
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA practical and sustainable two-component Minisci alkylation via photo-induced EDA-complex activation†
Mohammed
Sharique
 ,
Jadab
Majhi‡
,
Roshan K.
Dhungana‡
,
Lisa Marie
Kammer
,
Matthias
Krumb
,
Alexander
Lipp
,
Eugénie
Romero
and
Gary A.
Molander
*
,
Jadab
Majhi‡
,
Roshan K.
Dhungana‡
,
Lisa Marie
Kammer
,
Matthias
Krumb
,
Alexander
Lipp
,
Eugénie
Romero
and
Gary A.
Molander
*
Roy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, Pennsylvania 19104-6323, USA. E-mail: gmolandr@sas.upenn.edu
First published on 26th April 2022
Abstract
An operationally simple, open-air, and efficient light-mediated Minisci C–H alkylation method is described, based on the formation of an electron donor–acceptor (EDA) complex between nitrogen-containing heterocycles and redox-active esters. In contrast to previously reported protocols, this method does not require a photocatalyst, an external single electron transfer agent, or an oxidant additive. Achieved under mildly acidic and open-air conditions, the reaction incorporates primary-, secondary-, and tertiary radicals, including bicyclo[1.1.1]pentyl (BCP) radicals, along with various heterocycles to generate Minisci alkylation products in moderate to good yields. Additionally, the method is exploited to generate a stereo-enriched, hetereoaryl-substituted carbohydrate.
Introduction
The extensive occurrence of nitrogen-containing heterocycles in biologically active natural products and pharmaceutical drugs makes them an attractive target in the development of new synthetic methods. In particular, reactions that can directly functionalize such heterocyclic substructures to generate medicinally valuable structural analogues are highly desirable.1–4One way to access these heterocyclic building blocks is to perform a C(sp2)–C(sp3) bond construction via an alkyl radical addition to heteroarenes.5–9 In this context, the venerable Minisci alkylation reaction developed by F. Minisci in 1971 has proven to be a powerful synthetic strategy for the C–H functionalization of a diverse range of heteroarenes.10,11 Since its discovery, extensive follow-up protocols have been identified by synthetic chemists to improve the applicability and scope of the reaction; however, the synthetic utility of this reaction has often been limited because of the use of stoichiometric, strong oxidants, highly acidic conditions, and/or high temperatures.12–21
In recent years, the emergence of photoredox catalysis has transformed the Minisci reaction by offering milder conditions, concomitantly circumventing the obligatory requirement of a strong oxidant to generate radicals under harsh conditions, thus making this process amenable to a wide range of heterocycles and radical precursors (Fig. 1a).22–25 However, despite contemporary advances in this field, some of the existing methods still require strong acids for the activation of heterocycles. In addition, a stoichiometric strong oxidant is often needed in conjunction with the photocatalyst, particularly when the reaction is not net-redox neutral. External electron transfer agents (e.g., Hantzsch ester) have also been required in some instances, diminishing the convenience and sustainability of the methods developed. Thus, further improvements are desirable to make Minisci C–H alkylation reactions more tolerant of sensitive functional groups, more convenient, and more efficient.
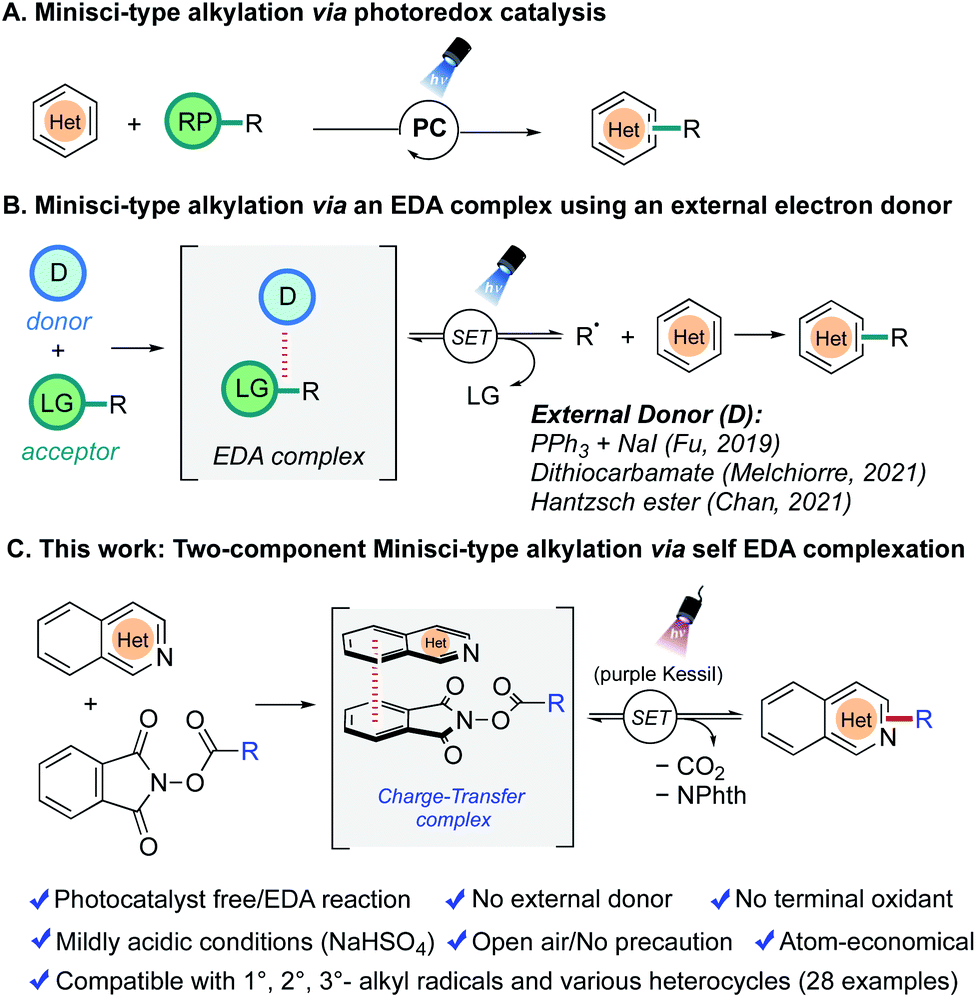 | ||
| Fig. 1 Strategies for photoinduced Minisci alkylations. (A) Via photo-redox catalysis (B) via EDA-activation using an external donor. (C) Developed two-component Minisci-alkylation via an EDA method. | ||
As an alternative, the photoinduced electron donor–acceptor (EDA) complex strategy is gaining considerable attention because of its ability to manifest photocatalyst-free, oxidant-free reaction conditions.26–30 The radicals are generated via a synergistic non-covalent interaction between an electron-rich donor and an electron-deficient acceptor. Although there are already many transformations reported based on this paradigm to construct new C–C, C–N, C–S, and C–B bonds efficiently, only a handful of reports are available in the realm of Minisci reactions (Fig. 1b). In all of these reported to date, an external single electron transfer agent is required. Thus, in 2019, Fu and coworkers disclosed an enantioselective variant of the Minisci alkylation by identifying a unique combination of catalytic sodium iodide and triphenylphosphine (NaI/PPh3) as an effective electron donor to participate in an EDA-complex formation with redox-active esters (RAEs).31 Another report by Melchiorre et al. (2021) demonstrated that a catalytic dithiocarbamate anion possesses a similar capability as an electron donor toward the same acceptor, a redox-active ester.32 During the course of this investigation, Chan et al. reported that the commercially available Hantzsch ester, an established organo-reductant in EDA chemistry, could be used in the presence of trifluoroacetic acid to perform Minisci alkylations on heterocycles by generating alkyl radicals under photocatalyst-free conditions.33
Discussion
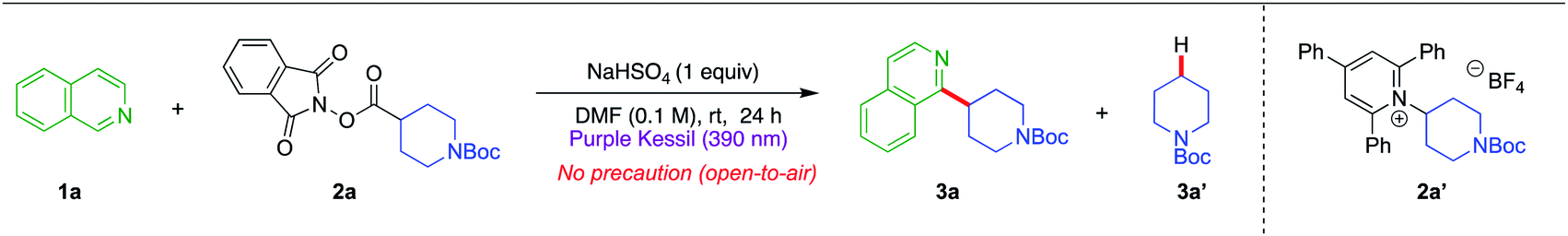
In an effort to make the Minisci alkylation reactions operationally simpler and applicable in the presence of acid-sensitive groups, we envisioned exploring a two-component Minisci reaction (with no photocatalyst and no exogenous electron transfer agents) by investigating the feasibility of having nitrogen-containing heterocycles such as isoquinoline 1a themselves serve as electron donors in a charge-transfer complex with suitable electron-accepting partners, using a very mild acid, NaHSO4, as an activating agent (Fig. 1c). Both redox-active ester 2a and pyridinium salt 2a′ were probed in optimization studies, owing to their pervasive utilization as effective electron-acceptor partners in EDA chemistry (Table 1).31–38 Gratifyingly, treatment of isoquinoline 1a (1 equiv.) with redox-active ester 2a (1.5 equiv.) generated the desired alkylation product 3a in good yield (62% NMR, 58% isolated) after 24 h of purple light irradiation in DMF solvent (0.1 M) in the presence of NaHSO4 (1 equiv.) under open-to-air conditions (entry 1). On the other hand, the reaction with pyridinium salt 2a′ under the same conditions failed to produce any desired product (entry 2). Notably, in entry 1, the yield of the Minisci alkylation product 3a is somewhat compromised because of the formation of the decarboxylative hydrogenation side product 3a′ (33% NMR yield). A quick examination of other reaction solvents such as MeCN, DMA, and THF resulted in lower yields of the alkylation product 3a and a higher amount of the side product 3a′ (entries 3–5), while dichloromethane did not support the desired reaction (entry 6). Typically, there is a strict requirement for a stoichiometric strong acid in Minisci alkylations, mainly because of the sluggishness in the addition of alkyl radicals possessing nucleophilic character to electrophilic heterocycles in their non-protonated form. However, under the conditions developed, the reaction occurs under relatively benign acidic conditions, permitting incorporation of some highly acid-sensitive functional groups. Thus, making a direct comparison with the most-utilized Brønsted acids in Minisci alkylation, p-toluenesulfonic acid (pKa = −2.8) or trifluoracetic acid (pKa = 0.23), the current method presents much milder acidic conditions with the use of NaHSO4 (pKa = 1.99).39 Although the process works best when NaHSO4 is used in a stoichiometric (1.0 equiv.) quantity, it also produces a significant amount of product with catalytic NaHSO4 (entry 7).40 Surprisingly, we still observed the desired product formation, albeit in low yield, when the acid (NaHSO4) was completely removed from the reaction conditions (entry 8). This might be attributed to the formation of an isoquinoline radical cation species after an electron transfer event between isoquinoline 1a and redox-active ester 2a during the EDA complex formation, which is sufficiently electrophilic to react with the nucleophilic radical species. We also investigated the effect of light sources on the reaction. Replacing the purple Kessil lamp (λ = 390 nm) by a blue Kessil lamp (λ = 427 nm) delivered the Minisci alkylation product in a slightly lower yield (entry 9), although longer reaction times with a blue Kessil lamp compensated for the low yields. Next, changing the reaction environment from an open-air to an inert (argon) atmosphere provided only a slight improvement in the yield, suggesting the method to be tolerant of oxygen and moisture (entry 10). Finally, as expected, the exclusion of a light source from the reaction conditions resulted in no conversion of the starting materials to the products (entry 11).|
|
||
|---|---|---|
| Entry | Deviation from standard condition | %Yieldb |
| a Reaction conditions: isoquinoline 1a (0.1 mmol, 1 equiv.), RAE 2a (0.15 mmol, 1.5 equiv.), NaHSO4 (0.1 mmol, 1 equiv.) in DMF (1.0 mL, 0.1 M), 24 h irradiation with purple LEDs (390 nm). b Yields were determined by 1H NMR analysis using trimethoxybenzene as internal standard. | ||
| 1 | None | 62(58) |
| 2 | Pyridinium salt 2a′ instead of RAE 2a | 0 |
| 3 | MeCN instead of DMF | 15 |
| 4 | DMA instead of DMF | 46 |
| 5 | THF instead of DMF | 20 |
| 6 | CH2Cl2 instead of DMF | 0 |
| 7 | 0.5 equiv. NaHSO4 | 49 |
| 8 | No NaHSO4 | 24 |
| 9 | Blue Kessil instead of purple | 55 |
| 10 | Argon condition | 65 |
| 11 | Dark | 0 |
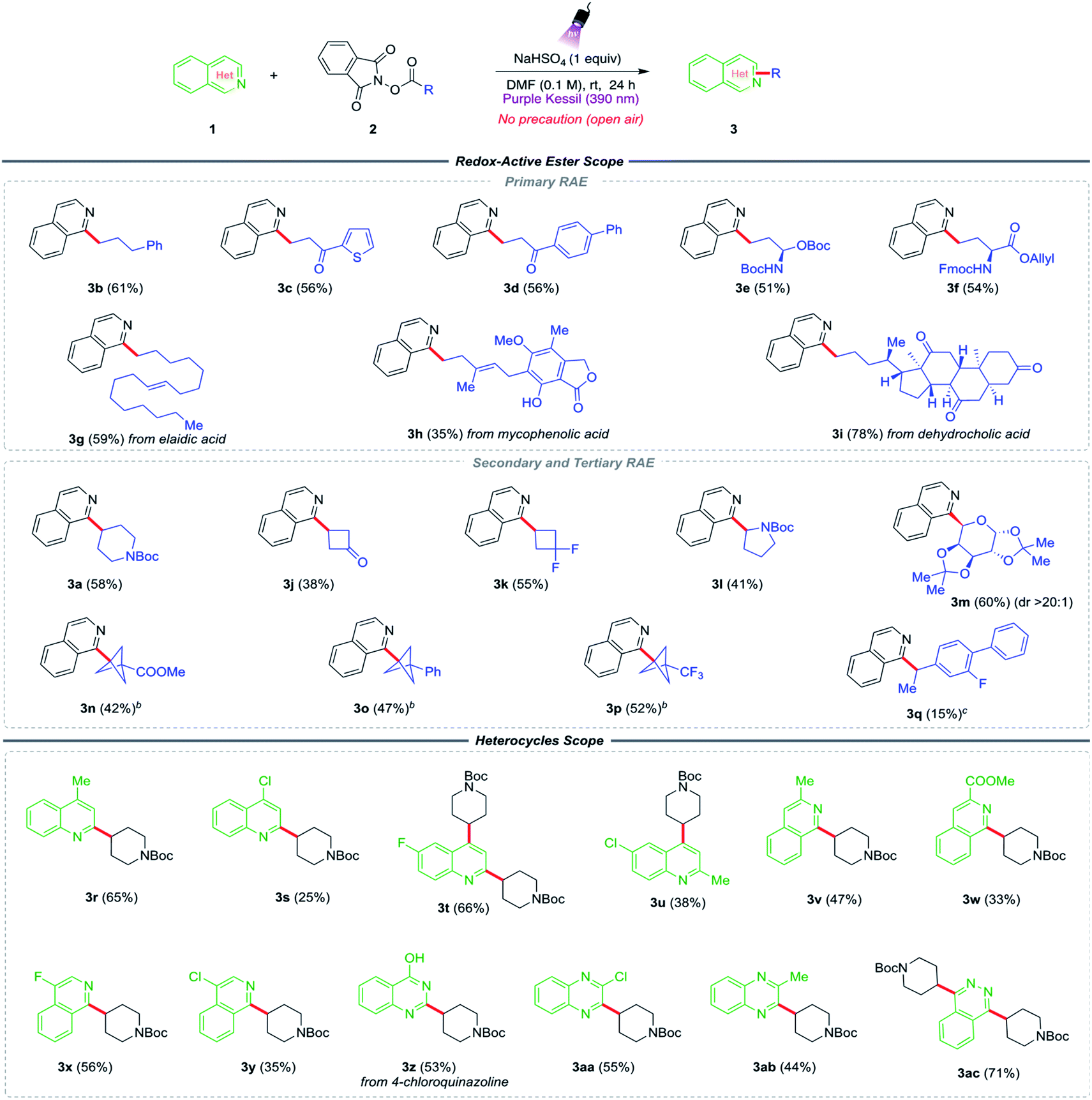
With suitable conditions in hand, the generality of the two-component Minisci alkylation method was evaluated (Table 2). First, the scope of redox-active esters was examined by employing various primary-, secondary-, and tertiary radical precursors. The method works well with relatively unstable primary radicals embellished with various functional groups. Redox-active esters with phenyl-, thienyl-, and biphenyl ketones on the side chain generated the desired products in good yields (3b–3d). Notably, because of the mildly acidic conditions, the reaction efficiently tolerated a substrate containing an acid-sensitive amine protecting group (3e). Further, the primary radical scope was extended to the modification of some biologically active and medicinally relevant compounds such as substrates derived from L-glutamate (3f), long-chain fatty acid elaidic acid (3g), an immunosuppressive drug mycophenolic acid (3h), and a steroid, dehydrocholic acid (3i).
| a Reaction conditions: isoquinoline 1 (0.3 mmol, 1 equiv.), RAE 2 (0.45 mmol, 1.5 equiv.), NaHSO4 (0.3 mmol, 1 equiv.) in DMF (3.0 mL, 0.1 M), 24 h irradiation with purple Kessil (390 nm). b Trifluroacetic acid (TFA) was used instead of NaHSO4. c Yield was determined by 1H NMR analysis using trimethoxybenzene as internal standard. |
|---|
|
Next, we explored the viability of secondary- and tertiary redox-active esters under the reaction conditions. Secondary radical precursors such as N-Boc piperidine (3a), cyclobutanone (3j), and 1,1-difluorocyclobutane (3k) facilitated the alkylation reaction, with products being formed in moderate to good yields. Some heteroatom-containing secondary radicals were also examined, such as an α-amino alkyl radical from N-Boc pyrrolidine RAE (3l), which provided the desired product in modest yield. More importantly, the Minisci alkylation product (3m) from α-D-galactopyranose sugar was isolated with excellent diastereoselectivity (dr > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, NMR analysis). This particular result points to another feature of the developed method, in that in some cases high diastereoselectivity can be achieved at radical reaction centers in carbohydrate substrates. Because of the importance of bicyclo[1.1.1]pentanes (BCPs) in drug development programs, there was an interest in exploring the feasibility of incorporating these substructures in the current protocol. Being identified as sp3-rich aryl bioisosteres, the incorporation of BCPs into drug candidates has increased considerably in recent years as they exhibit enhanced pharmacokinetic properties, including improved aqueous solubility and membrane permeability over that of their aryl congeners.41 Stimulated by this, BCP-redox active esters containing –COOMe, –Ph, and –CF3 groups were examined, all of which provided the desired alkylated products (3n–3p) in reasonable yields. Lastly, the addition of the benzylic radical derived from a flurbiprofen precursor to the isoquinoline core under the standard conditions provided the expected product in a slightly diminished yield (3q).
:1, NMR analysis). This particular result points to another feature of the developed method, in that in some cases high diastereoselectivity can be achieved at radical reaction centers in carbohydrate substrates. Because of the importance of bicyclo[1.1.1]pentanes (BCPs) in drug development programs, there was an interest in exploring the feasibility of incorporating these substructures in the current protocol. Being identified as sp3-rich aryl bioisosteres, the incorporation of BCPs into drug candidates has increased considerably in recent years as they exhibit enhanced pharmacokinetic properties, including improved aqueous solubility and membrane permeability over that of their aryl congeners.41 Stimulated by this, BCP-redox active esters containing –COOMe, –Ph, and –CF3 groups were examined, all of which provided the desired alkylated products (3n–3p) in reasonable yields. Lastly, the addition of the benzylic radical derived from a flurbiprofen precursor to the isoquinoline core under the standard conditions provided the expected product in a slightly diminished yield (3q).
Next, attention was focused on the scope of nitrogen-containing heterocycles. Some functionally elaborated quinolines were assessed in the presence of the N-Boc piperidine radical precursor (2a). Lepidine (3r) and 4-chloroquinolines (3s) led to good yields of the mono-alkylated Minisci products by substituting the most electrophilic 2-position of the quinoline. However, when both the 2- and 4-positions were unoccupied, such as in the case of 6-fluoroquinoline (3t), the reaction afforded disubstituted alkyl product. With 6-chloro-2-methylquinoline (3u), the alkylation occurred at the 4-position. Similarly, isoquinoline-based heterocycles with methyl-, ester-, and halide substitutions at the 3- and 4- positions also furnished the desired products in good yields (3v–3y). Some other medicinally important heterocycles, such as substituted quinazolines and quinoxalines, also proved to be equally effective under the established reaction conditions (3z–3ac).
To investigate the reaction mechanism of the two-component Minisci alkylation, ultraviolet/visible (UV/vis) and radical trapping experiments were performed (Fig. 2a). The UV/vis absorption spectrum of each individual reaction component was measured, along with that of the combined reaction mixture to provide evidence for the formation of an electron donor–acceptor (EDA) complex. When isoquinoline 1a and redox-active ester 2a were mixed along with NaHSO4 in DMF (0.1 M), the obtained absorption spectrum clearly depicted a red (bathochromic) shift, with a visible-light absorption tailing to the 410–420 nm region, indicating the formation of a charge-transfer molecular aggregate. To probe the generation of an alkyl radical species, a radical trapping agent, TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy), was added under standard conditions (Fig. 2b). This additive inhibited the alkylation reaction, with none of the Minisci alkylated product observed. Instead, TEMPO adduct 4 formed as confirmed via LC-MS analysis (see ESI†).
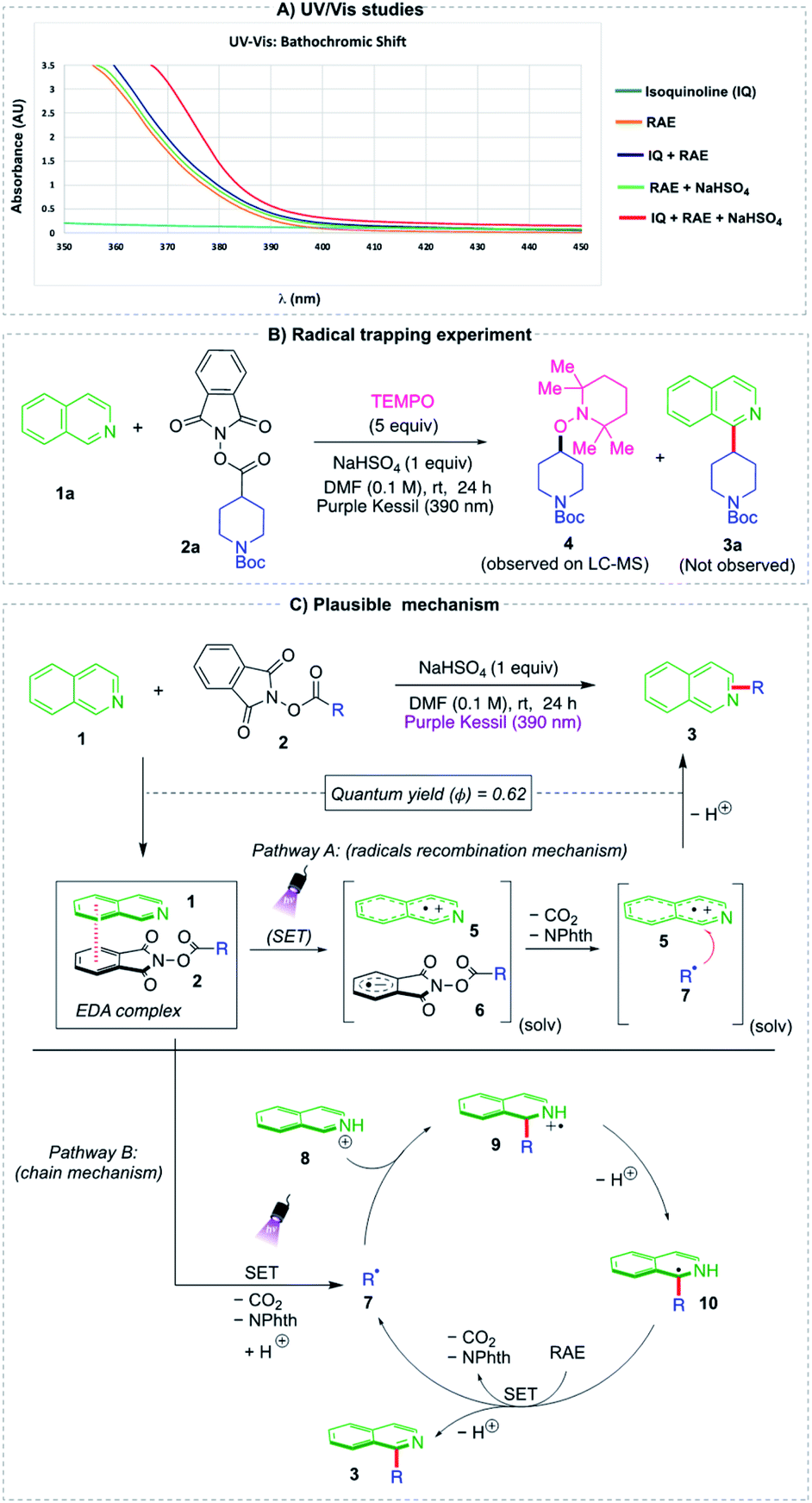 | ||
| Fig. 2 (A) Chromatogram of UV-vis absorption study (B) TEMPO-radical trapping experiment (C) our proposed mechanisms. | ||
Based on these findings, two mechanisms appear reasonable (Fig. 2). Thus, considering pathway A, the heterocycle 1 and redox-active ester 2 form an EDA complex in the ground state. Upon photoirradiation of this molecular aggregate at 390 nm, a single-electron transfer (SET) event from 1 to 2 occurs, which leads to the formation of radical ion pair 5/6 in a solvent cage. Fragmentation of reduced RAE 6 generates phthalimidate ion, CO2, and alkyl radical. This newly generated radical 7 subsequently undergoes a radical recombination with the aromatic radical cation 5,42 followed by deprotonation to afford the desired product 3. Alternatively, a chain mechanism pathway B could also be operative, where the alkyl radical 7 escapes from the solvent cage and reacts with protonated N-heterocyclic intermediate 8 to yield an alkyl-substituted, nitrogen-centered radical cation 9. The adduct formed subsequently undergoes deprotonation followed by oxidation via another molecule of redox-active ester 2 (ref. 24 and 33) to furnish the desired product 3 along with another alkyl radical species 7 to participate in the radical chain propagation step. Given that the experimentally determined quantum yield is high, but <1, (Φ = 0.62, see ESI†), it seems likely that the two pathways coexist, with the radical ion pair recombination mechanism prevailing.
Conclusions
In summary, an operationally simple yet highly practical two-component Minisci alkylation method has been developed. This protocol avoids the requirement for expensive metal-based photocatalysts, strong oxidants, an external single electron transfer agent, and harsh acidic conditions, enabling the formation of medicinally relevant alkylated heterocycles from commercially available and/or readily synthesizable starting materials. Consistent with the spectroscopic studies and radical trapping experiment, a photoactivated EDA complexation between heterocycles and redox-active esters forms the basis of the reaction mechanism for the generation of alkyl radicals under open-air-conditions.Data availability
Source/synthesis of N-(acyloxy)phthalimides and heterocycles used. General procedure for the two-component Minisci reactions. Description of mechanistic investigations. Quantum yield measurements. NMR spectra of all materials synthesized.Author contributions
Lisa Marie Kammer, Matthias Krumb, Alexander Lipp, and Eugénie Romero carried out initial optimization studies, which were subsequently refined by Mohammed Sharique, Jadab Majhi, and Roshan K. Dhungana. All of the examples presented in the paper were carried out by Mohammed Sharique, Jadab Majhi, and Roshan K. Dhungana, with Mohammed Sharique leading the effort. Mohammed Sharique, Jadab Majhi, and Roshan K. Dhungana wrote the original draft of the manuscript with input from Gary A. Molander. All authors contributed to reaction development and discussion of results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Albert Granados for assisting in UV-Vis studies and Ms. Shivani Patel for providing redox-active esters. The authors are grateful for financial support provided by NIGMS (R35 GM 131680 to G. A. M.). The NSF Major Research Instrumentation Program (award NSF CHE-1827457), the NIH supplement awards 3R01GM118510-03S1 and 3R01GM087605-06S1, as well as the Vagelos Institute for Energy Science and Technology supported the purchase of the NMRs used in this study. We thank Dr Charles W. Ross, III (UPenn) for mass spectral data. Kessil is acknowledged for the donation of lamps.Notes and references
- J. A. Joule, in Advances in Heterocyclic Chemistry, ed. E. F. V. Scriven and C. A. Ramsden, Academic Press, 2016, vol. 119, pp. 81–106 Search PubMed.
- J. C. Lewis, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2008, 41, 1013–1025 CrossRef CAS PubMed.
- J. Liu, J. Jiang, L. Zheng and Z.-Q. Liu, Adv. Synth. Catal., 2020, 362, 4876–4895 CrossRef CAS.
- J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed.
- J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Acc. Chem. Res., 2016, 49, 1911–1923 CrossRef CAS PubMed.
- F. O'Hara, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2013, 135, 12122–12134 CrossRef PubMed.
- J.-T. Yu and C. Pan, Chem. Commun., 2016, 52, 2220–2236 RSC.
- Y. Tan, J. Wang, H.-Y. Zhang, Y. Zhang and J. Zhao, Front. Chem., 2020, 8, 582 CrossRef CAS PubMed.
- Y. Ji, T. Brueckl, R. D. Baxter, Y. Fujiwara, I. B. Seiple, S. Su, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14411–14415 CrossRef CAS PubMed.
- F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Tetrahedron, 1971, 27, 3575–3579 CrossRef CAS.
- F. Minisci, F. Fontana and E. Vismara, J. Heterocycl. Chem., 1990, 27, 79–96 CrossRef CAS.
- M. A. J. Duncton, MedChemComm, 2011, 2, 1135–1161 RSC.
- R.-J. Tang, L. Kang and L. Yang, Adv. Synth. Catal., 2015, 357, 2055–2060 CrossRef CAS.
- D. R. Sutherland, M. Veguillas, C. L. Oates and A.-L. Lee, Org. Lett., 2018, 20, 6863–6867 CrossRef CAS PubMed.
- D. N. Mai and R. D. Baxter, Org. Lett., 2016, 18, 3738–3741 CrossRef CAS PubMed.
- A. P. Antonchick and L. Burgmann, Angew. Chem., Int. Ed., 2013, 52, 3267–3271 CrossRef CAS PubMed.
- I. B. Seiple, S. Su, R. A. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 13194–13196 CrossRef CAS PubMed.
- G. A. Molander, V. Colombel and V. A. Braz, Org. Lett., 2011, 13, 1852–1855 CrossRef CAS PubMed.
- M. Presset, N. Fleury-Brégeot, D. Oehlrich, F. Rombouts and G. A. Molander, J. Org. Chem., 2013, 78, 4615–4619 CrossRef CAS PubMed.
- Y. Fujiwara, J. A. Dixon, R. A. Rodriguez, R. D. Baxter, D. D. Dixon, M. R. Collins, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 1494–1497 CrossRef CAS PubMed.
- Á. Gutiérrez-Bonet, C. Remeur, J. K. Matsui and G. A. Molander, J. Am. Chem. Soc., 2017, 139, 12251–12258 CrossRef PubMed.
- R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS PubMed.
- R. S. J. Proctor, P. Chuentragool, A. C. Colgan and R. J. Phipps, J. Am. Chem. Soc., 2021, 143, 4928–4934 CrossRef CAS PubMed.
- L. M. Kammer, A. Rahman and T. Opatz, Molecules, 2018, 23, 764–780 CrossRef PubMed.
- M. Krumb, L. M. Kammer, S. O. Badir, M. J. Cabrera-Afonso, V. E. Wu, M. Huang, A. Csakai, L. A. Marcaurelle and G. A. Molander, Chem. Sci., 2022, 13, 1023–1029 RSC.
- Z. Yang, Y. Liu, K. Cao, X. Zhang, H. Jiang and J. Li, Beilstein J. Org. Chem., 2021, 17, 771–799 CrossRef CAS PubMed.
- Y.-q. Yuan, S. Majumder, M.-h. Yang and S.-r. Guo, Tetrahedron Lett., 2020, 61, 151506 CrossRef CAS.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- L. Zheng, L. Cai, K. Tao, Z. Xie, Y.-L. Lai and W. Guo, Asian J. Org. Chem., 2021, 10, 711–748 CrossRef CAS.
- C. G. S. Lima, T. de M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS.
- M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429–1434 CrossRef CAS PubMed.
- E. de Pedro Beato, D. Spinnato, W. Zhou and P. Melchiorre, J. Am. Chem. Soc., 2021, 143, 12304–12314 CrossRef CAS PubMed.
- J. Li, S. Siang Tan, S. H. Kyne and P. Wai Hong Chan, Adv. Synth. Catal., 2022, 364, 802–810 CrossRef CAS.
- V. C. Polites, S. O. Badir, S. Keess, A. Jolit and G. A. Molander, Org. Lett., 2021, 23, 4828–4833 CrossRef CAS PubMed.
- M. Elkhalifa, M. B. Elbaum, D. M. Chenoweth and G. A. Molander, Org. Lett., 2021, 23, 8219–8223 CrossRef CAS PubMed.
- L. M. Kammer, S. O. Badir, R.-M. Hu and G. A. Molander, Chem. Sci., 2021, 12, 5450–5457 RSC.
- M. Yang, T. Cao, T. Xu and S. Liao, Org. Lett., 2019, 21, 8673–8678 CrossRef CAS PubMed.
- F. J. R. Klauck, M. J. James and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 12336–12339 CrossRef CAS PubMed.
- International Union of Pure and Applied Chemistry, E. P. Serjeant and B. Dempsey, Ionisation constants of organic acids in aqueous solution, Pergamon Press, Oxford, 1979 Search PubMed.
- W.-M. Cheng, R. Shang and Y. Fu, ACS Catal., 2017, 7, 907–911 CrossRef CAS.
- M. R. Bauer, P. Di Fruscia, S. C. C. Lucas, I. N. Michaelides, J. E. Nelson, R. I. Storer and B. C. Whitehurst, RSC Med. Chem., 2021, 12, 448–471 RSC.
- S. R. Kandukuri, A. Bahamonde, I. Chatterjee, I. D. Jurberg, E. C. Escudero-Adan and P. Melchiorre, Angew. Chem., Int. Ed., 2015, 54, 1485–1489 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and mechanistic studies details, as well as spectral data. See https://doi.org/10.1039/d2sc01363k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |