
Evaluation of CH4 oxidation activity of high-valent iron-oxo species of a μ-nitrido-bridged heterodimer of iron porphycene and iron phthalocyanine†
Yasuyuki
Yamada
 *ab,
Yusuke
Miwa
a,
Yuka
Toyoda
b,
Quan Manh
Phung
ac,
Kin-ichi
Oyama
b and
Kentaro
Tanaka
*a
*ab,
Yusuke
Miwa
a,
Yuka
Toyoda
b,
Quan Manh
Phung
ac,
Kin-ichi
Oyama
b and
Kentaro
Tanaka
*a
aDepartment of Chemistry, Graduate School of Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan. E-mail: kentaro@chem.nagoya-u.ac.jp; yy@chem.nagoya-u.ac.jp
bResearch Center for Materials Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan
cInstitute of Transformative Bio-Molecules (ITbM), Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8601, Japan
First published on 31st January 2023
Abstract
A μ-nitrido-bridged dimer of iron phthalocyanine is one of the most potent molecule-based CH4 oxidation catalysts reported to date. The reactive intermediate is a high-valent iron-oxo species generated through reaction with H2O2 in an acidic aqueous solution. However, there are few reports on the synthesis and catalytic CH4 oxidation activity of a μ-nitrido-bridged heterodimer of two different iron porphyrinoids, despite that there are a variety of iron porphyrinoids with coordination and electronic structures different from those of iron phthalocyanines or iron porphyrins. Herein, we report the synthesis of a novel μ-nitrido-bridged heterodimer of an iron phthalocyanine and iron porphycene and examine its CH4 oxidation activity. Porphycenes are an important class of porphyrinoids with a smaller coordination sphere than phthalocyanines or porphyrins. Single crystal structural analyses revealed that the heterodimer possessed a Fe–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Fe core structure similar to that of the phthalocyanine homodimer. The heterodimer showed catalytic CH4 oxidation activity in an acidic aqueous solution in the presence of H2O2 at 60 °C through the high-valent iron-oxo species as in the case with the phthalocyanine homodimer. This was in clear contrast to the result that the high-valent iron-oxo species of a μ-nitrido-bridged iron porphycene dimer was so unstable that it decomposed quickly in the same reaction conditions.
Fe core structure similar to that of the phthalocyanine homodimer. The heterodimer showed catalytic CH4 oxidation activity in an acidic aqueous solution in the presence of H2O2 at 60 °C through the high-valent iron-oxo species as in the case with the phthalocyanine homodimer. This was in clear contrast to the result that the high-valent iron-oxo species of a μ-nitrido-bridged iron porphycene dimer was so unstable that it decomposed quickly in the same reaction conditions.
Introduction
Several decades have passed since some methane monooxygenases (MMOs) were found to utilize high-valent iron-oxo species as reaction intermediates for the catalytic conversion of CH4 into CH3OH under ambient reaction conditions.1–4 CH4 has long been expected to be the next-generation carbon resource because it is abundant in nature as natural gas or methane hydrate.5,6 However, CH4 is a stable organic compound with a particularly high C–H bond dissociation energy of ca. 105 kcal mol−1.7 Therefore, the development of an efficient catalyst is indispensable for low-cost and efficient conversion of CH4 into industrially useful chemical raw materials such as CH3OH.6,8 Therefore, it is quite understandable that the fact that some MMOs achieve efficient catalytic conversion of CH4 into CH3OH at ambient temperature by utilizing high-valent iron-oxo species has stimulated the interest of chemists to fabricate a large variety of biomimetic iron-oxo-based molecular catalysts.9–17 The detailed reaction mechanisms of natural MMOs have been clarified as a result of utilizing these biomimetic catalysts. The CH4 oxidation reaction by high-valent iron-oxo species is facilitated by the proton-coupled electron transfer (PCET) pathway, enabling low-temperature and efficient CH4 oxidation.18,19However, from the viewpoint of catalytic activity, a very limited number of artificial molecular iron-oxo-based biomimetic catalysts exist that activate the C–H bonding of CH4 at temperatures lower than 100 °C because of the high stability of CH4.3,18,20 A. B. Sorokin et al. found that a μ-nitrido-bridged iron phthalocyanine (Pc) dimer (1 or 1+) reacts with H2O2 in an acidic aqueous solution to afford a high-valent iron-oxo species 1oxo, which shows a particularly high CH4 oxidation activity among various molecular CH4 oxidation catalysts at a lower temperature than 100 °C (Fig. 1a).20–22 Similarly, μ-nitrido-bridged iron porphyrin dimer 2 was reported to indicate CH4 oxidation activity comparable to that of μ-nitrido-bridged iron phthalocyanine dimer (Fig. 1b).23 According to some theoretical estimates, the core μ-nitrido-bridged dinuclear iron structure (Fe–NFe) is believed to be crucial in the reaction of these high CH4 oxidation activities.24–27
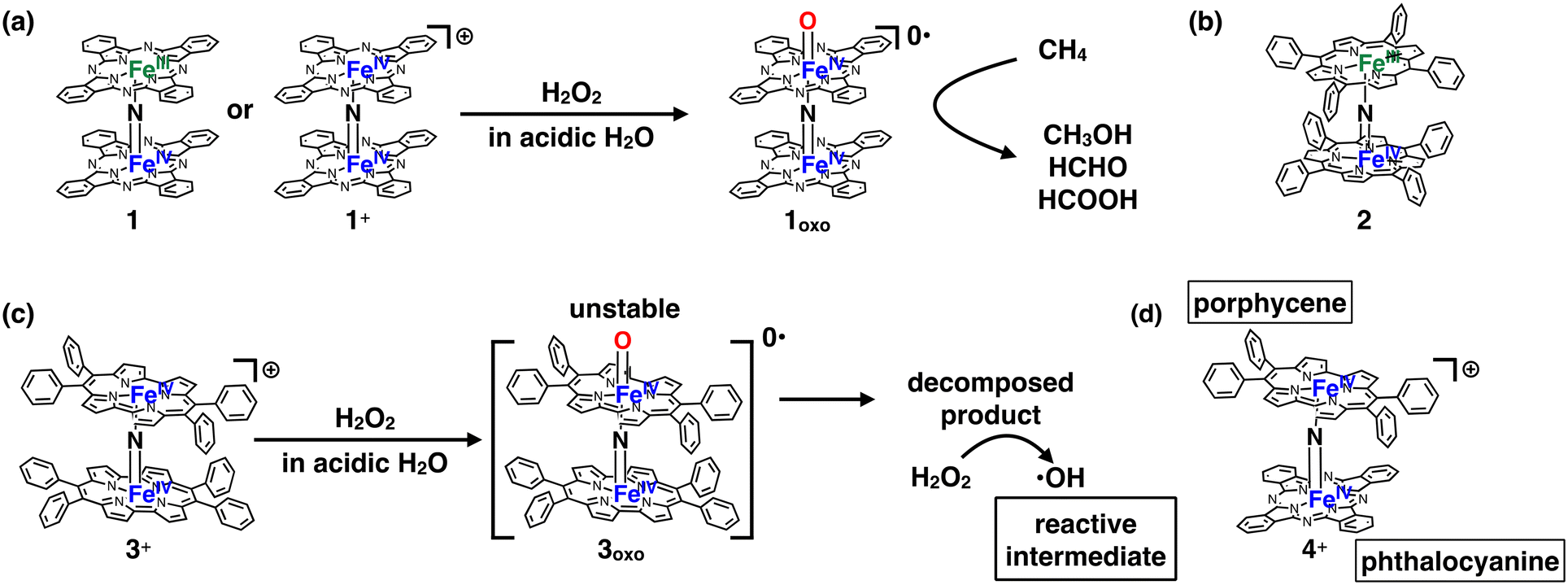 | ||
| Fig. 1 (a) Formation of high-valent iron oxo species 1oxo having potent CH4 oxidation ability, from 1 or 1+ through the reaction with H2O2. (b) A μ-nitrido-bridged iron porphyrin dimer (2) showing potent CH4 oxidation ability. (c) Structure of a monocationic μ-nitrido-bridged iron porphycene dimer (3+) and its proposed CH4 oxidation mechanism. (d) A monocationic μ-nitrido-bridged heterodimer of an iron porphycene and an iron phthalocyanine (4+) synthesized in this work. | ||
Inspired by these previous works, we recently synthesized μ-nitrido-bridged dinuclear iron porphycene (Ppc) dimer 3+, a structural analog of 1+, and investigated its CH4 oxidation activity (Fig. 1c).28 Porphycene is an important class of porphyrinoids and has a rectangular-shaped narrower cavity than phthalocyanine, which renders the coordination structures and reactivities of the metal complexes different from those of phthalocyanine.29,30
Although 3+ has a μ-nitrido-bridged dinuclear iron core (Fe–NFe) similar to that of 1+, it was found that 3+ showed less CH4 oxidation activity, and the reaction proceeded through a different mechanism (Fenton-type reaction; the reaction intermediate is ˙OH). This is presumably because the high-valent iron-oxo species 3oxo is less stable than 1oxo and decomposes during the CH4 oxidation reaction. The decomposed product could act as a catalyst for ˙OH production. These results indicate that the difference in the structure of the porphyrinoid moiety strongly affects the reactivity of the high-valent iron-oxo species.
In this study, we report the synthesis of a μ-nitrido-bridged heterodimer of an iron phthalocyanine and an iron porphycene (4+, Fig. 1d) and its CH4 oxidation activity. Thus far, there have been very few reports investigating the CH4 oxidation activity of μ-nitrido-bridged heterodimers of iron porphyrinoids. We previously confirmed that a supramolecular μ-nitrido-bridged heterodimer of an iron porphyrin and an iron phthalocyanine provided sufficiently stable high-valent iron-oxo species for catalytic CH4 or CH3CH3 oxidation reactions.31,32 It has also been reported that H2O2 preferentially reacts with the electron-rich iron phthalocyanine moiety of the μ-nitrido-bridged heterodimer of an electron-rich iron phthalocyanine and an electron-deficient iron phthalocyanine to afford a high-valent iron-oxo species, although the reactivity of the heterodimer has not been reported.33 Therefore, the fact that a μ-nitrido-bridged iron phthalocyanine dimer 1 (or 1+) indicated potent CH4 oxidation activity through the reaction of high-valent iron-oxo species, whereas a μ-nitrido-bridged dinuclear iron porphycene dimer 3+ showed a different CH4 oxidation mechanism, prompted us to clarify the relationship between the structure and reactivity of 4+.
Results and discussion
Synthesis and spectroscopic characterization of 4+
A mixture of iron porphycene 5 and iron phthalocyanine 6 was heated in 1-chloronaphthalene in the presence of excess NaN3 at 280 °C, followed by oxidation with iodine to yield a monocationic μ-nitrido-bridged heterodimer of an iron phthalocyanine and iron porphycene (4+·I3−, Fig. 2a). When an equimolar mixture of iron porphycene 5 and iron phthalocyanine 6 was heated, the isolated yield of the desired compound, 4+·I3−, was relatively low (ca. 10%). This is because a significant amount of the μ-nitrido-bridged iron phthalocyanine dimer was obtained (ca. 40%), suggesting that the formation of the μ-nitrido-bridged iron phthalocyanine dimer was faster than the formation of the desired heterodimer under these reaction conditions. Therefore, heating a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of iron porphycene 5 and iron phthalocyanine 6 with NaN3 improved the isolated yield of 4+·I3− to 71%. Elemental analysis suggested that the counter anion of the monocationic heterodimer was triiodide, which was further confirmed using single-crystal X-ray structural analysis.
:1 mixture of iron porphycene 5 and iron phthalocyanine 6 with NaN3 improved the isolated yield of 4+·I3− to 71%. Elemental analysis suggested that the counter anion of the monocationic heterodimer was triiodide, which was further confirmed using single-crystal X-ray structural analysis.
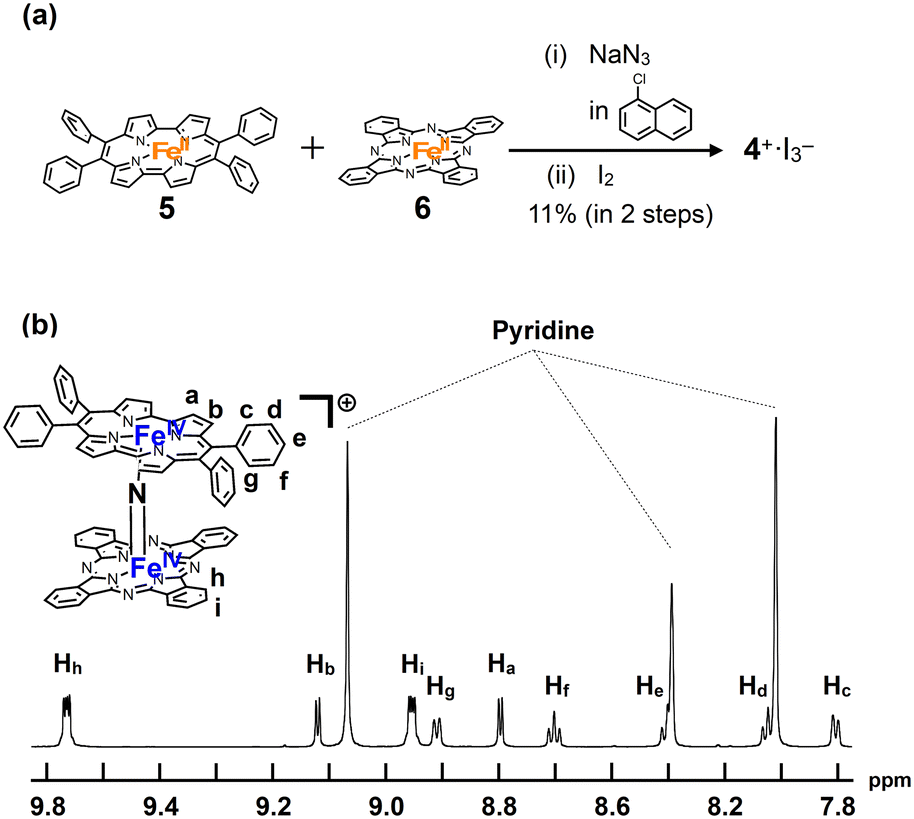 | ||
| Fig. 2 (a) Synthesis of a monocationic heterodimer 4+·I3−. (b) 1H-NMR spectrum of 4+·I3− in pyridine-d5. | ||
1H-NMR and MALDI-TOF MS analyses were performed to characterize 4+·I3−. In the MALDI-TOF MS spectrum shown in Fig. S1 in the ESI,† a signal corresponding to 4+ was observed at m/z = 1250.26, which corresponds to the theoretical isotope distribution pattern of 4+. As shown in Fig. 2b, all the 1H-NMR peaks of 4+·I3− were observed as sharp signals in the range of 6–10 ppm. A neutral μ-nitrido-bridged FeTPP dimer containing Fe(III) and Fe(IV) was reported to indicate a significant broadening of the 1H-NMR signals, whereas its monocationic species containing two Fe(IV) centers showed sharp 1H-NMR signals because the two Fe(IV) centers interacted with each other in an antiferromagnetic manner.34–36 Therefore, it is considered that 4+·I3− includes two Fe(IV) ions interacting with each other in an antiferromagnetic manner. Moreover, considering that two sets of signals corresponding to those of the o- and m-protons of the peripheral phenyl groups were observed, the rotation of the peripheral phenyl groups of the porphycene ring was restricted owing to steric repulsion between the phenyl groups and the porphycene ring, as in the case of the previously reported μ-nitrido-bridged dinuclear iron porphycene dimer 3+·PF6−. However, the rotation of the porphycene ring along the Fe–NFe axis was sufficiently fast compared to the NMR timescale because the porphycene ring was observed as a C2 symmetrical structure.
A comparison of the UV-vis spectrum of 4+·PF6− with those of μ-nitrido-bridged iron phthalocyanine dimer 1+·I− and μ-nitrido-bridged iron porphycene dimer 3+·PF6− is shown in Fig. 3. The bands at 357 and 399 nm were assignable to the absorptions by the phthalocyanine and porphycene units, respectively, both of which showed significant shifts compared to those of the homoleptic dimers 1+·I− and 3+·PF6−.28,37 The Q-band of the phthalocyanine unit of 4+·PF6− at 634 nm showed an apparent hypochromic shift compared to that of the phthalocyanine dimer 1+·I−, whereas the Q-band of the phthalocyanine unit of 4+·PF6− at 705 nm appeared at a higher wavelength than that of 1+·I−. These results implied an apparent electronic interaction between the two porphyrinoids in 4+·PF6−.
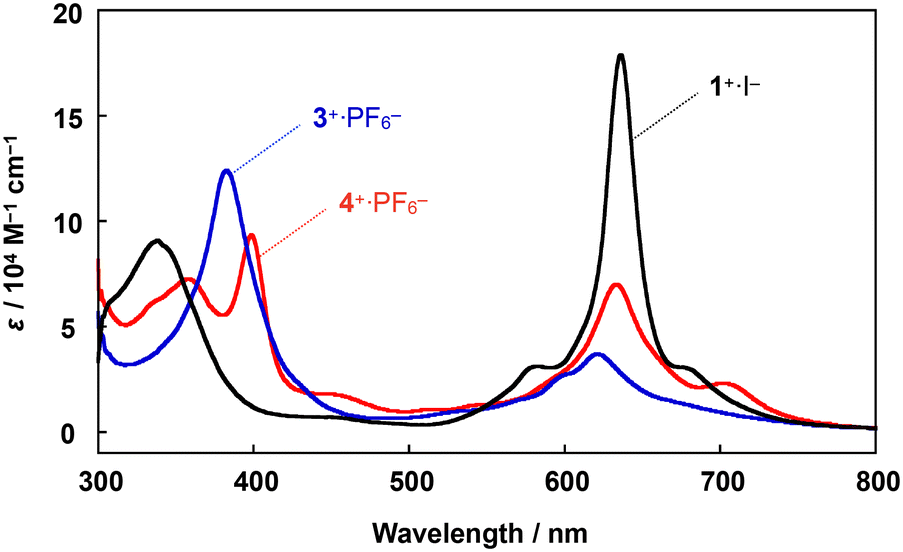 | ||
| Fig. 3 Comparison of UV-vis spectra of a heterodimer 4+·PF6− and two homodimers, 1+·I− and 3+·PF6−, in pyridine at 20 °C. | ||
Single crystal X-ray structural analysis
Crystal structural analysis of 4+·I3− demonstrated that 4+·I3− crystallized in the monoclinic space group P21/c from a solution of 1:1 (v/v) mixture of pyridine and CHCl3. The molecular structures of 4+·I3− are shown in Fig. 4a and b.
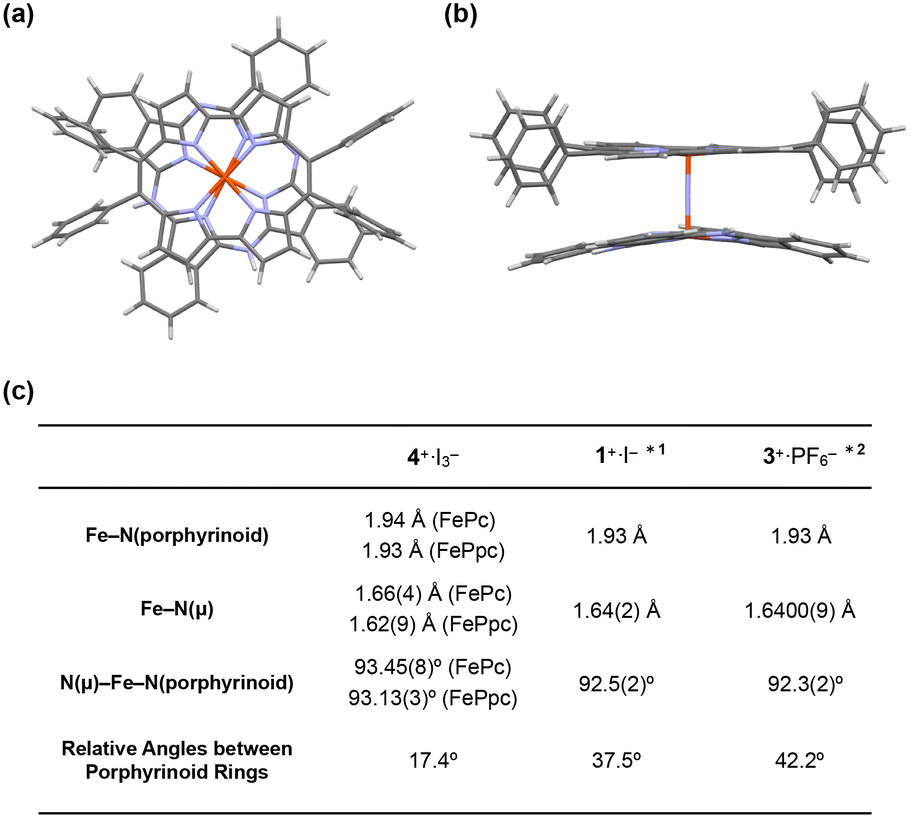 | ||
| Fig. 4 (a) Side and (b) top views of the single crystal X-ray structure of 4+·I3−. I3−, coordinating pyridines, and crystalline solvents were omitted for clarity. (c) Comparison of some mean bond lengths and angles of 4+·I3−, 1+·I−, and 3+·PF6−. *1: ref. 28, *2: ref. 38. | ||
The distance between the μ-nitrogen and Fe(IV) ion in porphycene (1.62(9) Å) is almost identical to that of the μ-nitrogen and Fe(IV) ion in phthalocyanine (1.66(4) Å), suggesting that the Fe–μ-N–Fe core has a π-conjugated structure. The mean bond distances between the four porphycene nitrogens and the coordinated Fe(IV) ion (1.93 Å) are almost identical to those between the four phthalocyanine nitrogens and the coordinated Fe(IV) ion (1.94 Å). The average bond angles of the μ-nitrogen–Fe–nitrogens(porphyrinoid) in 4+·I3− were 93.45(8)° for iron phthalocyanine and 93.13(3)° for iron porphycene, both of which were slightly larger than 90°. The core-coordinating N4 planes of the two porphyrinoid rings are located in an almost parallel configuration. The phthalocyanine ring was slightly distorted from the planar structure, presumably because of steric repulsion between the phthalocyanine ring and peripheral phenyl groups of the porphycene ring. As suggested by 1H NMR spectroscopy, the rotation of the peripheral phenyl groups of the porphycene ring appeared to be significantly suppressed by steric repulsion between the phenyl groups and porphycene rings.
A comparison of the single-crystal structure of 4+·I3− with those of 1+·I− and 3+·PF6− is summarized in a table shown in Fig. 4c.28,38 Although slight differences were observed in the distance between nitrogen and Fe(IV) and the average bond angles of the μ-nitrogen–Fe–nitrogen (porphyrinoid), these differences are presumably derived from the heteroleptic structure of 4+·I3−. Overall, it could be concluded that 4+·I3− has a similar Fe–N–Fe core structure to those of 1+·I− and 3+·PF6−. The relative angle between the phthalocyanine ring and the long axis of the porphycene ring was calculated to be 17.4°, which was much smaller than the relative angles between the two porphyrinoid rings in 1+·I− (37.5°) and 3+·PF6− (42.2°). We attributed this difference in the relative angle between the two porphyrinoids to the difference in the packing structure, because both rings in 4+·I3− are rotatable along the Fe–NFe axis.
Cyclic voltammetric analysis
Cyclic voltammograms of 4+·PF6− were recorded in a pyridine solution (200 μM) containing 100 mM nBu4N+·PF6− (Fig. 5a). A comparison of the voltammograms of the μ-nitrido-bridged iron phthalocyanine dimer 1+·I− and μ-nitrido-bridged iron porphycene dimer 3+·PF6− is shown in Fig. 5b and c. 4+·PF6− showed four quasi-reversible redox waves at −0.75, −1.37, −1.69 and −1.92 V vs. Fc/Fc+ and one irreversible oxidation wave at 0.50 V.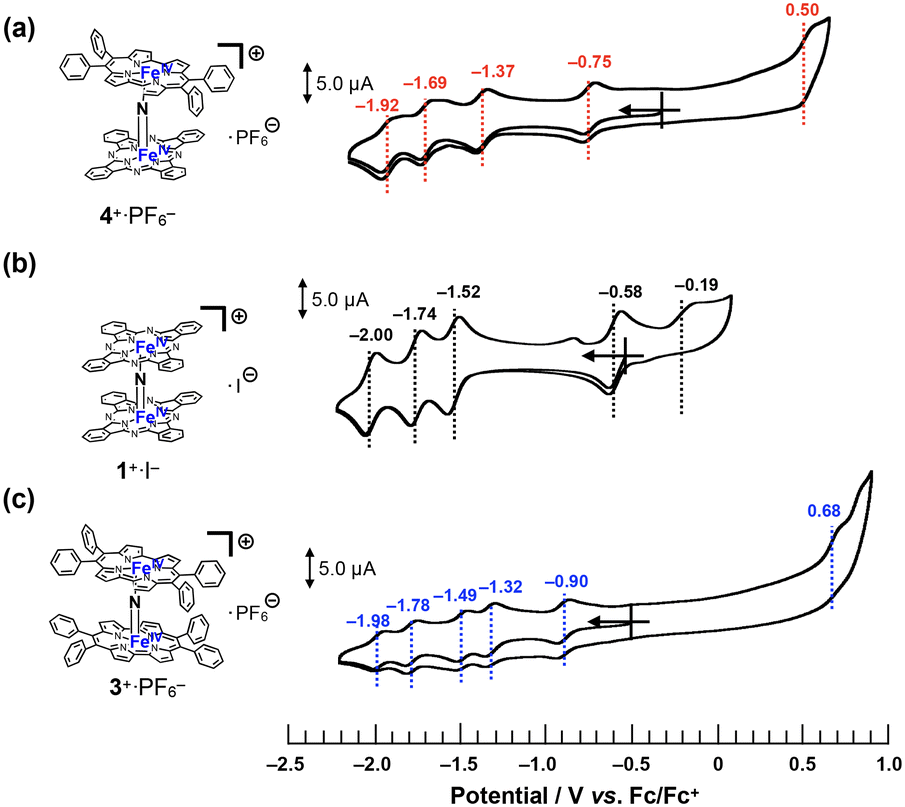 | ||
| Fig. 5 Comparison of cyclic voltammograms of (a) 4+·PF6−, (b) 1+·I−, and (c) 3+·PF6− in a pyridine solution containing 100 mM of nBu4N+·PF6− at room temperature. [Substrate] = 200 μM, [scan rate] = 100 mV s−1. | ||
In the case of a voltammogram of a μ-nitrido-bridged iron phthalocyanine dimer 1+·I− (shown in Fig. 5b), Ercolani et al. carefully investigated the redox processes to conclude that the four redox waves at −0.58, −1.52, −1.74 and −2.00 V should be assignable to the redox in the iron cores of 1+·I−.39 For the voltammogram of a μ-nitrido-bridged iron porphycene dimer 3+·PF6− shown in Fig. 5c, we assigned the 1st (−0.90 V vs. Fc+/Fc), 2nd (−1.32 V), 3rd (−1.49 V), and 4th (−1.78 V) reduction waves of the porphycene dimer to those of Fe(IV)Fe(IV)/Fe(III)Fe(IV), Fe(III)Fe(IV)/Fe(III)Fe(III), Fe(III)Fe(III)/Fe(II)Fe(III), and Fe(II)Fe(III)/Fe(II)Fe(II), respectively. This is based on the fact that Gross et al. investigated the detailed redox properties of a 2,7,12,17-tetra-n-propylporphycene iron(III) chloride (ClFeTprPpc), a monomeric porphycene, by electrochemical and spectroelectrochemical measurements to report that the reduction of the iron center (Fe(III)/Fe(II)) occurred at more positive potential than that of the porphycene center (TPrPpc/TPrPpc˙−).40 Similarly, it is assumed that at least the four reduction waves of 4+·PF6− at −0.75, −1.37, −1.69, and −1.92 V could be assignable to those of the iron centers of 4+·PF6−. Thus, it was demonstrated that 4+·PF6− has a completely different electronic structure from those of 1+·I− and 3+·PF6− owing to the different types of porphyrinoid rings.38
As for the oxidation of 4+·PF6−, theoretical calculations suggested that the phthalocyanine ring is more easily oxidized than the porphycene ring (vide infra, see “DFT calculations” section). In addition, the μ-nitrido-bridged iron phthalocyanine dimer 1+·I− showed a lower oxidation potential at −0.19 V than that of a μ-nitrido-bridged iron porphycene dimer 3+·PF6− at 0.68 V, both of which are assignable to the oxidations of the porphyrinoid cores. Therefore, the oxidation wave at 0.50 V is assignable to that of the phthalocyanine ring. Overall, 4+·PF6− showed a first reduction wave higher than that of 1+·I− and lower than that of 3+·PF6−, whereas the first oxidation wave of 4+·PF6− was observed at potentials higher than 1+·I− and lower than 3+·PF6−.
ESI-TOF MS analysis of high-valent iron-oxo species
To confirm the generation of the high-valent iron-oxo species, the reactive intermediate for the oxidation reaction, by treating 4+·I3− with H2O2 in an aqueous solution, ESI-TOF MS measurement of an acetonitrile solution of 4+·I3− in the presence of H2O2 was performed.As shown in Fig. 6, the signals assignable to those of the high-valent iron-oxo species ([4 + O]+) were observed at approximately m/z = 1266 after the addition of aqueous H2O2 at room temperature, with its isotopic distribution pattern corresponding to that of the calculated pattern. The signals observed at approximately 1284 correspond to those of the hydroperoxo species ([4 + HOOH]+). We also confirmed that treatment of 4+·I3− with H218O2 resulted in the generation of the peaks assignable to [4 + 18O] as shown in the ESI.† These results demonstrate that high-valent iron-oxo species were generated after the threat of 4+ with H2O2via the corresponding hydroperoxo species, as in the case with the other μ-nitrido-bridged iron phthalocyanine dimers. Sorokin et al. reported that treatment of heteroleptic μ-nitrido-bridged dimer of iron phthalocyanine and iron porphyrazine with m-chloroperbenzoic acid (mCPBA) resulted in the attachment of oxo moiety either on the iron phthalocyanine and iron porphyrazine. In this case, they concluded that the attachment of oxo moiety occurred with slight preference for the iron phthalocyanine unit based on the results of cryospray collision induced dissociation MS/MS technique.41 However, in the case ESI-TOF MS analysis of [4 + O]+, it was difficult to determine the accurate position of the FeO moiety (whether it is on the iron porphycene or the iron phthalocyanine in 4+) even after MS/MS analysis of the [4 + O]+ peak because the ionization efficiency of the Fe-phthalocyanine fragments was much smaller than that of the Fe-porphycene fragments.
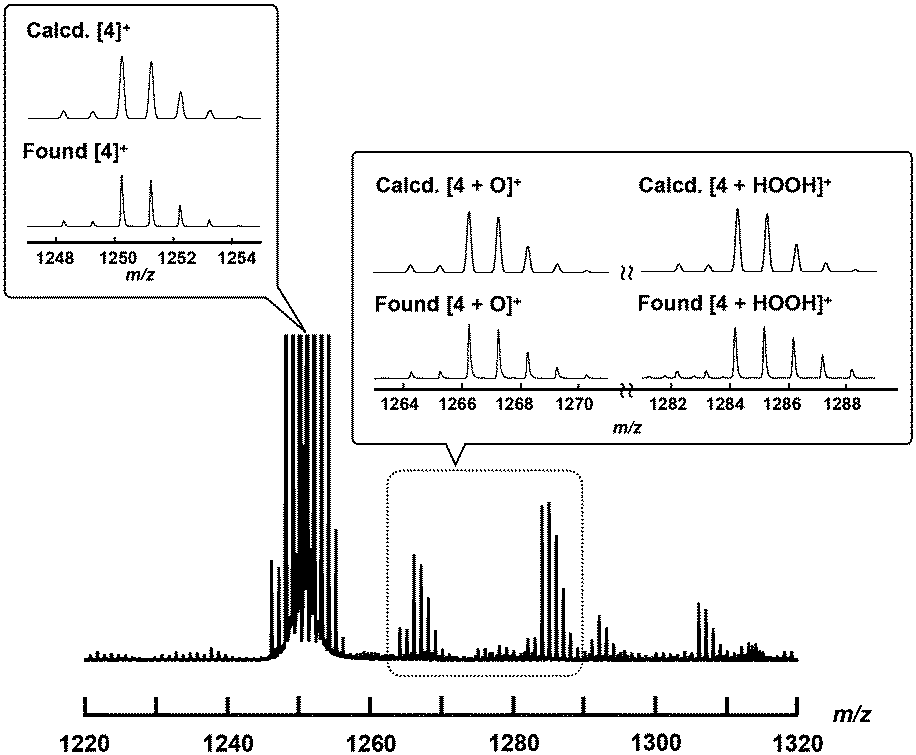 | ||
| Fig. 6 ESI-TOF MS spectrum of 4+·I3− in an aqueous CH3CN solution containing excess amount of H2O2 at room temperature (the detail of the experimental procedure is shown in the Experimental section). Insets: Comparison of calculated (top) and experimental (bottom) isotopic distribution patterns of molecular peaks assignable to [4]+, [4 + O]+, and [4 + HOOH]+. | ||
DFT calculations
Density functional theory (DFT) calculations were performed to gain further insight into experimental observations. The calculation details are presented in the Experimental section. We first focused on the electronic structure of 4+ as well as its oxidized and reduced species. The electronic structure of 4+ is very similar to that of [{(porphyrin)Fe}2N]+, as described by Ghosh et al.42 (Fig. 7). The predicted Fe–N(Pc) and Fe–N(Ppc) average bond distance is 1.948 and 1.939 Å, respectively, which reasonably agrees with the experimental values of 1.94 and 1.93 Å, respectively (see Tables S1 and S2†). The short Fe–N bond distances were consistent with the low-spin character of the Fe centers. The predicted Fe–N(μ) bond lengths are 1.582 and 1.607 Å, respectively, which are systematically smaller than the X-ray data by approximately 0.05 Å.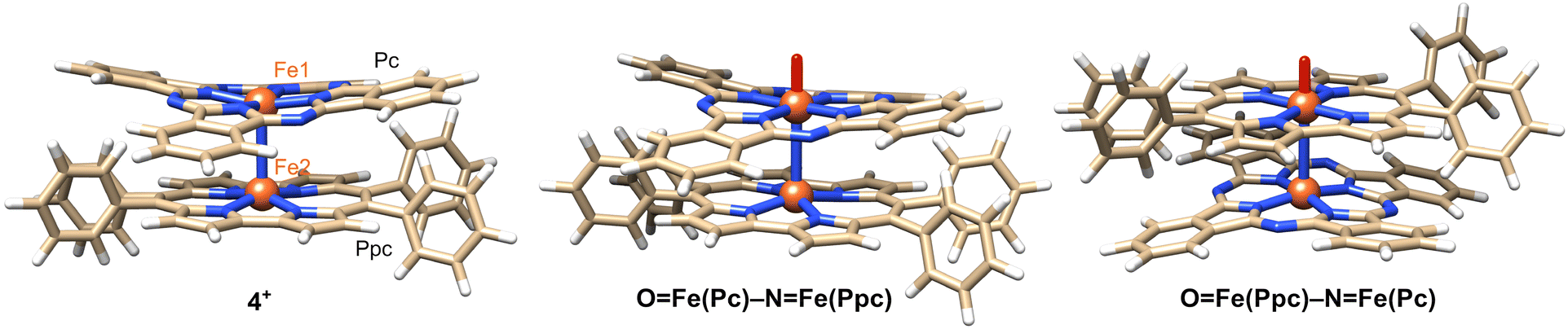 | ||
| Fig. 7 DFT structures of 4+ and its isomeric oxo complexes. | ||
Upon reduction, both Fe–N(μ) bonds were lengthened as an antibonding orbital comprising Fe(3dz2) and N(2s) orbitals. The difference between the two Fe–N(μ) bonds is only 0.03 Å, indicating that the two Fe centers are similar and the complex could be formally characterized as Fe3.5–N–Fe3.5.20 Spin populations also reveal that both Fe centers are involved in the reduction, with Fe(Pc) being slightly more reduced than Fe(Ppc). Upon oxidation, the Fe–N(μ) bond lengths change by less than 0.01 Å, indicating that the oxidation occurs mainly at the macrocycles. The spin population of Pc is 0.76, whereas Ppc has a spin population of 0.15. This difference in the spin density implies that oxidation primarily occurs at Pc. The higher tendency of phthalocyanine to be oxidized is related to its longer conjugation length compared to that of porphycene.
We now compare the two oxo isomers, OFe(Pc)–N–Fe(Ppc) and OFe(Ppc)–N–Fe(Pc) (DFT structures are shown in Fig. 7). Similar to a previous work,27 two doublet states were calculated: the ground state in which one of the macrocycles was oxidized FeIVFeIV(P˙+) and a low-lying excited state FeIVFeV (Fig. S4†). Selected bond lengths are listed in Table S3.† The Fe–N–Fe fragment is no longer symmetric. The general trend is that the formation of Fe-oxo significantly increases the length of the adjacent Fe–N(μ) bond above 1.7 Å. DFT predicts that OFe(Pc)–N–Fe(Ppc) and OFe(Ppc)–N–Fe(Pc) are close in energy. The former isomer (in pyridine at 298 K) was slightly more stable (2–4 kcal mol−1) than the latter (Table S5†).
Considering that the phthalocyanine ring is more easily oxidized than the porphycene ring, H2O2 might coordinate to Fe-porphycene more strongly than to Fe-phthalocyanine because the porphycene ring is more electron deficient than the phthalocyanine ring. This suggests that the oxo species of 4+ could be preferentially generated on the Fe-porphycene moiety. This prompted us to compare the stability of the hydroperoxo species on the Fe-porphycene ring (HOOFePpc–NFePc) with that of the Fe-phthalocyanine ring (HOOFePc–NFePpc). We found that the difference between these two species is quite small (ca. 3 kcal mol−1; HOOFePpc–NFePc is more stable than HOOFePc–NFePpc). Moreover, we compared the O–O bond dissociation free energy in HOOFePpc–NFePc and HOOFePc–NFePpc because dissociation of the O–O bond is indispensable for the generation of iron-oxo species from hydroperoxo species. However, it was demonstrated that both hydroperoxo species have similar O–O bond dissociation free energy (37.2 kcal mol−1 for HOOFePpc–NFePc and 36.7 kcal mol−1 for HOOFePc–NFePpc in pyridine).
Considering the uncertainty in DFT calculations, the results obtained from DFT calculation propose that both oxo isomers (OFe(Pc)–N–Fe(Ppc) and OFe(Ppc)–N–Fe(Pc)) could be produced. However, it is also important to consider that the formation of oxo isomers should be kinetically controlled. The complete reaction mechanisms for the production of high-valent iron-oxo species are outside the scope of this study.
CH4 oxidation activity
We adopted the method developed by Sorokin et al. to evaluate the catalytic CH4 oxidation activity of 4+·I3−.22 H2O was used as the solvent for CH4 oxidation because it is sufficiently stable against oxidation by the high-valent iron-oxo species. For this purpose, we first prepared a silica gel-supported catalyst because 4+·I3− is not soluble in H2O. The CH4 oxidation reaction was performed in an acidic aqueous solution containing 189 mM H2O2 and 51 mM trifluoroacetic acid (TFA) at 60 °C under a CH4 atmosphere of 1.0 MPa. After the oxidation reaction, MeOH, HCHO, and HCOOH were observed and quantified by GC-MS spectroscopy. The time dependence of each oxidized product is shown in Fig. 8a and Table S7.† The amount of oxidized products was significantly larger than that observed in the absence of CH4, suggesting that the observed MeOH, HCHO, and HCOOH were mostly derived from CH4. The small amount of oxidized products observed in the absence of CH4 (under N2 atmosphere) was mainly derived from the organic solvents adsorbed on the SiO2 surface of the catalysts.21,28,31,32,43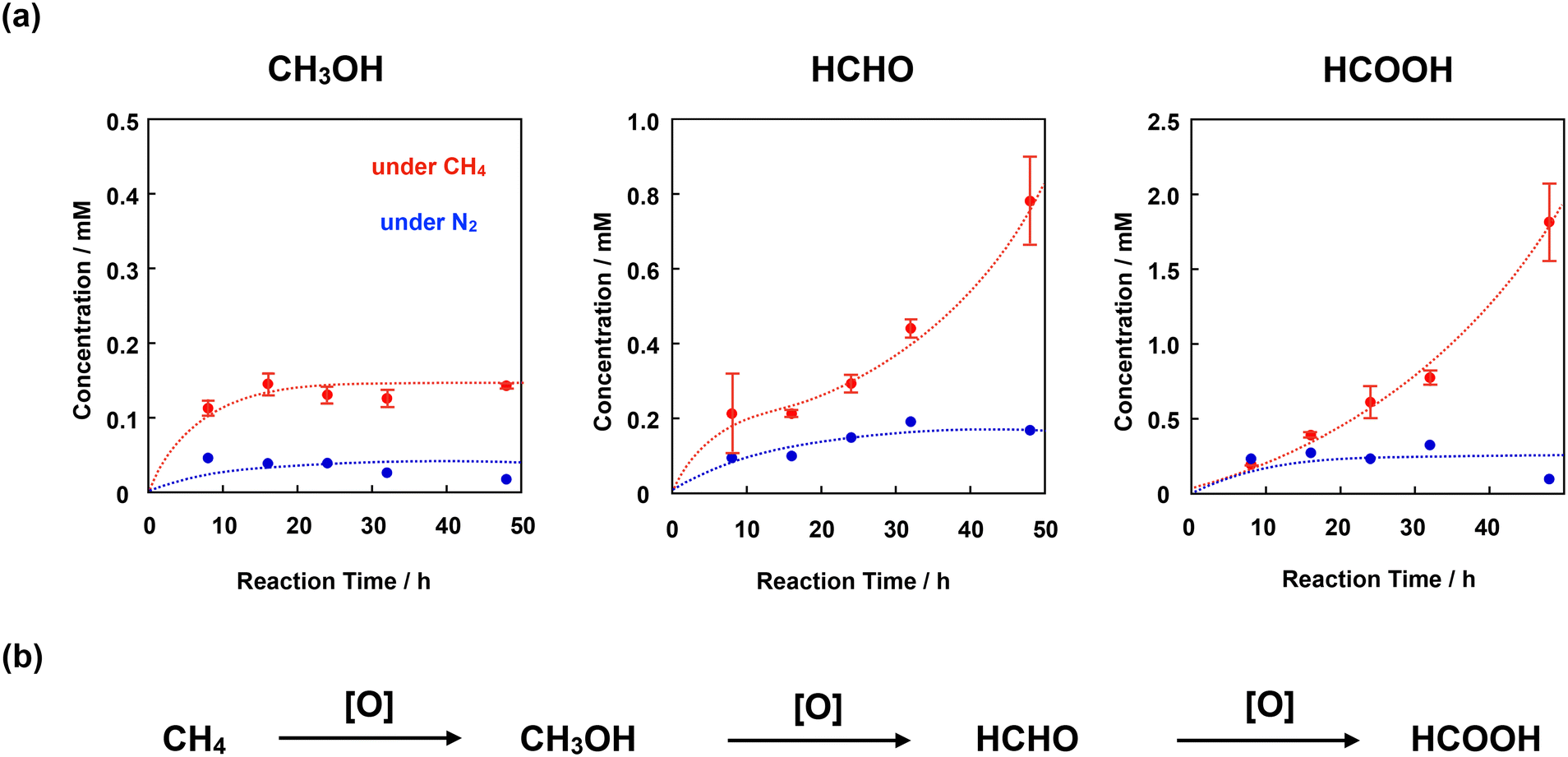 | ||
| Fig. 8 (a) Time dependence of the concentrations of each oxidized product (MeOH, HCHO, and HCOOH) observed in the reaction under a CH4 atmosphere of 1.0 MPa (red filled circle) or in the absence of CH4 (under 1.0 MPa of N2, blue filled circle) in an aqueous solution (3.0 mL) containing 4+·I3−/SiO2 (55 μM as 4+·I3−), H2O2 (189 mM), and TFA (51 mM) at 60 °C. Error bars indicate standard deviations of three independent oxidation reactions. (b) Stepwise CH4 oxidation reaction. | ||
To appropriately evaluate the catalytic CH4 oxidation activity, the effective total turnover number (TTNeff) and effective methane conversion number (MCNeff) were defined according to eqn (i)–(iv), based on the idea that CH4 is oxidized in a stepwise manner, as shown in Fig. 8b.
| TTNeff = TTN(CH4) − TTN(N2) | (i) |
| TTN(CH4) or TTN(N2) = (CMeOH + 2 × CHCHO + 3 × CHCOOH)/CCat | (ii) |
| MCNeff = MCN(CH4) − MCN(N2) | (iii) |
| MCN(CH4) or MCN(N2) = (CMeOH + CHCHO + CHCOOH)/CCat | (iv) |
Fig. 9a shows the time dependence of TTNeff. Although TTNeff increased almost linearly at the initial stage, the rate of increase increased, particularly after 32 h of oxidation. It should also be mentioned that the bluish-green color of the catalyst apparently became thinner, especially after 32 h, and almost colorless after 48 h of oxidation (Fig. S5†), suggesting that the porphyrinoid core of 4+·I3− gradually decomposed during the reaction. A similar bleaching of the color of the catalyst was observed in the oxidation reaction using the porphycene dimer 3+·PF6−. However, the bleaching speed was much slower for 4+·I3−, implying that the stability of 4+·I3− is higher than that of 3+·PF6− under these reaction conditions. Considering that apparent bleaching was not observed in the case of the phthalocyanine dimer 1+·I−, the higher stability of 4+·I3− than 3+·PF6− might be because the phthalocyanine unit in 4+·I3− contributed to the stable generation of the high-valent iron-oxo species after the reaction with H2O2.
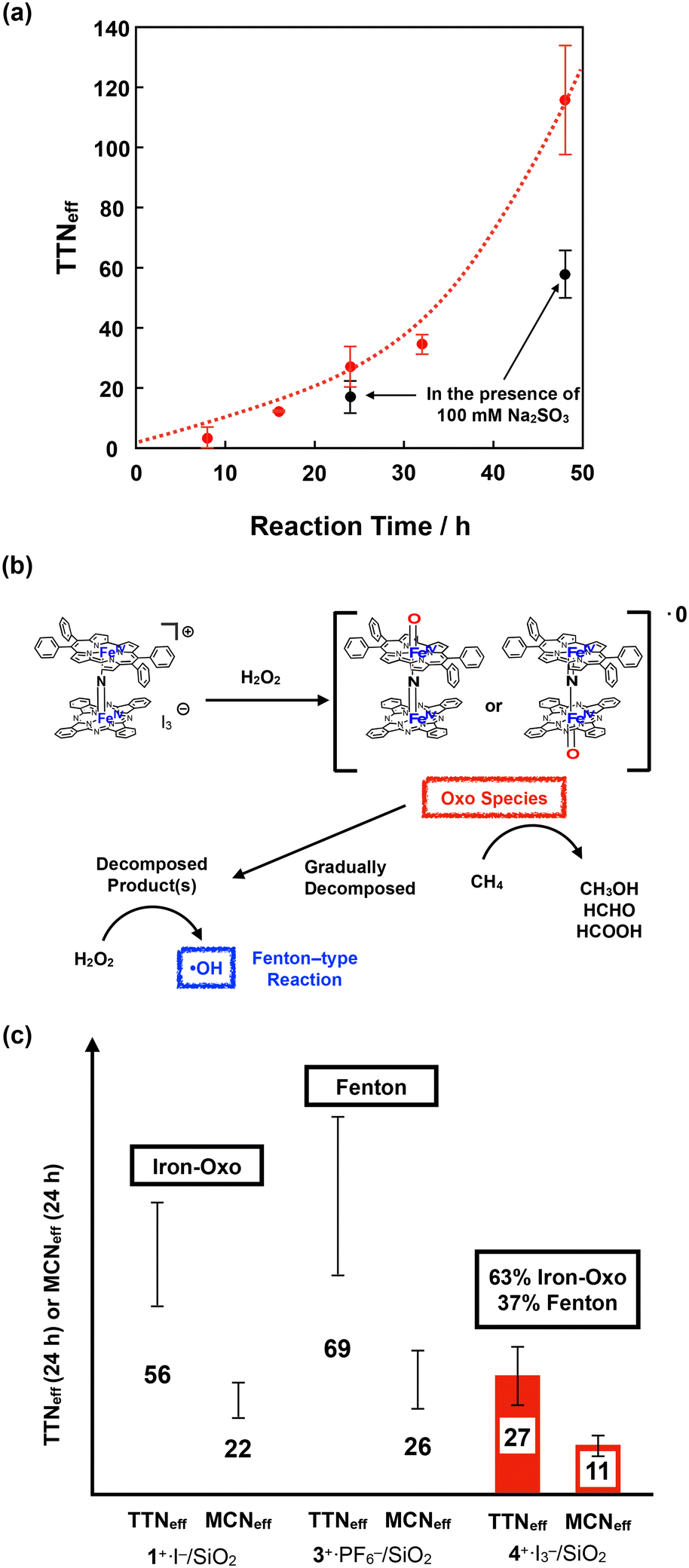 | ||
| Fig. 9 (a) Time dependence of TTNeff for CH4 oxidation by 4+·I3− in the absence (red circle) and presence (black circle) of 100 mM Na2SO3. Error bars indicate standard deviations of three independent oxidation reactions. (b) Proposed reaction mechanism for the CH4 oxidation by 4+·I3−. (c) Comparison of TTNeff and MCNeff for the CH4 oxidation for 24 h by 1+·I−, 3+·PF6−, and 4+·I3−. | ||
To further investigate the reaction mechanism of CH4 oxidation by 4+·I3−, we performed the CH4 oxidation in the presence of Na2SO3, a radical scavenger (Fig. 9a and Table S7,† entries 6 and 7).44 It was demonstrated that the reaction after 24 h of oxidation was suppressed by approximately 63%, whereas the reaction after 48 h of oxidation was suppressed to 50%. This indicates that the oxidation reaction mainly proceeded through the high-valent iron-oxo species, at least in its initial stage. However, considering that the color of 4+·I3− gradually bleached, particularly after 32 h, oxidation by the Fenton-type reaction via ˙OH gradually increased, presumably because the decomposed product acted as the catalyst for the Fenton-type reaction, as summarized in Fig. 9b. Because ˙OH is so reactive that it can even decompose the porphyrinoid cores of 4+·I3−, the accelerated decomposition of the catalyst is not only because of the intrinsic instability of the catalyst, but also because of the decomposition by ˙OH.
Fig. 9c shows a comparison of TTNeff and MCNeff of 1+·I−, 3+·PF6−, and 4+·I3− after 24 h of oxidation under the same reaction conditions. The reason why 4+·I3− showed less CH4 oxidation activity than the porphycene dimer 3+·PF6− is that 3+·PF6− decomposed more rapidly and CH4 oxidation for 24 h mostly proceeded via a Fenton-type reaction. However, both TTNeff and MCNeff for oxidation by 4+·I3− were much less than in the case of oxidation by 1+·I−, which mostly proceeded via the high-valent iron-oxo species. Therefore, it can be concluded that the catalytic methane oxidation activity via the high-valent iron-oxo species of 4+·I3− (OFePpc–NFePc and/or OFePc–NFePpc) is lower than that of the high-valent iron-oxo species of 1+·I− (OFePc–NFePc). This result indicates that the reactivity of the μ-nitrido-bridged iron porphyrinoid dimer strongly reflects the difference in the porphyrinoid skeleton, although the structures of the Fe–NFe cores are quite similar. We consider that one possible explanation for the lower catalytic activity of 4+·I3− than 1+·I− could be the slow generation of the high-valent iron-oxo species of 4+·I3− than that of 1+·I− because of the lower efficiency of the push effect, which facilitates the heterolytic O–O bond cleavage of the hydroperoxo species (Fe–O–OH) owing to the lower electron-donating ability of the porphyrinoid ring of 4+, as can be estimated from the fact that the oxidation potential of the phthalocyanine ring of 4+·I3− is higher than that of 1+·I−.
Conclusions
In this study, we synthesized a monocationic μ-nitrido-bridged heterodimer of an iron phthalocyanine and an iron porphycene (4+·I3−) and investigated its electrochemical, spectroscopic, and catalytic properties. Single-crystal X-ray structural analysis demonstrated that the Fe–NFe core structure of 4+·I3− is similar to that of a monocationic μ-nitrido-bridged iron phthalocyanine dimer (1+·I−) and a monocationic μ-nitrido-bridged iron porphycene dimer (3+·PF6−). ESI-TOF MS study indicated that treatment of 4+·I3− with H2O2 gave the high-valent iron-oxo species of 4+. DFT calculations suggested that the high-valent iron-oxo species of 4+·I3− could be generated on both iron-porphycene and iron-phthalocyanine rings through the reaction with H2O2. The catalytic CH4 oxidation reaction by 4+·I3− in an acidic aqueous solution proceeded in the presence of excess H2O2 to afford a mixture of MeOH, HCHO, and HCOOH through the high-valent iron-oxo species at its initial stage as in the case with 1+·I−. It was demonstrated that the catalytic activity of the high-valent iron-oxo species of 1+·I− was higher than that of 4+·I3−. However, as the reaction time was elongated, it was found that 4+·I3− was gradually decomposed. This implies that the high-valent iron-oxo species of 4+·I3− was less stable than that of 1+·I−. Considering that the high-valent iron-oxo species of 3+·PF6− is much less stable and readily decomposes under the same reaction conditions, the lower stability of the high-valent iron-oxo species of 4+·I3− compared to that of 1+·I− is derived from the difference in the porphyrinoid ring. Thus, it was confirmed that the difference in the porphyrinoid ring of the monocationic μ-nitrido-bridged iron porphyrinoid dimer strongly affected the stability of the high-valent iron-oxo species as well as the catalytic activity of the monocationic μ-nitrido-bridged iron porphyrinoid dimer. We believe that these findings would provide some valuable insights for the future development of molecular CH4 oxidation catalysts based on μ-nitrido-bridged iron porphyrinoid dimers.
Experimental
General
All reagents and solvents were purchased at the highest commercial quality available and used without further purification, unless otherwise stated. A metal-free porphycene was synthesized according to the reported procedure.451H NMR spectra were recorded on a JEOL JNM-ECS400 (400 MHz for 1H) spectrometer at a constant temperature of 298 K. Elemental analysis was performed on a Yanaco MT-6 analyzer. The absorption spectrum was recorded with a Hitachi U-4100 spectrophotometer in pyridine solutions at 20 ± 0.1 °C in 1.0 cm quartz cells. MALDI-TOF MS was performed on Bruker Daltonics ultrafleXtreme using α-CHCA as a matrix. MALDI-TOF MS was performed on Bruker Daltonics compact.Synthesis of 4+·I3−
A mixture of 5 (201 mg, 0.30 mmol), 6 (85 mg, 0.15 mmol), NaN3 (300 mg, 4.6 mmol) and 1-chloronaphtalene (7.5 mL) was heated under air at 280 °C for 2 h. After cooling to room temperature, the reaction mixture was diluted with hexane (200 mL), the precipitate was collected by suction filtration, washed with hexane (400 mL) H2O (300 mL), successively, dried under vacuum to give blackish green crude solid (392.3 mg). The crude product was dissolved in pyridine (400 mL). After iodine (70.2 mg, 0.55 mmol as I) was added, the resulting solution was stirred for 1 h at room temperature, followed by evaporation of the volatile compounds. The residue was washed with Et2O (300 mL) to remove the remaining iodine, and then, dried under reduced pressure to give dark green solid. The crude was purified by silica gel column chromatography (4.5 cm ϕ × 15.0 cm, CH2Cl2/AcOEt = 10/10, then CH2Cl2/AcOEt/MeOH = 10/10/1) and recrystallization from CH2Cl2/Et2O to yield 4+·I3− as a bluish green solid (121 mg, 89% in 2 steps from a metal-free porphycene). 1H-NMR (400 MHz, pyridine-d5/TMS): δ = 9.73 (dd, J = 5.4, 3.0 Hz, 8H), 8.84 (d, J = 4.8 Hz, 4H), 8.51 (dd, J = 6.0, 2.8 Hz, 8H), 8.42 (d, J = 7.2 Hz, 4H), 8.19 (d, J = 4.8 Hz, 4H), 8.00 (t, J = 7.8 Hz, 4H), 7.59–7.62 (m, 4H), 7.22–7.26 (m, 4H), 7.01 (d, J = 7.6 Hz, 4H). MALDI-TOF MS: m/z = 1250.25: calcd for C76H44Fe2N13 ([M]+) found: 1250.26. Anal. calcd for C164H122Fe4I6N26O5 ((4+·I3−)2·3Et2O·2H2O): C; 55.93, H; 3.49, N; 10.34, found: C; 55.66, H; 3.32, N; 10.04 (0.30% error).Synthesis of 4+·PF6−
4 +·I3− (61 mg, 38 μmol) was dissolved in 10 mL of acetonitrile. After KPF6 (691 mg, 3.8 mmol) in CH3CN (10 mL) was added to the solution, H2O (80 mL) was added. The resulting precipitate was collected by centrifugation, followed by decantation to yield dark green solid (54 mg). The crude product was further purified by PTLC (CH2Cl2/MeOH = 50/1) and recrystallization from CH2Cl2/Et2O to give the desired product 4+·PF6− as a bluish green solid (27.8 mg, 53%). 1H-NMR (400 MHz, pyridine-d5/TMS): δ = 9.72 (dd, J = 5.6, 2.8 Hz, 8H), 8.84 (d, J = 4.8 Hz, 4H), 8.50 (dd, J = 5.6, 2.8 Hz, 8H), 8.42 (d, J = 7.6 Hz, 4H), 8.20 (d, J = 4.8 Hz, 4H), 8.00 (t, J = 7.2 Hz, 4H), 7.59–7.62 (m, 4H), 7.22–7.26 (m, 4H), 7.00 (d, J = 7.6 Hz, 4H). MALDI-TOF MS: m/z = 1250.25: calcd for C76H44Fe2N13 ([M]+) found: 1250.26. Anal. calcd for C76H44F6Fe2N13P (4+·PF6−): C; 65.39, H; 3.18, N; 13.04, found: C; 65.45, H; 3.49, N; 13.14 (0.31% error).Single-crystal X-ray structural analysis of 4+·I3−
A 4+·I3− crystal suitable for single-crystal X-ray structural analysis was obtained by vapor diffusion of Et2O into a 4+·I3− solution in a 1:1 (v/v) mixture of pyridine and CHCl3. Single-crystal X-ray diffraction measurements were performed using a Rigaku X-ray diffractometer equipped with a molybdenum MicroMax-007 and Saturn 70 CCD detector. The measurement was performed at 123 K. The structure was solved via the direct method (SHELXT) and refined via full-matrix least-squares on F2 (SHELX-2018) using Olex2-1.3 program. All non-hydrogen atoms were refined anisotropically. Geometrical restraints were applied: DFIX, SADI, SIMU, ISOR, and OMIT. The crystal data are as follows: formula C74H49.6Cl15.6Fe1.6I2.4N12, FW = 2053.882, crystal size 0.19 × 0.20 × 0.22 mm3, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 20.6469(2) Å, b = 23.6627(3) Å, c = 24.8965(3) Å, α = 64.380(1)°, β = 69.270(1)°, γ = 68.880(1)°, V = 9942.9(2) Å3, Z = 5, R1 = 0.1207 (I > 2(I)), wR2 = 0.3227 (all), GOF = 1.110. CCDC identification code 2217367. All the checkCIF Level-B alerts are due to the low crystal quality.
, a = 20.6469(2) Å, b = 23.6627(3) Å, c = 24.8965(3) Å, α = 64.380(1)°, β = 69.270(1)°, γ = 68.880(1)°, V = 9942.9(2) Å3, Z = 5, R1 = 0.1207 (I > 2(I)), wR2 = 0.3227 (all), GOF = 1.110. CCDC identification code 2217367. All the checkCIF Level-B alerts are due to the low crystal quality.
Cyclic voltammogram of 4+·PF6−
Cyclic voltammograms were measured with a BAS Electrochemical Analyzer Model 750Ds at room temperature in pyridine solutions containing 100 mM TBAPF6 in a standard one-component cell under an N2 atmosphere equipped with a 3 mm-O.D. glassy carbon disk working electrode, platinum wire counter electrode, and Ag/AgCl reference electrode. All solutions were deoxygenated by N2 bubbling for at least 20 min. Obtained E°′ vs. Ag/AgCl were converted to those vs. Fc/Fc+ based on measured redox potential of ferrocene. Tetrabutylammonium hexafluorophosphate (TBA+PF6−) was recrystallized from 95% EtOH and dried under vacuum overnight at 100 °C.ESI-TOF MS measurement of 4+·I3− in CH3CN in the presence of H2O2
To a solution of 4+·I3− in CH3CN (1.0 μM, 2.0 mL) was added 35% aqueous H2O2 (0.1 mL, 1.16 mmol). After addition, the resulting mixture was subjected to ESI-TOF MS measurement immediately.DFT calculation
DFT calculations were done with the BP86-D3(BJ)/def2-TZVP and B3LYP-D3(BJ)/def2-TVZP levels of theory implemented in the Turbomole software package.46 The former functional was chosen based on the studies of Sorokin et al.20 Only BP86 results were discussed, whereas the results calculated with B3LYP can be found in the ESI.† Frequency calculations were done to confirm the character of the optimized structures and to obtain Gibbs free energies. In some calculations, the solvent effect (pyridine) was determined using COSMO with a dielectric constant of 12.5.47Preparation of silica-supported catalyst (4+·I3−/SiO2)
4 +·I3− (10.00 mg, 6.2 μmol) was dissolved in 50 mL of mixture of pyridine and CHCl3 (1:1 (v/v)). After the addition of silica gel (1082 mg) to the solution, solvent was evaporated. CHCl3 (10 mL) was added to the residue. After solvent was evaporated, the residue was dried under vacuum at 60 °C over night. The catalyst was suspended in an aqueous TFA (TFA 10.0 mL + H2O 100 mL). The mixture was sonicated for 30 min and then the solid was filtered. This TFA wash procedure was repeated twice. Finally, the resulting solid was washed with H2O (300 mL) and dried under vacuum at 60 °C over night. The silica-supported catalyst was obtained quantitatively.
CH4 oxidation reactions
Heterogeneous CH4 oxidation was performed in a stainless-steel autoclave with a glass tube. A mixture containing 4+·I3−/SiO2 (30 mg, 55 μM as 4+·I3−), 35% aqueous H2O2 (50 μL, 189 mM), and TFA (12 μL, 51 mM) in H2O (3.0 mL) was heated at 60 °C under 1.0 MPa of CH4 for 8–48 h with continuous stirring (900 rpm). After the autoclave was opened, the reaction mixture was filtrated through a disposable membrane filter. The filtrate was analyzed by GC-MS (system: Agilent 7890A equipped with JEOL JMS-T100GCV, detection: EI, column: Agilent DB-WAX UI, external standard: isovaleric acid (5 mM), temperature conditions: initial: 70 °C to 220 °C (10 °C min−1) – hold (5 min)). The yields of CH3OH and formic acid were determined based on the results of GC-MS. The yield of formaldehyde was examined using the method reported in our previous paper.21,28,31,42Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by a JSPS KAKENHI Grant-in-Aid for Scientific Research (A) (19H00902) and (B) (22H02094) awarded to KT, a JST PRESTO (number 14J04135), a JSPS KAKENHI Grant-in-Aid for Challenging Exploratory Research (number 22K19045), and a Grant-in-Aid for Scientific Research (B) (number 19H02787 and 22H02156) awarded to YY. YY thanks to the financial supports by Tatematsu Foundation, Iwatani Naoji Foundation, and Toyoaki Scholarship Foundation.Notes and references
- M. H. Sazinsky and S. J. Lippard, Acc. Chem. Res., 2006, 39, 558–566 CrossRef CAS PubMed.
- S. Sirajuddin and A. C. Rosenzweig, Biochemistry, 2015, 54, 2283–2294 CrossRef CAS PubMed.
- V. C.-C. Wang, S. Maji, P. P.-Y. Chen, H. K. Lee, S. S.-F. Yu and S. I. Chan, Chem. Rev., 2017, 117, 8574–8621 CrossRef CAS PubMed.
- C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44(4), 280–288 CrossRef CAS PubMed.
- P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2013, 52, 11980–11987 CrossRef CAS PubMed.
- Z. Guo, B. Liu, Q. Zhang, W. Deng, Y. Wang and Y. Yang, Chem. Soc. Rev., 2014, 43, 3480–3524 RSC.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef CAS PubMed.
- F. T. de Oliveira, A. Chanda, D. Banerjee, X. Shan, S. Mondal, L. Que Jr., E. L. Bominaar, E. Münck and T. J. Collins, Science, 2007, 315, 835–838 CrossRef PubMed.
- G. Xue, R. D. Hont, E. Münck and L. Que Jr., Nat. Chem., 2010, 2, 400–405 CrossRef CAS PubMed.
- Y. Hitomi, K. Arakawa and M. Kodera, Chem. Commun., 2014, 50, 7485–7487 RSC.
- E. Y. Tshuva and S. J. Lippard, Chem. Rev., 2004, 104, 987–1012 CrossRef CAS PubMed.
- X. Shan and L. Que Jr., J. Inorg. Biochem., 2006, 100, 421–433 CrossRef CAS PubMed.
- W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS PubMed.
- A. R. McDonald and L. Que Jr., Coord. Chem. Rev., 2013, 257, 414–428 CrossRef CAS.
- W. Nam, Acc. Chem. Res., 2015, 48, 2415–2423 CrossRef CAS PubMed.
- M. Guo, T. Corona, K. Ray and W. Nam, ACS Cent. Sci., 2019, 5, 13–28 CrossRef CAS PubMed.
- J. E. M. N. Klein and G. Knizia, Angew. Chem., Int. Ed., 2018, 57, 11913–11917 CrossRef CAS PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- P. Afanasiev and A. B. Sorokin, Acc. Chem. Res., 2016, 49, 583–593 CrossRef CAS PubMed.
- Y. Yamada, J. Kura, Y. Toyoda and K. Tanaka, Dalton Trans., 2021, 50, 6718–6724 RSC.
- A. B. Sorokin, E. V. Kudrik and D. Bouchu, Chem. Commun., 2008, 2562–2564 RSC.
- E. V. Kudrik, P. Afanasiev, L. X. Alvarez, P. Dubourdeaux, M. Clémancey, J.-M. Latour, G. Blondin, D. Bouchu, F. Albrieux, S. E. Nefedov and A. B. Sorokin, Nat. Chem., 2012, 4, 1024–1029 CrossRef CAS PubMed.
- C. Colomban, E. V. Kudrik, V. Briois, J. C. Shwarbrick, A. B. Sorokin and P. Afanasiev, Inorg. Chem., 2014, 53, 11517–11530 CrossRef CAS PubMed.
- M. G. Quesne, D. Senthilnathan, D. Singh, D. Kumar, P. Maldivi, A. B. Sorokin and S. P. de Visser, ACS Catal., 2016, 6, 2230–2243 CrossRef CAS.
- M. Ansari, N. Vyas, A. Ansari and G. Rajaraman, Dalton Trans., 2015, 44, 15232–15243 RSC.
- Q. M. Phung and K. Pierloot, Chem. – Eur. J., 2019, 25, 12491–12496 CrossRef CAS PubMed.
- Y. Yamada, Y. Miwa, Y. Toyoda, T. Yamaguchi, S. Akine and K. Tanaka, Dalton Trans., 2021, 50, 16775–16781 RSC.
- G. Anguera and D. Sánchez-García, Chem. Rev., 2017, 117, 2481–2516 CrossRef CAS PubMed.
- J. Waluk, Chem. Rev., 2017, 117, 2447–2480 CrossRef CAS PubMed.
- Y. Yamada, K. Morita, N. Mihara, K. Igawa, K. Tomooka and K. Tanaka, New J. Chem., 2019, 43, 11477–11482 RSC.
- N. Mihara, Y. Yamada, H. Takaya, Y. Kitagawa, K. Igawa, K. Tomooka, H. Fujii and K. Tanaka, Chem. – Eur. J., 2019, 25, 3369–3375 CrossRef CAS PubMed.
- Ü. Íşci, A. S. Faponle, P. Afanasiev, F. Albrieux, V. Briois, V. Ahsen, F. Dumoulin, A. B. Sorokin and S. P. de Visser, Chem. Sci., 2015, 6, 5063–5075 RSC.
- K. M. Kadish, R. K. Rhodes, L. A. Bottomley and H. M. Goff, Inorg. Chem., 1981, 20, 3195–3200 CrossRef CAS.
- L. A. Bottomley and B. B. Garrett, Inorg. Chem., 1982, 21, 1260–1263 CrossRef CAS.
- M. Li, M. Shang, N. Ehlinger, C. E. Schulz and W. R. Scheidt, Inorg. Chem., 2000, 39, 580–583 CrossRef CAS PubMed.
- Y. Yamada, T. Sugiura, K. Morita, H. Ariga-Miwa and K. Tanaka, Inorg. Chim. Acta, 2019, 489, 160–163 CrossRef CAS.
- T. Shimizu, K. Wakamatsu, Y. Yamada, Y. Toyoda, S. Akine, K. Yoza and H. Yoshikawa, ACS Appl. Mater. Interfaces, 2021, 13, 40612–40617 CrossRef CAS PubMed.
- L. A. Bottomley, J.-N. Gorce, V. L. Goedken and C. Ercolani, Inorg. Chem., 1985, 24, 3733–3737 CrossRef CAS.
- C. Bernard, J. P. Gisselbrecht, M. Gross, E. Vogel and M. Lausmann, Inorg. Chem., 1994, 33, 2393–2401 CrossRef CAS.
- C. Colomban, E. V. Kudrik and A. B. Sorokin, J. Porphyrins Phthalocyanines, 2017, 21, 346–353 CrossRef.
- A. Ghosh, E. Tangen, E. Gonzalez and L. Que, Angew. Chem., 2004, 116, 852–856 CrossRef.
- Y. Yamada, J. Kura, Y. Toyoda and K. Tanaka, New J. Chem., 2020, 44, 19179–19183 RSC.
- C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, D. M. Murphy, A. F. Carley, S. H. Taylor, D. J. Willock, E. E. Stangland, J. Kang, H. Hagen, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 51, 5129–5133 CrossRef CAS PubMed.
- T. Ono, N. Xu, D. Koga, T. Ideo, M. Sugimoto and Y. Hisaeda, RSC Adv., 2018, 8, 39269–39273 RSC.
- S. G. Balasubramani, G. P. Chen, S. Coriani, M. Diedenhofen, M. S. Frank, Y. J. Franzke, F. Furche, R. Grotjahn, M. E. Harding, C. Hättig, A. Hellweg, B. Helmich-Paris, C. Holzer, U. Huniar, M. Kaupp, A. Marefat Khah, S. Karbalaei Khani, T. Müller, F. Mack, B. D. Nguyen, S. M. Parker, E. Perlt, D. Rappoport, K. Reiter, S. Roy, M. Rückert, G. Schmitz, M. Sierka, E. Tapavicza, D. P. Tew, C. van Wüllen, V. K. Voora, F. Weigend, A. Wodyński and J. M. Yu, J. Chem. Phys., 2020, 152, 184107 CrossRef CAS PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2217367. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cy01980a |
| This journal is © The Royal Society of Chemistry 2023 |