's native phases and cause a variety of transformations and reactions in the material, such as disorder and amorphization, grain boundary disorder, and polycrystalline transformations.173,177 In addition, the use of mechanical forces can also generate a large number of surface defects in situ on the material, such as edges, cavities, structural collapse, and swelling,97 and contribute to the formation of porous structures. Researchers have also used ball milling to modify catalysts.178–181 Shu et al. mechanically mixed NaCl and metal salts by ball milling and washed with deionised water to remove NaCl-crystal support to obtain porous metal oxides Co3O4 (ref. 182) (Fig. 10A) and NiO (ref. 183) (Fig. 10B) with an abundant oxygen vacancy concentration, which showed excellent activity and stability in methane combustion catalytic reactions. The high entropy oxides (HEOs) can also be used as efficient catalysts in the field of thermal catalysis due to their multi-element composition and the lattice distortions arising from their special solid solution states.184,185 Thus, Nie et al.186 prepared defect-enriched high-entropy MgAl2O4 using a ball milling method to create more grain boundaries and interfaces in the metal precursors (Fig. 10C), which gave the material excellent catalytic activity and hydrothermal stability. Furthermore, Bao et al.178 synthesized CoxMn1−xOy catalysts using cobalt chloride, sodium hydroxide, and potassium permanganate as raw materials by ball-milling under solvent-free conditions to drive the reaction between the raw materials (Fig. 10D). As shown in Fig. 10E, He et al.82 used a lattice refinement strategy with urea-assisted ball milling to effectively implant numerous oxygen vacancies in transition metal oxide materials (TMOs), as well as surface lattice disorder layers that significantly increased the specific surface area, with the modified catalysts having higher catalytic performance.
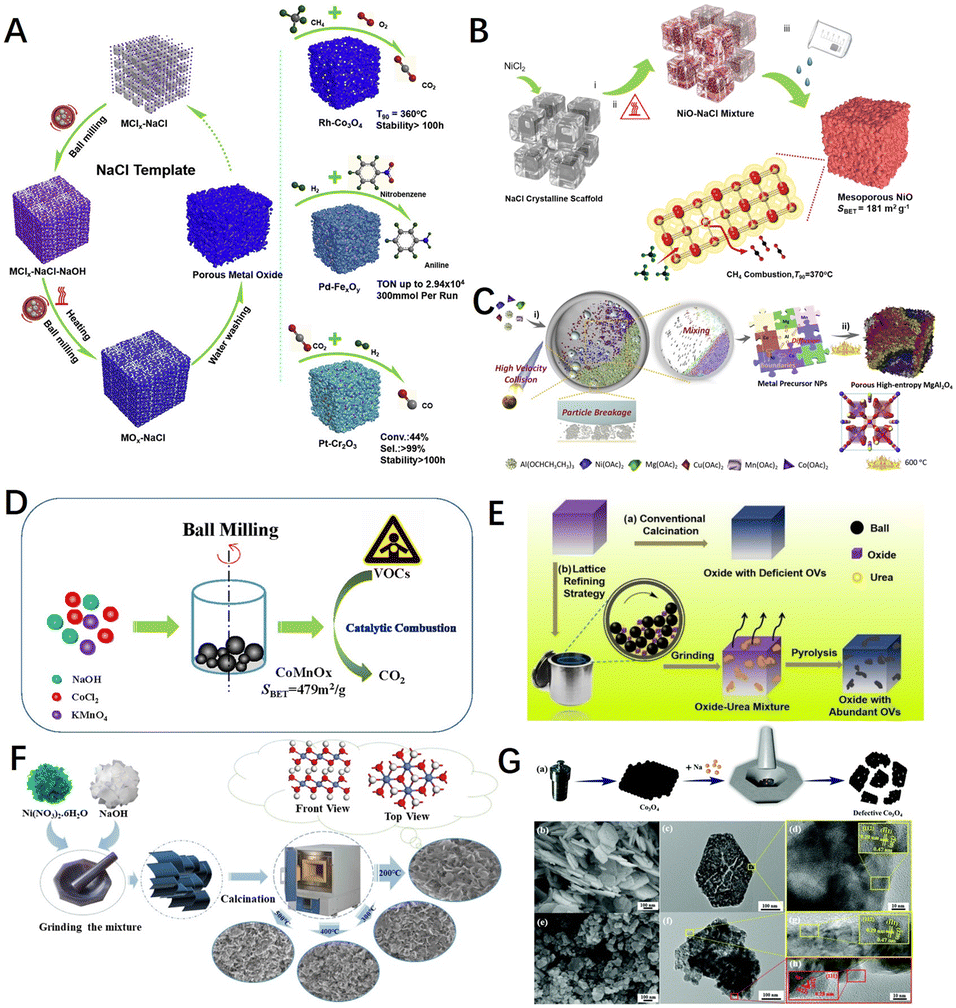 | ||
| Fig. 10 (A) Synthesis schematic diagram of porous metal oxides synthesized through NaCl-based solid solution. Reproduced from ref. 182 with permission from Elsevier. (B) A typical synthesis of porous NiO materials through NaCl crystalline scaffolds. Reproduced from ref. 183 with permission from the American Institute of Chemical Engineers. (C) Mechanochemical method for preparing porous HE–MgAl2O4 catalyst. Reproduced from ref. 186 with permission from Elsevier. (D) A synthesis scheme of porous CoxMn1−xOy–M via a mechanochemical redox process. Reproduced from ref. 178 with permission from Elsevier. (E) Schematic diagram of the transition metal oxide materials with abundant Ovs prepared by the ball milling method. Reproduced from ref. 82 with permission from the American Chemical Society. (F) Schematic illustration of the sample synthesis process by a direct manual grinding strategy. Reproduced from ref. 187 with permission from the American Chemical Society. (G) Schematic of the catalyst preparation of defective Co3O4, and SEM, (HR)TEM according to the selected part for Co3O4 and D-Co3O4. Reproduced from ref. 188 with permission from the Royal Society of Chemistry. | ||
In addition, the researchers have developed a manual grinding method. As shown in Fig. 10F, Chen et al.187 obtained β-Ni(OH)2 flakes by grinding a mixture of sodium hydroxide and nickel nitrate hexahydrate crystals and mixing them well. As the Ni(OH)2 was treated at 200 °C it showed a more disordered nano-sheet structure, more oxygen and hydroxyl groups adsorbed on its surface facilitating the dissociation of methane on the catalyst and reducing the activation energy of methane. Zhang et al.188 produced defective Co3O4 catalysts by hand-milling method with the aid of Na at room temperature (Fig. 10G). The methane conversion temperatures for 50% (T50) and 90% (T90) on the defective Co3O4 catalysts were 47 °C and 35 °C, respectively, lower than those of the pristine Co3O4 catalysts. The improved catalytic performance was attributed to a significant increase in the surface-active oxygen content and an increase in the oxygen mobility due to in build defects. The effects of crushing the volume and creating a high number of oxygen vacancies played an influential role in the surface exposure and intrinsic reactivity enhancement through a simple mechanochemical process. In conclusion, it is vital to control the reaction conditions for mechanochemistry.
4.2 Introduction of defects in pre-synthesized materials
The route of introducing defects into a pre-synthesized catalytic material (which already has an initial structure) does not involve the growth reaction process of the catalyst material in the vast majority of cases, only changing the properties of its surface and subsurface. Defects such as vacancies (oxygen or cationic vacancies), dopants (generally more doped anions), and steps are easily introduced into the surface or the subsurface of catalyst materials. The most common methods in this pathway are etching and quenching.Transition metal oxide materials can be etched with acid or alkali solutions, enabling the modification of the catalyst surfaces and the construction of specific defects. As shown in Fig. 11A, Yoo et al.193 treated mesoporous cobalt oxide catalysts with different concentrations of nitric acid, and the treated catalysts had a rough surface containing more surface chemisorbed oxygen and oxygen vacancies. The results showed a marked enhancement in the catalytic activity for methane combustion and a reduction in T50 by nearly 70 °C. Analogously, Guo et al.191 introduced rich oxygen vacancy and electron-deficient Lewis acid site Con+ on the surface of the macroporous perovskite La0.8Sr0.2CoO3 (LSCO) by taking advantage of the special corrosion properties of oxalic acid. The local electron cloud migration between them effectively regulated the electronic properties and coordination structure of the catalyst surface. The interaction between CH4 molecules and the LSCO surface was improved, and the activation of reactive oxygen species as well as lattice oxygen mobility were improved. Chen et al.194 performed alkaline etching of the electrospun LaCoO3 nanorods with NaOH solutions (Fig. 11B), where the La–O–Co bond was attacked and weakened the Co–O bond, contributing to more surface adsorbed oxygen species and exposed Co3+.
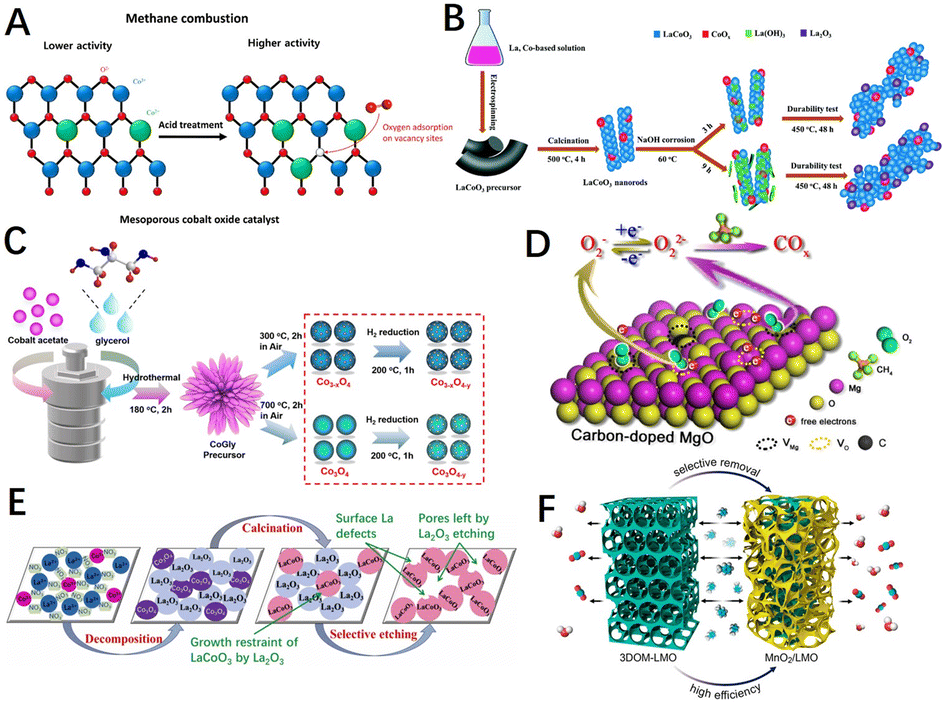 | ||
| Fig. 11 (A) Diagram of HNO3 etching mesoporous cobalt oxide catalysts before and after. Reproduced from ref. 193 with permission from Elsevier. (B) Scheme of the structural evolution of NaOH-treated LaCoO3. Reproduced from ref. 194 with permission from the Royal Society of Chemistry. (C) Synthetic schematic for H2 reduction-treated Co3O4 catalyst. Reproduced from ref. 195 with permission from the American Chemical Society. (D) Diagram of carbon-doped MgO and the proposed mechanism of oxygen transfer in methane oxidation. Reproduced from ref. 87 with permission from the American Chemical Society. (E) Preparation of porous LaCoO3 perovskite oxides by a selective etching method. Reproduced from ref. 198 with permission from Elsevier. (F) Synthesis route of the MnO2/LMO sample. Reproduced from ref. 196 with permission from the American Chemical Society. | ||
The reduction etching strategy utilizes the reaction of reducing agents such as H2 and NaHB4 with the lattice oxygen of materials, creating defects such as oxygen vacancies, cationic vacancies, or low valence states in metal oxides. For example, Qiu et al.195 introduced Co defects and oxygen vacancies on the surface of Co3O4 by hydrogen reduction reactions (Fig. 11C), which weakened the Co–O bond strength and enhanced the oxygen mobility as well as the redox properties of the material. As shown in Fig. 11D, Hao et al.87 introduced a carbon source (citric acid monohydrate) into the material matrix to form an in situ reductive combustion atmosphere, releasing the lattice oxygen by the solution combustion method. Moreover, the carbon substitution for magnesium induced the formation of magnesium vacancies, while embedding oxygen–magnesium double vacancies in the MgO catalyst simultaneously. Hence, the migration of charge from the material surface to the adsorbed oxygen species was accelerated, leading to an enrichment of peroxide species, which enhanced the activation and oxidation of methane on the material surface.
In comparison to other chemical etching methods, selective dissolution etching, which is characterized by well-defined etching targets and directivity, has an excellent performance in VOCs removal,189,196 CH4 and CO oxidation190,197 as a new way to induce defects on the material surface. For example, Wang et al.198 selectively etched La2O3 with acetic acid (HAc) (Fig. 11E), which not only produced surface La defects but also improved the porosity, active site exposure, and redox properties of the material. Thus, porous LaCoO3 could effectively contact and activate the reactants and accelerate the reaction rate. As shown in Fig. 11F, Si et al.196 used nitric acid to selectively remove some La cations from three-dimensional chain-ordered macroporous (3DOM) LaMnO3 materials to obtain MnO2/LaMnO3 samples with a large specific surface area and good dispersion, with a significantly higher number of surface oxygen radicals and lattice oxygen mobility. However, chemical etching at industrial-scale production generates a large amount of acid and alkaline waste solution that pollutes the environment, as well as etching depth and time still need a lot of research results to support.
800 mL g−1 h−1). Tian et al.199 used plasma etching to successfully construct defects in perovskite LaMnO3 for catalytic methane combustion, as shown in Fig. 12B. The surface defects caused by plasma etching improved the oxygen utilization capacity of perovskite LaMnO3 and increased the number of oxygen vacancies, which acted as adsorption and activation centres. The plasma technology will provide a novel and universal strategy for the synthesis of composite oxide catalysts with rich surface defects.199,200
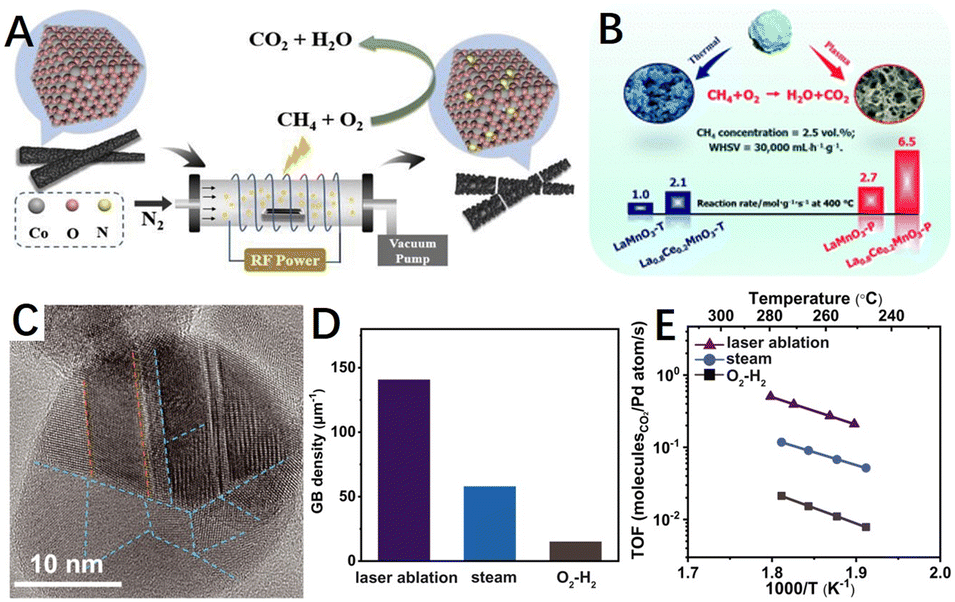 | ||
| Fig. 12 (A) Schematic diagram of N2 plasma-treated Co3O4 catalyst. Reproduced from ref. 140 with permission from Elsevier. (B) Diagram of O2 atmosphere glow discharge plasma-treated LaMnO3 and Ce–LaMnO3 catalyst. Reproduced from ref. 199 with permission from the Royal Society of Chemistry. (C) High-resolution TEM image of laser ablation-generated Pd NPs. The orange dashed lines highlight ∑3(111) TBs, and the blue dash lines highlight GBs; (D) GB density statistical histogram of laser-generated Pd/Al2O3 and Pd/Al2O3 after steam (600 °C) and O2–H2 pretreatments; (E) Arrhenius plots of methane combustion of laser-generated Pd/Al2O3 and Pd/Al2O3 after steam (600 °C) and O2–H2 pretreatments. TOF, turnover frequency. Reproduced from ref. 77 with permission from the American Association for the Advancement of Science. | ||
Besides, laser ablation is also widely used for material modification as another physical etching route. After the laser energy is absorbed by the target, the energy first excites electron leaps within the target, which are then converted into heat energy, etc., causing the target vaporisation to form a plasma state. As mentioned in Section 3.2, Huang et al.77 demonstrated that twin/grain boundaries are excellent active sites for CMC (Fig. 12C), significantly improving the catalytic activity. Also, Huang et al.77 applied the liquid laser ablation technique reported by Li et al.,201 where the laser ablation treatment was able to significantly increase the concentration of grain boundaries, as shown in Fig. 12D. The turnover frequency of the Pd/Al2O3 catalyst prepared by laser ablation was approximately 25 times higher than that of a conventional catalyst pretreated in just oxygen and hydrogen (Fig. 12E).
Metal oxide catalysts are often doped with heteroatoms to form solid solutions with the aim of improving the catalytic activity. The disordered solid solution undergoes structural ordering, called superstructure, during slow cooling. However, the ordered structure is not conducive to the migration of electrons and lattice oxygen,205,206 which is not beneficial to the catalytic activity of CMC, either from an in-plane or surface mechanism point of view. The disordered solid solution phase can be formed when the solid solution is quenched, i.e., rapidly cooled. For example, Vereshchagin et al.207 showed that quenching the Sr0.8Gd0.2CoO3−δ perovskite from 1200 °C to 900 °C (30 °C s−1) leads to the preservation of the Gd/Sr disordered cubic phase. The results showed that the cubic perovskite phase with randomly (disordered) distributed Gd3+/Sr2+ ions was about 5 times more active in the CMC than the tetragonal phase Sr0.8Gd0.2CoO3−δ with ordered Gd3+/Sr2+ ions and oxygen vacancies (Fig. 13A). As such, maintaining the disordered structure in the solid solution materials by controlling the quenching rate without changing the chemical properties of catalysts. It is an approach to improve the activity of methane combustion catalysts.
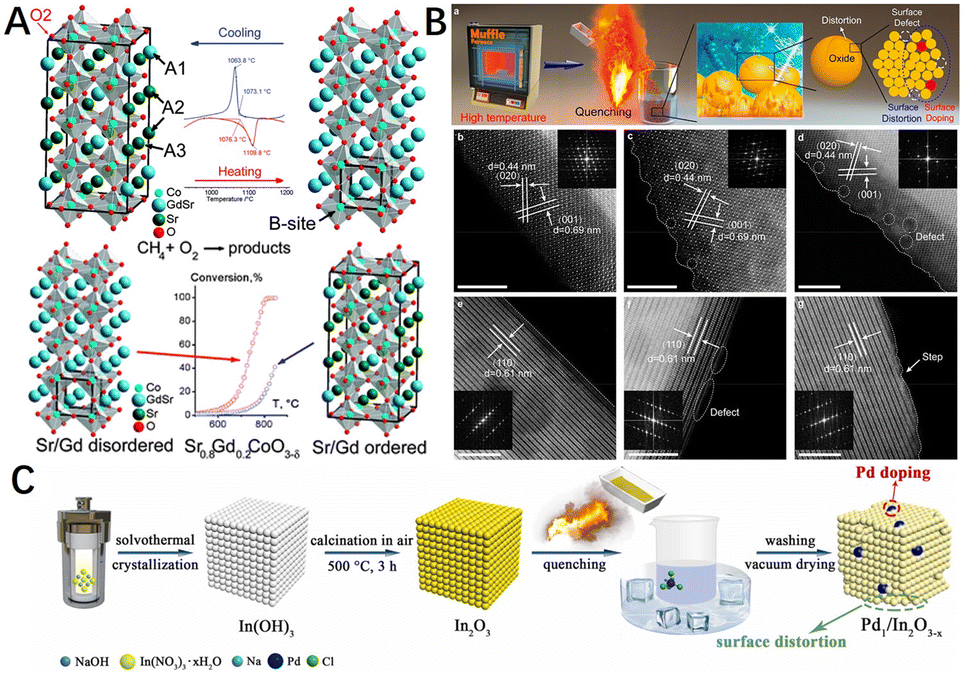 | ||
| Fig. 13 (A) DSC curves near the transition and the structure of ordered (left) and disordered (right) Sr0.8Gd0.2CoO3−δ. The heating (red) and cooling (blue) ramp rate of 10 °C min−1, 20% O2–Ar. Reproduced from ref. 207 with permission from the Royal Society of Chemistry. (B) Schematic illustration of the quenching method used to prepare the electrocatalysts and aberration-corrected HAADF-STEM images of different electrocatalysts. Reproduced from ref. 208 with permission from the American Chemical Society. (C) Schematic illustration for the construction of Pd1/In2O3−x by a simple quenching strategy. Reproduced from ref. 83 with permission from Elsevier. | ||
In addition to quenching by modulating the cooling rate during calcination of the material (rapid cooling), it is also possible to quench the catalyst directly in low-temperature water or metal salt solution. Recently, Ye et al.208 subjected NiMoO4 nanoparticles (NPs) and NiMoO4 nanorods (NRs) to rapid cooling in aqueous solutions with different concentrations of Cr, Mn, Fe, or Co salts, and HAADF-STEM images (Fig. 13B) showed that the quenched nanorods (NRs) had surface areas with jagged, stepped, and abundant defects such as atomic absences and disordered internal atomic structures, which may be the result of the synergistic effect of rapid cooling and cation doping. Furthermore, the authors successfully extended this quenching strategy to other metal oxides such as spinel-type Co3O4, Fe2O3, LaMnO3, and CoSnO3. Besides, Li et al.83 prepared Pd1/In2O3−x single-atom catalysts with surface oxygen vacancies by a simple quenching strategy, as shown in Fig. 13C. Owing to quenching and palladium atom doping, the smooth surface of cubic In2O3 was transformed into a jagged (step-like) surface rich in defects.
4.3 Other methods
In addition to the above methods, researchers have induced the formation of defects in materials through several special synthetic methods and conditions. For example, Wang et al.78 synthesized highly defective cobalt oxide nanocrystals through the controlled oxidation of carbon-coated cobalt nanoparticles, which exploited the principle that nanoscale voids are created inside the nanocrystals motivated by the Kirkendall effect (Fig. 14A). The strain gradients elicited in the nanoconfined carbon shell favored the generation of abundant active sites and also prevented the heavy aggregation of metal nanoparticles. The HRTEM images of the catalysts after reaction at different temperatures showed that the defects are severe on the catalyst surface, showing a large number of steps and kinks, for multiphase catalysis, and these active sites have considerable catalytic activity for the combustion of various hydrocarbons (methane, propane, and substituted benzene). Thus, for methane combustion, the catalyst exhibited a unique activity comparable to or even better than that of Pd catalysts. Yu et al.209 synthesized an effective manganese oxide (MnO2) from LaMnO3 by ball milling combined with acid etching-selective atom removal (Fig. 14B), a material with high specific surface area and structural defects. In addition, Liu et al.210 synthesized ultra-small MnOx nanoparticles with fully exposed defects by two-step pyrolysis of metal–organic backbone materials (MOFs) via in situ carbon-limited oxidation, using their inert atmosphere-derived carbon species to limit the growth of metal oxides (Fig. 14C).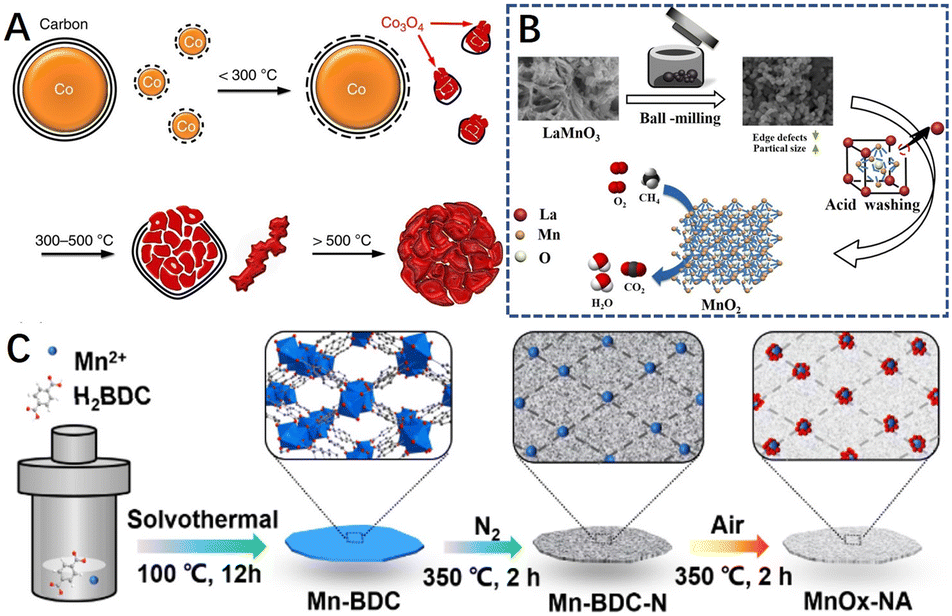 | ||
| Fig. 14 (A) Scheme for the transformation process of the Co3O4 catalyst. Reproduced from ref. 78 with permission from Springer Nature. (B) The preparation process of the MnO2 catalyst by a combined ball-milling and selective atom removal method. Reproduced from ref. 209 with permission from Elsevier. (C) Preparation process of the MnOx–NA catalyst. Reproduced from ref. 210 with permission from the American Chemical Society. | ||
The above-mentioned modification routes for the construction and modulation of defects in catalysts can effectively boost the performance of methane combustion catalysts, but the mechanism of action and the effect of defects on the stability of the catalyst remain to be explored. In addition, some of the modification pathways are cumbersome, costly, and environmentally friendly from the point of view of industrial applications. Therefore, a lot of research needs to be done on the modification of methane low-temperature combustion catalysts based on defect engineering, to find a defect-induced modification technology that can be produced on a large scale, with simple technology, low cost, and excellent catalytic activity.
5. Machine learning prediction and exploration
The above modification methods based on defect chemistry provide rich ways for material regulation. However, mining the feasibility and stability of material defects through experiments is time-consuming and expensive, which requires a large amount of experimental data and time to screen and prove. As an excellent tool, data-driven machine learning has gradually emerged in the field of material mining with excellent performance.211–214 Machine learning has been widely used in the screening and mining of heterogeneous catalysts.215–218For example, Yang et al.219 employed a neuro-genetic machine learning framework (ANN-GA) to optimize and predict the optimal preparation parameters for the precipitation synthesis of highly efficient silver-doped manganese oxides (Ag/MnOx) for the total oxidation of toluene, by (i) conducting a central composite design (CCD) and experimental design based on the critical conditions of the catalyst preparation process to establish an experimental matrix; (ii) applying an ML framework consisting of feed-forward neural networks and genetic optimizer was used to search potential Ag/MnOx catalysts and their preparation conditions from the virtual catalyst space; (iii) multilayer perceptron neural network (MLPNN) was employed for tracking the correlation between the independent variables and the activity of catalysts (Fig. 15). Due to the small dataset size of this study, one cross-validation (LOOCV) was used. That is, when the first data sample was designated as the test subset, the second to 30th data samples were designated as the training subset, and this process was repeated for all 30 data sets; (iv) the experiments were conducted to verify the mining results of the ML model. Finally, the relative importance of each factor in the preparation process was obtained, among which the silver loading had the most significant effect on the catalytic activity. Additionally, Sharma et al.220 applied ML techniques to identify the factors affecting the defect formation energy in perovskite oxides. Using 13 element attributes as features and random forest regression, a systematic approach was used to select those important features downward from a larger feature set to establish a framework for accurate prediction of defect formation energy. These results demonstrate the effectiveness of ML tools in identifying and quantifying the dependence of catalysts on different characteristics and provide a promising approach for rapid prediction and screening of the catalysts. Moreover, a general framework can potentially be applied to other material classes.
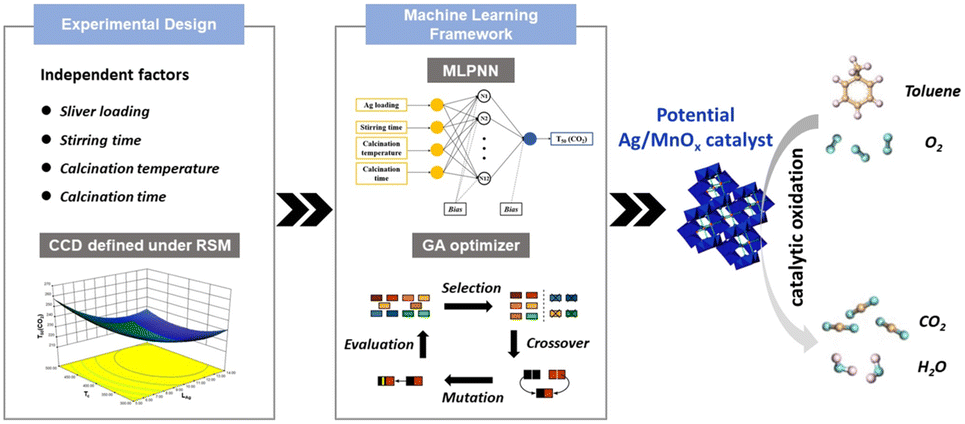 | ||
| Fig. 15 Machine learning methodology for the discovery of Ag/MnOx catalysts consists of (i) experimental design, (ii) machine learning framework, (iii) composition exploration and prediction, and (iv) experimental verification and feedback. Reproduced from ref. 219 with permission from Elsevier. | ||
Machine learning employs a different approach to model development, relying on flexible nonlinear models that require large amounts of data to train and optimize.221 According to the data source, it is mainly from two perspectives, “top-down”: a large number of experimental data of defect engineering modified catalysts in the literature are screened, and the relationship between characteristic quantities such as defect type and quantity and catalytic performance is constructed to guide the experimental applications. “Bottom-up”: starting from the theory-defect site as the decisive factor-training model-output to improve the optimization and prediction ability of high-performance catalysts based on defect chemistry. Thus, mathematical models trained with data can learn stepwise and facilitate prediction, guided optimization, and fundamental understanding of the performance response of defects in methane combustion catalysts. Machine learning has not yet been realized in the development of catalytic methane combustion catalysts based on defect engineering, and has great research space and prospects.
6. Summary and outlook
Due to the increasing global warming effect, gradual and more attention is being provided to the emission–reduction of lean methane. The researchers have performed a lot of related research on the preparation and modification of methane combustion catalysts, reaction mechanisms, and simulation calculations. Due to the rapid development of techniques for the preparation and characterization of catalysts, the influence of defects on catalytic methane combustion has also attracted extensive attention from researchers. This paper reviewed the research advancement of utilizing defects to promote the catalytic performance of methane combustion in recent years, mainly from the following four respects: (1) the model of the catalytic combustion reaction mechanism of methane; (2) the role of defects in the catalytic combustion of methane; (3) the main methods of constructing defects in existing research; (4) the prediction and exploration of machine learning. Despite some attractive results, some challenges and limitations remain in the understanding and application of defects in the catalytic methane combustion reaction.First, whether the performance of defect-mediated catalysts for methane combustion at low-temperature could meet the practical requirements. From this perspective, it is necessary to rationally design the defects in the catalyst, such as combining different types of defects with different types of catalysts to improve the adsorption and activation of methane/oxygen molecules, surface reactivity, and product desorption. Second, controlling precisely the distribution and number of defects in the catalysts. Since defects may exist in different locations in the catalyst, such as surface, subsurface, or volume equivalent locations, it will have different effects on the performance of the methane combustion catalysts. In addition, the reactivity of the defect sites is much higher than that of the non-defect sites, but excessive content of defects will damage the structure of catalysts, thereby affecting the catalyst stability.
Third, this paper explored the structure–activity link between defects (type, content, distribution)-properties in catalysts of methane combustion. The catalytic active site evolves dynamically due to the possible active site transitions (agglomeration or redispersion) and redox cycles during the catalytic reaction of methane combustion. The complexity of the reactions involved in regulating defective structures in materials also made this work difficult. Therefore, the precise and controllable structural defect type, defect concentration, defect distribution, and the regulatory relationship between the catalytic activity in materials still require more in-depth research and understanding. The progressive development of characterization techniques has allowed researchers to reveal the dynamic evolution of defect structures and reactant molecules during the catalytic reaction of methane combustion at the molecular level, with more advanced equipment and technologies. Finally, optimizing the strategies for building defects. From the perspective of the production application, and combined with the structure and type of catalyst, it is crucial to explore and develop simpler, low-cost, and commercial-friendly synthetic techniques based on defect engineering. Despite these challenges, defect engineering still has a promising future in methane combustion catalysis.
Conflicts of interest
The authors declare no competing financial interest.References
- M. Cargnello, J. J. D. Jaen, J. C. H. Garrido, K. Bakhmutsky, T. Montini, J. J. C. Gamez, R. J. Gorte and P. Fornasiero, Science, 2012, 337, 713–717 CrossRef CAS PubMed.
- W. Huang, X. Zhang, A.-C. Yang, E. D. Goodman, K.-C. Kao and M. Cargnello, ACS Catal., 2020, 10, 8157–8167 CrossRef CAS.
- K. Murata, D. Kosuge, J. Ohyama, Y. Mahara, Y. Yamamoto, S. Arai and A. Satsuma, ACS Catal., 2019, 10, 1381–1387 CrossRef.
- X. Feng, L. Jiang, D. Li, S. Tian, X. Zhu, H. Wang, C. He and K. Li, J. Energy Chem., 2022, 75, 173–215 CrossRef CAS.
- D. Jiang, K. Khivantsev and Y. Wang, ACS Catal., 2020, 10, 14304–14314 CrossRef CAS.
- R. J. Farrauto, Science, 2012, 337, 659–660 CrossRef CAS PubMed.
- A. K. Datye and M. Votsmeier, Nat. Mater., 2021, 20, 1049–1059 CrossRef CAS PubMed.
- J. Yang and Y. Guo, Chin. Chem. Lett., 2018, 29, 252–260 CrossRef CAS.
- L. He, Y. Fan, J. Bellettre, J. Yue and L. Luo, Renewable Sustainable Energy Rev., 2020, 119, 109589 CrossRef CAS.
- X. Feng, W. Li, D. Liu, Z. Zhang, Y. Duan and Y. Zhang, Small, 2017, 13, 1700941 CrossRef.
- L. He, Y. Fan, L. Luo, J. Bellettre and J. Yue, Chem. Eng. J., 2020, 380, 122424 CrossRef CAS.
- J. J. Willis, E. D. Goodman, L. Wu, A. R. Riscoe, P. Martins, C. J. Tassone and M. Cargnello, J. Am. Chem. Soc., 2017, 139, 11989–11997 CrossRef CAS PubMed.
- N. Nunotani, N. Moriyama, K. Matsuo and N. Imanaka, ACS Appl. Mater. Interfaces, 2017, 9, 40344–40350 CrossRef CAS PubMed.
- Y. Wang, H. Arandiyan, J. Scott, M. Akia, H. Dai, J. Deng, K.-F. Aguey-Zinsou and R. Amal, ACS Catal., 2016, 6, 6935–6947 CrossRef CAS.
- K. Murata, Y. Mahara, J. Ohyama, Y. Yamamoto, S. Arai and A. Satsuma, Angew. Chem., Int. Ed., 2017, 56, 15993–15997 CrossRef CAS PubMed.
- H. Arandiyan, H. Dai, J. Deng, Y. Wang, S. Xie and J. Li, Chem. Commun., 2013, 49, 10748–10750 RSC.
- J. Chen, X. Wang, L. Zhang and Z. Rui, Appl. Catal., B, 2021, 297, 120410 CrossRef CAS.
- Z. Liang, T. Li, M. Kim, A. Asthagiri and J. F. Weaver, Science, 2017, 356, 299–303 CrossRef CAS PubMed.
- J. Lin, X. Chen, Y. Zheng, Y. Xiao, Y. Zheng and L. Jiang, Appl. Surf. Sci., 2022, 587, 152835 CrossRef CAS.
- X. Li, X. Wang, K. Roy, J. A. van Bokhoven and L. Artiglia, ACS Catal., 2020, 10, 5783–5792 CrossRef CAS.
- Y. Ma, S. Li, T. Zhang, Y. Zhang, X. Wang, Y. Xiao, Y. Zhan and L. Jiang, Nanoscale, 2021, 13, 5026–5032 RSC.
- H. Xiong, D. Kunwar, D. Jiang, C. E. García-Vargas, H. Li, C. Du, G. Canning, X. I. Pereira-Hernandez, Q. Wan, S. Lin, S. C. Purdy, J. T. Miller, K. Leung, S. S. Chou, H. H. Brongersma, R. ter Veen, J. Huang, H. Guo, Y. Wang and A. K. Datye, Nat. Catal., 2021, 4, 830–839 CrossRef CAS.
- A. W. Petrov, D. Ferri, F. Krumeich, M. Nachtegaal, J. A. van Bokhoven and O. Krocher, Nat. Commun., 2018, 9, 2545 CrossRef PubMed.
- J. Yang, M. Peng, G. Ren, H. Qi, X. Zhou, J. Xu, F. Deng, Z. Chen, J. Zhang, K. Liu, X. Pan, W. Liu, Y. Su, W. Li, B. Qiao, D. Ma and T. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18522–18526 CrossRef CAS PubMed.
- P. J. Jodlowski, R. J. Jedrzejczyk, D. K. Chlebda, A. Dziedzicka, L. Kuterasinski, A. Gancarczyk and M. Sitarz, Nanomaterials, 2017, 7, 174 CrossRef PubMed.
- X. Shao, J. He, Q. Su, D. Zhao and S. Feng, RSC Adv., 2022, 12, 17490–17497 RSC.
- G. Águila, F. Gracia, J. Cortés and P. Araya, Appl. Catal., B, 2008, 77, 325–338 CrossRef.
- F. Wang, L. Zhang, L. Xu, Z. Deng and W. Shi, Fuel, 2017, 203, 419–429 CrossRef CAS.
- Z. Chen, S. Wang, W. Liu, X. Gao, D. Gao, M. Wang and S. Wang, Appl. Catal., A, 2016, 525, 94–102 CrossRef CAS.
- Z. Fei, S. He, L. Li, W. Ji and C. T. Au, Chem. Commun., 2012, 48, 853–855 RSC.
- E. Akbari, S. M. Alavi, M. Rezaei and A. Larimi, Ind. Eng. Chem. Res., 2021, 60, 7572–7587 CrossRef CAS.
- Y. He, F. Guo, K. R. Yang, J. A. Heinlein, S. M. Bamonte, J. J. Fee, S. Hu, S. L. Suib, G. L. Haller, V. S. Batista and L. D. Pfefferle, J. Am. Chem. Soc., 2020, 142, 17119–17130 CrossRef CAS PubMed.
- R. Spinicci, A. Delmastro, S. Ronchetti and A. Tofanari, Mater. Chem. Phys., 2003, 78, 393–399 CrossRef CAS.
- H. Chen, J. Li, W. Cui, Z. Fei, Q. Tian, Q. Liu, X. Chen, M. Cui, Z. Zhang, J. Tang and X. Qiao, Appl. Surf. Sci., 2020, 505, 144112 CrossRef CAS.
- Y. Zhu, Y. Sun, X. Niu, F. Yuan and H. Fu, Catal. Lett., 2010, 135, 152–158 CrossRef CAS.
- F. F. Tao, J. J. Shan, L. Nguyen, Z. Wang, S. Zhang, L. Zhang, Z. Wu, W. Huang, S. Zeng and P. Hu, Nat. Commun., 2015, 6, 7798 CrossRef CAS PubMed.
- T. Wang, L. Qiu, H. Li, C. Zhang, Y. Sun, S. Xi, J. Ge, Z. J. Xu and C. Wang, J. Catal., 2021, 404, 400–410 CrossRef CAS.
- Z. Zhang, J. Li, T. Yi, L. Sun, Y. Zhang, X. Hu, W. Cui and X. Yang, Chin. J. Catal., 2018, 39, 1228–1239 CrossRef CAS.
- A. Urdă, I. Popescu, T. Cacciaguerra, N. Tanchoux, D. Tichit and I.-C. Marcu, Appl. Catal., A, 2013, 464–465, 20–27 CrossRef.
- H. Liu, X. Fu, X. Weng, Y. Liu, H. Wang and Z. Wu, J. Chem., 2014, 2014, 1–6 Search PubMed.
- F. Huang, X. Wang, A. Wang, J. Xu and T. Zhang, Catal. Sci. Technol., 2016, 6, 4962–4969 RSC.
- L. Ma, B. Shi, M. Cui, L. Wang, D. Li and A. Chen, Sci. China, Ser. B: Chem., 2008, 51, 211–217 CrossRef CAS.
- J. Chen, H. Arandiyan, X. Gao and J. Li, Catal. Surv. Asia, 2015, 19, 140–171 CrossRef CAS.
- L. F. Liotta, H. Wu, G. Pantaleo and A. M. Venezia, Catal. Sci. Technol., 2013, 3, 3085–3102 RSC.
- T. Wang, C. Zhang, J. Wang, H. Li, Y. Duan, Z. Liu, J. Y. Lee, X. Hu, S. Xi, Y. Du, S. Sun, X. Liu, J.-M. Lee, C. Wang and Z. J. Xu, J. Catal., 2020, 390, 1–11 CrossRef CAS.
- S. Wang, P. Chu, J. Liu, C. Wang, E. Duan, J. Deng and L. A. Hou, Fuel, 2022, 316, 123358 CrossRef CAS.
- Y. Yao, Q. Dong, A. Brozena, J. Luo, J. Miao, M. Chi, C. Wang, I. G. Kevrekidis, Z. J. Ren, J. Greeley, G. Wang, A. Anapolsky and L. Hu, Science, 2022, 376, eabn3103 CrossRef CAS PubMed.
- H.-J. Qiu, G. Fang, Y. Wen, P. Liu, G. Xie, X. Liu and S. Sun, J. Mater. Chem. A, 2019, 7, 6499–6506 RSC.
- Y. Wang, J. Mi and Z.-S. Wu, Chem Catal., 2022, 2, 1624–1656 CrossRef CAS.
- T. Li, Y. Yao, Z. Huang, P. Xie, Z. Liu, M. Yang, J. Gao, K. Zeng, A. H. Brozena, G. Pastel, M. Jiao, Q. Dong, J. Dai, S. Li, H. Zong, M. Chi, J. Luo, Y. Mo, G. Wang, C. Wang, R. Shahbazian-Yassar and L. Hu, Nat. Catal., 2021, 4, 62–70 CrossRef CAS.
- P. Wang, X. Ma, X. Hao, B. Tang, A. Abudula and G. Guan, Catal. Rev.: Sci. Eng., 2022, 1–54, DOI:10.1080/01614940.2022.2078555.
- Y. Zheng, K. Fu, Z. Yu, Y. Su, R. Han and Q. Liu, J. Mater. Chem. A, 2022, 10, 14171–14186 RSC.
- L. Wang, Y. Li, J. Liu, Z. Tian and Y. Jing, Sep. Purif. Technol., 2021, 277, 119505 CrossRef CAS.
- S. Bai, N. Zhang, C. Gao and Y. Xiong, Nano Energy, 2018, 53, 296–336 CrossRef CAS.
- J. Xiong, J. Di, J. Xia, W. Zhu and H. Li, Adv. Funct. Mater., 2018, 28, 1801983 CrossRef.
- Y. Xu, X. Liu, N. Cao, X. Xu and L. Bi, Sustainable Mater. Technol., 2021, 27, e00229 CrossRef CAS.
- J. Zhu and S. Mu, Adv. Funct. Mater., 2020, 30, 2001097 CrossRef CAS.
- W. Li, D. Wang, Y. Zhang, L. Tao, T. Wang, Y. Zou, Y. Wang, R. Chen and S. Wang, Adv. Mater., 2020, 32, 1907879 CrossRef CAS PubMed.
- T. Liu, N. Peng, X. Zhang, R. Zheng, M. Xia, H. Yu, M. Shui, Y. Xie and J. Shu, Nano Energy, 2021, 79, 105460 CrossRef CAS.
- Z. Che, L. Ma, G. Luo, C. Xu, Z. Cen, Q. Feng, X. Chen, K. Ren and N. Luo, Nano Energy, 2022, 100, 107484 CrossRef CAS.
- J. Xiong, J. Yang, X. Chi, K. Wu, L. Song, T. Li, Y. Zhao, H. Huang, P. Chen, J. Wu, L. Chen, M. Fu and D. Ye, Appl. Catal., B, 2021, 292, 120201 CrossRef CAS.
- W. Hu, J. Lan, Y. Guo, X.-M. Cao and P. Hu, ACS Catal., 2016, 6, 5508–5519 CrossRef CAS.
- Z. Tang, T. Zhang, D. Luo, Y. Wang, Z. Hu and R. T. Yang, ACS Catal., 2022, 13457–13474, DOI:10.1021/acscatal.2c03321.
- Y. Gao, M. Jiang, L. Yang, Z. Li, F. X. Tian and Y. He, Front. Chem., 2022, 10, 959422 CrossRef CAS PubMed.
- K. Li, D. Xu, K. Liu, H. Ni, F. Shen, T. Chen, B. Guan, R. Zhan, Z. Huang and H. Lin, J. Phys. Chem. C, 2019, 123, 10377–10388 CrossRef CAS.
- J. Xiong, K. Wu, J. Yang, P. Liu, L. Song, J. Zhang, M. Fu, L. Chen, H. Huang, J. Wu and D. Ye, Appl. Surf. Sci., 2021, 539, 148211 CrossRef CAS.
- L. Song, J. Xiong, H. Cheng, J. Lu, P. Liu, M. Fu, J. Wu, L. Chen, H. Huang and D. Ye, Appl. Surf. Sci., 2021, 556, 149713 CrossRef CAS.
- C. Liu, H. Xian, Z. Jiang, L. Wang, J. Zhang, L. Zheng, Y. Tan and X. Li, Appl. Catal., B, 2015, 176–177, 542–552 CrossRef CAS.
- O. Demoulin, M. Navez and P. Ruiz, Appl. Catal., A, 2005, 295, 59–70 CrossRef CAS.
- F. Zasada, J. Janas, W. Piskorz, M. Gorczyńska and Z. Sojka, ACS Catal., 2017, 7, 2853–2867 CrossRef CAS.
- F. Zasada, J. Gryboś, C. Hudy, J. Janas and Z. Sojka, Catal. Today, 2020, 354, 183–195 CrossRef CAS.
- I. Popescu, Y. Wu, P. Granger and I.-C. Marcu, Appl. Catal., A, 2014, 485, 20–27 CrossRef CAS.
- M.-A. Mihai, D. C. Culita, I. Atkinson, F. Papa, I. Popescu and I.-C. Marcu, Appl. Catal., A, 2021, 611, 117901 CrossRef CAS.
- J. Ma, R. Long, D. Liu, J. Low and Y. Xiong, Small Struct., 2022, 3, 2100147 CrossRef CAS.
- D. Li, K. Li, R. Xu, X. Zhu, Y. Wei, D. Tian, X. Cheng and H. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 19227–19241 CrossRef CAS PubMed.
- L. Jiang, D. Li, G. Deng, C. Lu, L. Huang, Z. Li, H. Xu, X. Zhu, H. Wang and K. Li, Chem. Eng. J., 2023, 456, 141054 CrossRef CAS.
- W. Huang, C. Johnston-Peck Aaron, T. Wolter, D. Y. Wei-Chang, L. Xu, J. Oh, A. R. Benjamin, C. Zhou, E. H. Megan, A. H. Andrew, M. L. Aaron, M. Mavrikakis and M. Cargnello, Science, 2021, 373, 1518–1523 CrossRef CAS PubMed.
- H. Wang, C. Chen, Y. Zhang, L. Peng, S. Ma, T. Yang, H. Guo, Z. Zhang, D. S. Su and J. Zhang, Nat. Commun., 2015, 6, 7181 CrossRef CAS PubMed.
- Z. Han, H. Zhang, B. Dong, Y. Ni, A. Kong and Y. Shan, ChemistrySelect, 2016, 1, 979–983 CrossRef CAS.
- S. Royer, D. Duprez and S. Kaliaguine, J. Catal., 2005, 234, 364–375 CrossRef CAS.
- R. Wang, X. Chen, Z. Huang, J. Yang, F. Liu, M. Chu, T. Liu, C. Wang, W. Zhu, S. Li, S. Li, J. Zheng, J. Chen, L. He, L. Jin, F. Pan and Y. Xiao, Nat. Commun., 2021, 12, 3085 CrossRef CAS PubMed.
- J. He, P. Wu, L. Lu, H. Li, H. Ji, M. He, Q. Jia, M. Hua, W. Zhu and H. Li, ACS Appl. Mater. Interfaces, 2019, 11, 36666–36675 CrossRef CAS PubMed.
- Z. Li, M. Zhang, X. Dong, S. Ji, L. Zhang, L. Leng, H. Li, J. H. Horton, Q. Xu and J. Zhu, Appl. Catal., B, 2022, 313, 121462 CrossRef CAS.
- U. J. Etim, P. Bai, O. M. Gazit and Z. Zhong, Catal. Rev.: Sci. Eng., 2021, 1–187, DOI:10.1080/01614940.2021.1919044.
- Z. Su, W. Yang, C. Wang, S. Xiong, X. Cao, Y. Peng, W. Si, Y. Weng, M. Xue and J. Li, Environ. Sci. Technol., 2020, 54, 12684–12692 CrossRef CAS PubMed.
- Y. Wang, J. Yun, L. Zhu, B. Cao, J. Gao, X. Shi, Y. Huang, P. Liu and G. Zhu, Appl. Surf. Sci., 2022, 603, 154469 CrossRef CAS.
- Y. J. Hao, L. G. Tian, E. Duan, J. Liu, T. Y. Qi, W. Q. Kong, X. H. Qi, X. Liu, Y. Liu, J. Zhao and F. T. Li, ACS Appl. Mater. Interfaces, 2020, 12, 21761–21771 CrossRef CAS PubMed.
- Y. Shen, J. Deng, S. Impeng, S. Li, T. Yan, J. Zhang, L. Shi and D. Zhang, Environ. Sci. Technol., 2020, 54, 10342–10350 CrossRef CAS PubMed.
- P. Zhou, Y. Wang, C. Xie, C. Chen, H. Liu, R. Chen, J. Huo and S. Wang, Chem. Commun., 2017, 53, 11778–11781 RSC.
- J. Chen, W. Shi and J. Li, Catal. Today, 2011, 175, 216–222 CrossRef.
- G. Pecchi, M. G. Jiliberto, A. Buljan and E. J. Delgado, Solid State Ionics, 2011, 187, 27–32 CrossRef CAS.
- S. Wang, Y. Zhang, S. Zhao, Y. Zhang, Y. Wang, Y. Zhang, A. Kang, E. Duan and S. Qiao, Mol. Catal., 2022, 531, 112685 CrossRef CAS.
- T. Zhu, L. Hu, X. Zhao and J. He, Adv. Sci., 2016, 3, 1600004 CrossRef PubMed.
- Y. Zheng, T. J. Slade, L. Hu, X. Y. Tan, Y. Luo, Z.-Z. Luo, J. Xu, Q. Yan and M. G. Kanatzidis, Chem. Soc. Rev., 2021, 50, 9022–9054 RSC.
- Z. Su, W. Yang, C. Wang, S. Xiong, X. Cao, Y. Peng, W. Si, Y. Weng, M. Xue and J. Li, Environ. Sci. Technol., 2020, 54, 12684–12692 CrossRef CAS PubMed.
- S. Mo, Q. Zhang, J. Li, Y. Sun, Q. Ren, S. Zou, Q. Zhang, J. Lu, M. Fu, D. Mo, J. Wu, H. Huang and D. Ye, Appl. Catal., B, 2020, 264, 118464 CrossRef CAS.
- C. Xie, D. Yan, H. Li, S. Du, W. Chen, Y. Wang, Y. Zou, R. Chen and S. Wang, ACS Catal., 2020, 10, 11082–11098 CrossRef CAS.
- Z. Xu, Y. Zhang, L. Qin, Q. Meng, Z. Xue, L. Qiu, G. Zhang, X. Guo and Q. Li, Small, 2020, 16, e2002071 CrossRef PubMed.
- C. Vogt and B. M. Weckhuysen, Nat. Rev. Chem., 2022, 6, 89–111 CrossRef.
- L. Wang, Y. Zhang, Z. Zeng, H. Zhou, J. He, P. Liu, M. Chen, J. Han, D. J. Srolovitz, J. Teng, Y. Guo, G. Yang, D. Kong, E. Ma, Y. Hu, B. Yin, X. Huang, Z. Zhang, T. Zhu and X. Han, Science, 2022, 375, 1261–1265 CrossRef CAS PubMed.
- F. Polo-Garzon, V. Fung, X. Liu, Z. D. Hood, E. E. Bickel, L. Bai, H. Tian, G. S. Foo, M. Chi, D.-E. Jiang and Z. Wu, ACS Catal., 2018, 8, 10306–10315 CrossRef CAS.
- C. Li, B. Tang, W. Li, Q. Lu and L. Yuan, ACS Appl. Nano Mater., 2022, 5, 13055–13068 CrossRef CAS.
- C. Li, W. Li, K. Chen, A. T. Ogunbiyi, Z. Zhou, F. Xue and L. Yuan, ACS Appl. Nano Mater., 2020, 3, 12130–12138 CrossRef CAS.
- Q. Li, X. Chu, Y. Wang, Q. Yang, Z. Su, Y. Peng, W. Si and J. Li, ACS Catal., 2022, 12, 4430–4439 CrossRef CAS.
- S. Li, Y. Zhang, J. Shi, G. Zhu, Y. Xie, Z. Li, R. Wang and H. Zhu, Nanomaterials, 2019, 10, 31 CrossRef PubMed.
- W. Barrett, S. Nasr, J. Shen, Y. Hu, R. E. Hayes, R. W. J. Scott and N. Semagina, Catal. Sci. Technol., 2020, 10, 4229–4236 RSC.
- Y. Yang, L. Zhang, H. Guo, Z. Ding, W. Wang, J. Li, L. Zhou, X. Tu, Y. Qiu, G. Chen and Y. Sun, ACS Appl. Mater. Interfaces, 2022, 14(27), 30704–30713 CrossRef CAS PubMed.
- M. Tang, S. Li, S. Chen, Y. Ou, M. Hiroaki, W. Yuan, B. Zhu, H. Yang, Y. Gao, Z. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 22339–22344 CrossRef CAS PubMed.
- J. Song, S. Wang, Y. Xu, Q. Liu and Y. Zhao, Appl. Catal., A, 2021, 621, 118211 CrossRef CAS.
- M. Wu, W. Li, X. Zhang, F. Xue, T. Yang and L. Yuan, ChemCatChem, 2021, 13, 3490–3500 CrossRef CAS.
- D. Borodin, I. Rahinov, P. R. Shirhatti, M. Huang, A. Kandratsenka, D. J. Auerbach, T. Zhong, H. Guo, D. Schwarzer, T. N. Kitsopoulos and A. M. Wodtke, Science, 2020, 369, 1461–1465 CrossRef CAS PubMed.
- C. Xie, D. Yan, W. Chen, Y. Zou, R. Chen, S. Zang, Y. Wang, X. Yao and S. Wang, Mater. Today, 2019, 31, 47–68 CrossRef CAS.
- B. Jackson, J. Chem. Phys., 2020, 153, 034704 CrossRef CAS PubMed.
- Z.-F. Huang, J. Song, S. Dou, X. Li, J. Wang and X. Wang, Matter, 2019, 1, 1494–1518 CrossRef.
- X. Zhou, Y. Zhang, H. Guo and B. Jiang, Phys. Chem. Chem. Phys., 2021, 23, 4376–4385 RSC.
- G. R. Berdiyorov, E. Elbashier, G. Carchini, I. A. Hussein and A. Sakhaee-Pour, J. Mater. Res. Technol., 2021, 14, 3051–3058 CrossRef CAS.
- S. Wang, J. Liu, Y. Zhang, P. Chu, H. Liu, M. Wang and E. Duan, J. Rare Earths, 2021, 39, 51–57 CrossRef CAS.
- Y. Lou, J. Ma, W. Hu, Q. Dai, L. Wang, W. Zhan, Y. Guo, X.-M. Cao, Y. Guo, P. Hu and G. Lu, ACS Catal., 2016, 6, 8127–8139 CrossRef CAS.
- L. Bao, L. Chang, L. Yao, W. Meng, Q. Yu, X. Zhang, X. Liu, X. Wang, W. Chen and X. Li, New J. Chem., 2021, 45, 3546–3551 RSC.
- G. Liu, W. Huang, Y. Li, K. Ding, W. Chen, Y. Zhang and W. Lin, Appl. Surf. Sci., 2021, 555, 149728 CrossRef CAS.
- Z. Cheng, L. Qin, M. Guo, J. A. Fan, D. Xu and L. S. Fan, Phys. Chem. Chem. Phys., 2016, 18, 16423–16435 RSC.
- G. Li, K. He, F. Zhang, G. Jiang, Z. Zhao, Z. Zhang, J. Cheng and Z. Hao, Appl. Catal., B, 2022, 309, 121231 CrossRef CAS.
- C. Feng, Q. Gao, G. Xiong, Y. Chen, Y. Pan, Z. Fei, Y. Li, Y. Lu, C. Liu and Y. Liu, Appl. Catal., B, 2022, 304, 121005 CrossRef CAS.
- S. Wu, H. Liu, Z. Huang, H. Xu and W. Shen, Appl. Catal., B, 2022, 312, 121387 CrossRef CAS.
- J. Yang, S. Hu, Y. Fang, S. Hoang, L. Li, W. Yang, Z. Liang, J. Wu, J. Hu, W. Xiao, C. Pan, Z. Luo, J. Ding, L. Zhang and Y. Guo, ACS Catal., 2019, 9, 9751–9763 CrossRef CAS.
- M. D. Farahani, M. Wolf, T. P. O. Mkhwanazi, M. Claeys and H. B. Friedrich, J. Catal., 2020, 390, 184–195 CrossRef CAS.
- D. Li, K. Li, R. Xu, H. Wang, D. Tian, Y. Wei, X. Zhu, C. Zeng and L. Zeng, Catal. Today, 2018, 318, 73–85 CrossRef CAS.
- J. Zeng, H. Xie, Z. Liu, X. Liu, G. Zhou and Y. Jiang, Catal. Sci. Technol., 2021, 11, 6708–6723 RSC.
- Y. Wu, J. Chen, W. Hu, K. Zhao, P. Qu, P. Shen, M. Zhao, L. Zhong and Y. Chen, J. Catal., 2019, 377, 565–576 CrossRef CAS.
- N. D. Wasalathanthri, A. S. Poyraz, S. Biswas, Y. Meng, C.-H. Kuo, D. A. Kriz and S. L. Suib, J. Phys. Chem. C, 2015, 119, 1473–1482 CrossRef CAS.
- W. Zhu, J. Jin, X. Chen, C. Li, T. Wang, C. W. Tsang and C. Liang, Environ. Sci. Pollut. Res., 2018, 25, 5643–5654 CrossRef CAS PubMed.
- G. Li, N. Li, Y. Sun, Y. Qu, Z. Jiang, Z. Zhao, Z. Zhang, J. Cheng and Z. Hao, Appl. Catal., B, 2021, 282, 119512 CrossRef CAS.
- W. Zhu, X.-B. Wang, C. Li, X. Chen, W.-Y. Li, Z. Liu and C. Liang, J. Catal., 2022, 413, 150–162 CrossRef CAS.
- M. Jeong, N. Nunotani, N. Moriyama and N. Imanaka, Catal. Sci. Technol., 2017, 7, 1986–1990 RSC.
- D. Kim, M. Chung, S. Kim, K. Yun, W. Cha, R. Harder and H. Kim, Nano Lett., 2019, 19, 5044–5052 CrossRef CAS PubMed.
- Z. Ren, V. Botu, S. Wang, Y. Meng, W. Song, Y. Guo, R. Ramprasad, S. L. Suib and P. X. Gao, Angew. Chem., Int. Ed., 2014, 53, 7223–7227 CrossRef CAS PubMed.
- T. Wang, J. Wang, Y. Sun, Y. Duan, S. Sun, X. Hu, S. Xi, Y. Du, C. Wang and Z. J. Xu, Appl. Catal., B, 2019, 256, 117844 CrossRef CAS.
- Z. Pu, Y. Liu, H. Zhou, W. Huang, Y. Zheng and X. Li, Appl. Surf. Sci., 2017, 422, 85–93 CrossRef CAS.
- N. van Vegten, T. Baidya, F. Krumeich, W. Kleist and A. Baiker, Appl. Catal., B, 2010, 97, 398–406 CrossRef CAS.
- Q. Yu, C. Liu, X. Li, C. Wang, X. Wang, H. Cao, M. Zhao, G. Wu, W. Su, T. Ma, J. Zhang, H. Bao, J. Wang, B. Ding, M. He, Y. Yamauchi and X. S. Zhao, Appl. Catal., B, 2020, 269, 118757 CrossRef CAS.
- S. Peng, X. Yang, J. Strong, B. Sarkar, Q. Jiang, F. Peng, D. Liu and H. Wang, J. Hazard. Mater., 2020, 396, 122750 CrossRef CAS PubMed.
- L. Chen, W. Ren, C. Xu, Q. Chen, Y. Chen, W. Wang, W. Xu and Z. Hou, J. Alloys Compd., 2022, 894, 162038 CrossRef CAS.
- J. Du, H. Li, C. Wang, A. Zhang, Y. Zhao and Y. Luo, Catal. Commun., 2020, 141, 106012 CrossRef CAS.
- Y. Zhang, Z. Qin, G. Wang, H. Zhu, M. Dong, S. Li, Z. Wu, Z. Li, Z. Wu, J. Zhang, T. Hu, W. Fan and J. Wang, Appl. Catal., B, 2013, 129, 172–181 CrossRef CAS.
- X. Chen, Y. Zheng, F. Huang, Y. Xiao, G. Cai, Y. Zhang, Y. Zheng and L. Jiang, ACS Catal., 2018, 8, 11016–11028 CrossRef CAS.
- Z. Wang, J. Lin, H. Xu, Y. Zheng, Y. Xiao and Y. Zheng, ACS Appl. Nano Mater., 2021, 4, 11920–11930 CrossRef CAS.
- J. Li, X. Liang, S. Xu and J. Hao, Appl. Catal., B, 2009, 90, 307–312 CrossRef CAS.
- C. Shao, W. Li, Q. Lin, Q. Huang and D. Pi, Energy Technol., 2017, 5, 604–610 CrossRef CAS.
- J. Sun, H. Xue, Y. Zhang, X. L. Zhang, N. Guo, T. Song, H. Dong, Y. Kong, J. Zhang and Q. Wang, Nano Lett., 2022, 22, 3503–3511 CrossRef CAS PubMed.
- Y. Zheng, C. Wang, J. Li, F. Zhong, Y. Xiao and L. Jiang, ACS Appl. Nano Mater., 2020, 3, 9470–9479 CrossRef CAS.
- J. Chen, W. Shi, X. Zhang, H. Arandiyan, D. Li and J. Li, Environ. Sci. Technol., 2011, 45, 8491–8497 CrossRef CAS PubMed.
- V. Bashan and Y. Ust, Int. J. Energy Res., 2019, 43, 7755–7789 CAS.
- X. Tan, N. Han, H. Chen, L. Su, C. Zhang and Y. Li, Ceram. Int., 2021, 47, 8762–8768 CrossRef CAS.
- Y. Q. Su, J. X. Liu, I. A. W. Filot, L. Zhang and E. J. M. Hensen, ACS Catal., 2018, 8, 6552–6559 CrossRef CAS PubMed.
- K. Li, K. Liu, D. Xu, H. Ni, F. Shen, T. Chen, B. Guan, R. Zhan, Z. Huang and H. Lin, Chem. Eng. J., 2019, 369, 660–671 CrossRef CAS.
- X. Zhao, Y. Wang, Z. Zheng, Y. Zhang, K. Li, T. Chen, D. Guo, H. Cao, R. Zhan and H. Lin, Appl. Surf. Sci., 2021, 566, 150536 CrossRef CAS.
- X. Yuan, L. Meng, C. Zheng and H. Zhao, ACS Appl. Mater. Interfaces, 2021, 13, 52571–52587 CrossRef CAS PubMed.
- L. M. Toscani, P. A. Curyk, M. G. Zimicz, E. B. Halac, M. E. Saleta, D. G. Lamas and S. A. Larrondo, Appl. Catal., A, 2019, 587, 117235 CrossRef CAS.
- P. Mao, G. Lan, C. Liu, Z. Wang, Y. Liu, H. Sun and W. Huang, Sustainable Mater. Technol., 2021, 30, e00354 CrossRef CAS.
- M. Sadakiyo, Nanoscale, 2022, 14, 3398–3406 RSC.
- L. Li, S. Xue, M. Wei, H. Yao, Y. Dai and Z. Fei, Appl. Surf. Sci., 2022, 599, 153909 CrossRef CAS.
- Y. Li, W. Han, R. Wang, L.-T. Weng, A. Serrano-Lotina, M. A. Bañares, Q. Wang and K. L. Yeung, Appl. Catal., B, 2020, 275, 119121 CrossRef CAS.
- J. Hao, Z. Jiang, H. A. Khan, O. El Tall and A. Farooq, Fuel, 2022, 318, 123638 CrossRef CAS.
- M. Wu, M. Miao, W. Li, X. Zhang, L. Zhang, T. Zhen, Y. Fu, J. Jin and L. Yuan, Fuel, 2023, 331, 125575 CrossRef CAS.
- X. Fan, L. Li, X. Yang, Z. Guo, F. Jing and W. Chu, Catal. Today, 2020, 347, 23–30 CrossRef CAS.
- H. M. S. Al-Aani, E. Iro, P. Chirra, I. Fechete, M. Badea, C. Negrilă, I. Popescu, M. Olea and I.-C. Marcu, Appl. Catal., A, 2019, 586, 117215 CrossRef CAS.
- M. Răciulete, G. Layrac, F. Papa, C. Negrilă, D. Tichit and I.-C. Marcu, Catal. Today, 2018, 306, 276–286 CrossRef.
- S. Zhao and J. Li, ChemCatChem, 2015, 7, 1966–1974 CrossRef CAS.
- M. O'Connell, A. K. Norman, C. F. Hüttermann and M. A. Morris, Catal. Today, 1999, 47, 123–132 CrossRef.
- J. Jia, R. Ran, X. Wu, W. Chen, Z. Si and D. Weng, Mol. Catal., 2019, 467, 120–127 CrossRef CAS.
- Q. Meng, W. Wang, X. Weng, Y. Liu, H. Wang and Z. Wu, J. Phys. Chem. C, 2016, 120, 3259–3266 CrossRef CAS.
- Y. Xiao, W. Zhu, G. Cai, M. Chen, Y. Zheng, F. Zhong and L. Jiang, Phys. Chem. Chem. Phys., 2017, 19, 30418–30428 RSC.
- V. Sepelak, A. Duvel, M. Wilkening, K. D. Becker and P. Heitjans, Chem. Soc. Rev., 2013, 42, 7507–7520 RSC.
- M. Danielis, S. Colussi, C. de Leitenburg, L. Soler, J. Llorca and A. Trovarelli, Angew. Chem., Int. Ed., 2018, 57, 10212–10216 CrossRef CAS PubMed.
- H. Chen, S. Zhou, Z. Han, Y. Jiang, H. Yu, X. Zhou, R. Jiang, X. Liu and X. Li, J. Mater. Chem. A, 2016, 4, 17207–17214 RSC.
- M. Danielis, S. Colussi, C. de Leitenburg, L. Soler, J. Llorca and A. Trovarelli, Catal. Sci. Technol., 2019, 9, 4232–4238 RSC.
- P. Balaz, M. Achimovicova, M. Balaz, P. Billik, Z. Cherkezova-Zheleva, J. M. Criado, F. Delogu, E. Dutkova, E. Gaffet, F. J. Gotor, R. Kumar, I. Mitov, T. Rojac, M. Senna, A. Streletskii and K. Wieczorek-Ciurowa, Chem. Soc. Rev., 2013, 42, 7571–7637 RSC.
- J. Bao, H. Chen, S. Yang and P. Zhang, Chin. J. Catal., 2020, 41, 1846–1854 CrossRef CAS.
- M. Danielis, L. E. Betancourt, I. Orozco, N. J. Divins, J. Llorca, J. A. Rodríguez, S. D. Senanayake, S. Colussi and A. Trovarelli, Appl. Catal., B, 2021, 282, 119567 CrossRef CAS.
- A. Mussio, M. Danielis, N. J. Divins, J. Llorca, S. Colussi and A. Trovarelli, ACS Appl. Mater. Interfaces, 2021, 13, 31614–31623 CrossRef CAS PubMed.
- L. Yang, C. Fan, L. Luo, Y. Chen, Z. Wu, Z. Qin, M. Dong, W. Fan and J. Wang, Catalysts, 2021, 11, 725 CrossRef CAS.
- Y. Shu, H. Chen, N. Chen, X. Duan, P. Zhang, S. Yang, Z. Bao, Z. Wu and S. Dai, Chem, 2020, 6, 1723–1741 CAS.
- Y. Shu, M. Wang, X. Duan, D. Liu, S. Yang and P. Zhang, AIChE J., 2022, 68, e17664 CrossRef CAS.
- P. Xie, Y. Yao, Z. Huang, Z. Liu, J. Zhang, T. Li, G. Wang, R. Shahbazian-Yassar, L. Hu and C. Wang, Nat. Commun., 2019, 10, 4011 CrossRef PubMed.
- Y. Yao, Z. Huang, P. Xie, D. L. Steven, J. J. Rohit, H. Xie, F. Chen, A. Nie, T. Pu, M. Rehwoldt, D. Yu, R. Z. Michael, C. Wang, R. Shahbazian-Yassar, J. Li and L. Hu, Science, 2018, 359, 1489–1494 CrossRef CAS PubMed.
- S. Nie, L. Wu, L. Zhao, X. Zheng, S. Yang and P. Zhang, Chem Catal., 2021, 1, 648–662 CrossRef CAS.
- K. Chen, W. Li, G. Guo, C. Zhu, W. Wu and L. Yuan, ACS Omega, 2022, 7, 8536–8546 CrossRef CAS PubMed.
- X. Zhang, X. Jin, L. Bao, M. Zhang, R. Song, W. Yu, H. Zhang, W. Huang, W. Su and X. Li, New J. Chem., 2021, 45, 12655–12660 RSC.
- H. Chen, W. Cui, D. Li, Q. Tian, J. He, Q. Liu, X. Chen, M. Cui, X. Qiao, Z. Zhang, J. Tang and Z. Fei, Ind. Eng. Chem. Res., 2020, 59, 10804–10812 CrossRef CAS.
- Y. Liu, P. Zhao, L. Sun, N. Feng, L. Wang, H. Wan and G. Guan, ChemistrySelect, 2021, 6, 522–531 CrossRef CAS.
- J. Yang, S. Hu, L. Shi, S. Hoang, W. Yang, Y. Fang, Z. Liang, C. Pan, Y. Zhu, L. Li, J. Wu, J. Hu and Y. Guo, Environ. Sci. Technol., 2021, 55, 9243–9254 CrossRef CAS PubMed.
- X. Yang, Q. Gao, Z. Zhao, Y. Guo, Y. Guo, L. Wang, Y. Wang and W. Zhan, Appl. Catal., B, 2018, 239, 373–382 CrossRef CAS.
- S. Yoo, E. W. Lee and D. H. Kim, Mol. Catal., 2021, 511, 111728 CrossRef CAS.
- Y. Luo, K. Wang, J. Zuo, Q. Qian, Y. Xu, X. Liu, H. Xue and Q. Chen, Catal. Sci. Technol., 2017, 7, 496–501 RSC.
- Y. Li, T. Chen, S. Zhao, P. Wu, Y. Chong, A. Li, Y. Zhao, G. Chen, X. Jin, Y. Qiu and D. Ye, ACS Catal., 2022, 4906–4917, DOI:10.1021/acscatal.2c00296.
- W. Si, Y. Wang, S. Zhao, F. Hu and J. Li, Environ. Sci. Technol., 2016, 50, 4572–4578 CrossRef CAS PubMed.
- W. Si, Y. Wang, Y. Peng and J. Li, Angew. Chem., Int. Ed., 2015, 54, 7954–7957 CrossRef CAS PubMed.
- S. Wang, J. Zhu, S. A. C. Carabineiro, P. Xiao and Y. Zhu, Appl. Catal., A, 2022, 635, 118554 CrossRef CAS.
- Q. Tian, Z. Wang, M. Yuan, S. Zhao, H. Chen, L. Li, M. Cui, X. Qiao and Z. Fei, Environ. Sci.: Nano, 2021, 8, 2386–2395 RSC.
- X. Fan, L. Li, F. Jing, J. Li and W. Chu, Fuel, 2018, 225, 588–595 CrossRef CAS.
- Z. Li, J.-Y. Fu, Y. Feng, C.-K. Dong, H. Liu and X.-W. Du, Nat. Catal., 2019, 2, 1107–1114 CrossRef CAS.
- B. Liu, L. Yan and J. Wang, Appl. Surf. Sci., 2019, 494, 266–274 CrossRef CAS.
- C. Qiu, J. Shen, J. Lin, D. Liu, D. Li, J. Zhang, Z. Zhang, H. Lin, X. Wang and X. Fu, ACS Appl. Energy Mater., 2021, 4, 10172–10186 CrossRef CAS.
- X. Zhu, J. Yang, X. She, Y. Song, J. Qian, Y. Wang, H. Xu, H. Li and Q. Yan, J. Mater. Chem. A, 2019, 7, 5209–5213 RSC.
- J. Zhang, D. Wang, L. Lai, X. Fang, J. Xu, X. Xu, X. Zhang, J. Liu, H. Peng and X. Wang, Catal. Today, 2019, 327, 168–176 CrossRef CAS.
- X. Feng, J. Xu, X. Xu, S. Zhang, J. Ma, X. Fang and X. Wang, ACS Catal., 2021, 11, 12112–12122 CrossRef CAS.
- S. N. Vereshchagin, L. A. Solovyov, E. V. Rabchevskii, V. A. Dudnikov, S. G. Ovchinnikov and A. G. Anshits, Chem. Commun., 2014, 50, 6112–6115 RSC.
- C. Ye, J. Liu, Q. Zhang, X. Jin, Y. Zhao, Z. Pan, G. Chen, Y. Qiu, D. Ye, L. Gu, G. I. N. Waterhouse, L. Guo and S. Yang, J. Am. Chem. Soc., 2021, 143, 14169–14177 CrossRef CAS PubMed.
- Q. Yu, C. Wang, X. Li, Z. Li, L. Wang, Q. Zhang, G. Wu and Z. Li, Fuel, 2019, 239, 1240–1245 CrossRef.
- Y. Zheng, Q. Liu, C. Shan, Y. Su, K. Fu, S. Lu, R. Han, C. Song, N. Ji and D. Ma, Environ. Sci. Technol., 2021, 55, 5403–5411 CrossRef CAS PubMed.
- T. Toyao, Z. Maeno, S. Takakusagi, T. Kamachi, I. Takigawa and K.-I. Shimizu, ACS Catal., 2019, 10, 2260–2297 CrossRef.
- J. A. Esterhuizen, B. R. Goldsmith and S. Linic, Nat. Catal., 2022, 5, 175–184 CrossRef.
- Z. Yang and W. Gao, Adv. Sci., 2022, 9, e2106043 CrossRef PubMed.
- H. Chun, E. Lee, K. Nam, J.-H. Jang, W. Kyoung, S. H. Noh and B. Han, Chem Catal., 2021, 1, 855–869 CrossRef CAS.
- S. Ma and Z.-P. Liu, ACS Catal., 2020, 10, 13213–13226 CrossRef CAS.
- J. R. Kitchin, Nat. Catal., 2018, 1, 230–232 CrossRef.
- M. E. Günay and R. Yıldırım, Catal. Rev.: Sci. Eng., 2020, 63, 120–164 CrossRef.
- X. Liu, C. Cai, W. Zhao, H.-J. Peng and T. Wang, ACS Catal., 2022, 12, 4252–4260 CrossRef CAS.
- J. Yang, Z. Ye, G. Wang, L. Zhao, X. Zhang, J. Wang and Y. Jin, Appl. Catal., A, 2021, 622, 118221 CrossRef CAS.
- V. Sharma, P. Kumar, P. Dev and G. Pilania, J. Appl. Phys., 2020, 128, 034902 CrossRef CAS.
- B. R. Goldsmith, J. Esterhuizen, J. X. Liu, C. J. Bartel and C. Sutton, AIChE J., 2018, 64, 2311–2323 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |