
Mesoporous Pd@Pt core–shell nanoparticles supported on multi-walled carbon nanotubes as a sensing platform: application in simultaneous electrochemical detection of anticancer drugs doxorubicin and dasatinib†
Pramod K.
Kalambate
 ,
Yankai
Li
,
Yue
Shen
and
Yunhui
Huang
*
,
Yankai
Li
,
Yue
Shen
and
Yunhui
Huang
*
State Key Laboratory of Materials Processing and Die & Mould Technology, School of Materials Science and Engineering, Huazhong University of Science and Technology, Wuhan, Hubei 430074, P. R. China. E-mail: huangyh@hust.edu.cn; Tel: +86-27-87558241
First published on 11th December 2018
Abstract
In this work, a novel highly sensitive electrochemical sensing platform has been developed for simultaneous determination of anticancer drugs doxorubicin (DOX) and dasatinib (DAS) by means of mesoporous Pd@Pt core–shell nanoparticles supported on MWCNT (Pd@Pt/MWCNT). The electrochemical behaviour of the drugs was assessed through cyclic voltammetry, while simultaneous determination was carried out using adsorptive stripping square wave voltammetry (AdSSWV). Under optimized conditions, the method exhibits linear responses towards simultaneous determination of DOX and DAS in the concentration ranges of 4.4–8580 nM and 38–9880 nM, with detection limits of 0.86 and 6.72 nM, respectively. Furthermore, the proposed platform was successfully used for reliable determination of DOX and DAS in urine and blood serum samples with good accuracy and precision. The satisfactory recoveries obtained in blood serum and urine samples showed the potential for application of the proposed platform in quality control in the pharmaceutical industry, clinical analysis and optimization of chemotherapeutic treatment. We found that the established sensor has several advantages in terms of high sensitivity and selectivity, ease of preparation, short analysis time, long-term stability and fast response.
1. Introduction
Cancer is a leading cause of death in the modern world, and severely degrades the quality of life. According to GLOBOCAN 2012 about 14.1 million new cases and 8.2 million deaths occurred worldwide in 2012 due to a variety of cancers. Because of this massive death rate, researchers are trying to find effective therapies to treat cancers in an effective way. Doxorubicin (DOX) (also known as adriamycin) is an anthracycline antibiotic used extensively as a potent anticancer agent for the treatment of various cancers such as breast cancer, ovarian cancer, lung cancer, bladder cancer, cervical cancer, prostate cancer, sarcomas, and Hodgkin's and non-Hodgkin's lymphomas.1,2 DOX inhibits rapid growth of cancer cells by binding with DNA via intercalation between DNA base pairs in order to stop DNA replication.3 However, the effective treatment of DOX is limited by its ability to develop resistance and cardiotoxicity. Also, its excessive levels can cause marrow suppression, and gastrointestinal and dermatological disorders.4 A multi-drug therapy is an effective way to avoid such resistance and undesirable side effects, where the mechanism of action is unique for each drug. In recent years, it has been proved that DOX can be used in combination with other anticancer drugs including docetaxel, vinblastine, cyclophosphamide, 5-fluorouracil and bleomycin.5,6 On the other hand, dasatinib (DAS, sold as sprycel) is a tyrosine kinase inhibitor that targets various kinases including Src, BCR-Abl, FAK and c-Kit.7,8 It is largely recommended for the treatment of chronic myeloid leukemia (CML) and acute lymphocytic leukemia (ALL) in a patient with resistance or intolerance to imatinib therapy.9 In addition, DAS has also been successfully used for the treatment of lung cancer, prostate cancer, ovarian cancer and leukemia.10,11 A combination of DOX and DAS is a promising approach to decrease/inhibit migration, proliferation, cell metabolism and invasion of breast cancer cells.12–14 Furthermore, this combination therapy is also useful for minimizing the undesirable side effects and drug resistance. The determination of these drugs is important in a clinical environment to ensure adequate drug level and for the optimization of treatment. Considering the importance of these drugs in cancer treatment, we propose here a highly sensitive, simple and reliable voltammetric method for individual as well as simultaneous determination of DOX and DAS in urine and blood serum samples using Pd@Pt/MWCNT as the working electrode. The combination of a Pd@Pt core–shell structure and MWCNT leads to a highly efficient sensor with excellence electrocatalytic activity and enhanced electrochemical response towards DOX and DAS. Co-incidentally, there is a recent report published by Alavi-Tabari et al. for simultaneous determination of DOX and DAS based on an ionic liquid and ZnO nanoparticles.15 However, this sensor suffered seriously from sensitivity, reproducibility and stability issues. However, the proposed biosensing platform is way better than the published sensor in terms of low detection limits, wide linear working ranges, operational simplicity, excellent stability for 30 days, outstanding reproducibility and repeatability.To date, a large number of analytical methodologies have been developed for qualitative/quantitative determination of DOX and DAS individually, including high performance liquid chromatography,16,17 capillary electrophoresis18,19 and spectroscopy20,21 for DOX, and high performance liquid chromatography22,23 and spectroscopy24 for DAS. In addition, a few electrochemical methods based on chemically modified electrodes are also reported for individual determination of DOX25,26 and DAS.27,28 However, most of these conventional methods of analysis have limitations of complicated preconcentration steps, low sensitivity and time-consuming procedures. In contrast, modern electrochemical methods offer advantages of high sensitivity and selectivity, simplicity, low cost and user friendly instrumentation. During the past few years, detection methods based on biosensor technologies have attracted more and more attention because of their unique features. Despite the fact that HPLC methods offer the advantage of trace level determination, electrochemical methods can be successfully used as an alternative for the determination of compounds in pharmaceutical and biological samples.29–31
In the recent couple of years, intense research has been focused on development of electrochemical sensors and biosensors employing a range of nanomaterials.32 Electrochemical sensors based on such nanomaterials display characteristic properties such as high sensitivity, selectivity and stability. In present work, novel mesoporous Pd@Pt core–shell nanoparticles supported on MWCNT have been synthesized and employed as a highly sensitive electrochemical sensor for individual as well as simultaneous determination of DOX and DAS. Bimetallic nanoparticles have received immense attention due to their praiseworthy catalytic, electronic, magnetic and optical properties compared to bulk metals and monometallic particles.33 Unfortunately, these nanoparticles are susceptible to aggregation, which causes deterioration of their electrochemical response. Hence, a suitable support material is necessary to provide high surface area for distributing more nanoparticles and avoiding aggregation. In the present case highly conducting MWCNT has been used as a support material as well as a signal amplifier. Several analytical steps are involved in the present study including optimization of experimental parameters, electrochemical investigation of DOX and DAS, construction of calibration plots, real sample analysis, interference studies, and reproducibility and repeatability studies. All these steps have been explained systematically in their respective sections.
2. Experimental section
2.1. Reagents and apparatus
DOX (98%) and DAS (≥99%) were obtained from Aladdin Co., Ltd. (Shanghai, China). Phosphoric acid (≥85%), acetic acid (99.5%) and boric acid (≥99.5%) were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). MWCNT (OD = 20–30 nm, length = 10–30 μm) was purchased from Times Nano (Shanghai, China) and further purified by refluxing in HNO3 at 120 °C for 12 h. H2PtCl6·6H2O and PdCl2 were procured from Aladdin Co., Ltd. (Beijing, China). Pluronic® F127 employed as a structure directing agent was bought from Sigma-Aldrich (Beijing, China). A stock solution of H2PdCl4 (20 mM) was prepared by a published method34 in which 177 mg of PdCl2 was dissolved in 40 μL of HCl (36%) and diluted to 50 mL with ultrapure water. All selected reagents were of analytical-reagent grade and used as received without any further purification. Stock solutions of DOX and DAS were prepared in methanol. When not in use, these solutions were stored in a refrigerator. Electrochemical measurements were performed using PBS (0.1 M, pH 6.0) as a supporting electrolyte; its pH was adjusted with 0.5 M NaOH. The working standard solutions were prepared by appropriate dilution of the stock solution with PBS just before use. Ultra-purified water was used throughout the study (resistivity > 18 MΩ cm).All electrochemical measurements were performed on a CHI 660E electrochemical workstation (CH Instruments, China) with a three-electrode system consisting of Ag/AgCl/3 M KCl as the reference electrode, platinum as the counter electrode and Pd@Pt/MWCNT as the working electrode, respectively. The pH measurements were carried out employing a combined glass electrode with an Ag/AgCl (3 M KCl) reference electrode, which was calibrated time to time with standard buffer solutions. The surface morphologies, elemental mapping, line-scanning and structural studies of the materials were investigated on a Transmission Electron Microscope (TEM, JEOL JEM-2000F) equipped with an energy dispersive X-ray spectrometer (EDAX). X-ray diffraction (XRD) studies of the materials were performed on an X-ray diffractometer (PANalytical X'pert PRO-DY2198, Holland) with a Cu Kα radiation source (λ = 1.5406 Å). N2 adsorption–desorption isotherms were obtained on a Micromeritics ASAP 2010 Analyzer. X-ray photoelectron spectroscopy (XPS) was performed to study the surface chemistry of the materials on VG Multilab 2000 instruments using Al Kα radiation.
2.2. Synthesis of Pd@Pt/MWCNT
In a typical synthesis of Pd@Pt/MWCNT, 20.0 mg Pluronic® F127 and 10.0 mg MWCNT were dispersed in 5.0 mL water and sonicated for 1 h. To this solution, 1 mL 20 mM H2PtCl6 and 2.0 mL 20 mM H2PdCl4 aqueous solutions were added and mixed thoroughly. In the next step, 1 mL 0.25 mM ascorbic acid was added slowly and the reaction mixture was stirred for 2 h at 60 °C. Finally, a black precipitate was collected and washed several times with ultrapure water and acetone. The precipitate was dried at room temperature and preserved for further characterization. For comparison, Pd/MWCNT and Pt/MWCNT were prepared by the same method.2.3. Fabrication of the sensing platform
A glassy carbon disk electrode (GCE, 2 mm in diameter) was polished to a mirror like finish with 0.05 μm α-alumina powder on a micro-cloth polishing pad followed by sonication in absolute alcohol and ultrapure water for 5 min, respectively. The dispersion was prepared by suspending 2.0 mg Pd@Pt/MWCNT in 2.0 mL water and then sonicated for 30 min in order to obtain a uniform dispersion. 7.0 μL suspension was coated (drop-cast) onto the pre-cleaned GCE, which was dried at room temperature for 2 h. The excessive thick film of Pd@Pt/MWCNT could affect the electrochemical response due to increased interface resistance. Therefore, the film thickness was optimized by controlling the amount of coating in the range of 3–15 μL. The best response was obtained when 7.0 μL suspension was coated and therefore was chosen for further studies. For comparison, MWCNT, Pd/MWCNT and Pt/MWCNT were fabricated by following the same procedure.2.4. Measurement procedures
The electrochemical behaviour of the drugs was investigated by CV and AdSSWV in a cell with a three-electrode setup. CV was performed within the range of 0.25–1.1 V at a scan rate of 50 mV s−1. The AdSSWV measurements involve two steps: (i) accumulation of the two drugs at the electrode surface for 90 s (pre-concentration time) at OCP (pre-concentration potential) and (ii) measurement of the voltammetric signal within the range of 0.3–1.0 V. After each measurement, the surface of the electrode was renewed by cycling in PBS (0.1 M, pH 6.0) for five cycles at a scan rate of 100 mV s−1. The calibration plots were constructed by measuring the electrochemical response at various concentrations and used for the determination of the linear working range (LWR), limit of detection (LOD) and limit of quantification (LOQ). LOD and LOQ values were calculated using the following equations: LOD = 3 × (σ/m); LOQ = 10 × (σ/m), where σ stands for standard deviations of the blank for ten measurements and m is the slope of the calibration plot. Drug-free urine and blood serum samples were collected from healthy individuals from the Hospital of Huazhong University of Science and Technology, China. All experiments with human serum and urine were performed in compliance with the relevant laws in China and institutional guidelines, and after the approval of the ethical committee of the School of Materials Science and Engineering, Huazhong University of Science and Technology. Informed consent was obtained for any experimentation with human subjects. All samples were stored in a refrigerator for further use. The samples were centrifuged at 2000 rpm for 15 min and the supernatant was diluted with the supporting electrolyte (1 mL blood serum/urine in 10 mL PBS). The standard addition method was used for the determination of DOX and DAS in real samples.3. Results and discussion
3.1. Characterization of Pd@Pt/MWCNT
The morphologies of the materials were observed by TEM analysis. Fig. 1a depicts TEM micrographs of Pd@Pt/MWCNT in which the majority of the Pd@Pt nanoparticles are uniformly deposited on the MWCNT surface without any aggregation with an average diameter of 37.3 nm (Fig. S1†). Additionally, the HRTEM image (Fig. 1b) shows a highly crystalline structure with a lattice spacing of 0.225 nm corresponding to the (111) planes of face-centred cubic (fcc) Pd and Pt.35 It is worth highlighting that highly aggregated irregular nanoparticles were formed in the absence of MWCNT. Therefore, it is necessary to incorporate MWCNT into the Pd@Pt structure for uniform deposition and high mass loading. EDS analysis shows the presence of Pd, Pt and C in the Pd@Pt/MWCNT (Fig. 1c(i)). The core–shell structure is confirmed by high-angle annular dark-field scanning transmission electron microscopy-energy dispersive X-ray spectroscopy (HAADF-STEM-EDS) and line scanning data (Fig. 1c(iii)). It can be seen from Fig. 1c(iii) that the PdNPs are concentrated at the centre and PtNPs are at the outer edge of the PdNPs, forming a core–shell structure. Additionally, the core–shell structure is further confirmed by the HAADF-STEM-elemental line profile (Fig. 1d). The formation mechanism of Pd@Pt is explained in the ESI (1).†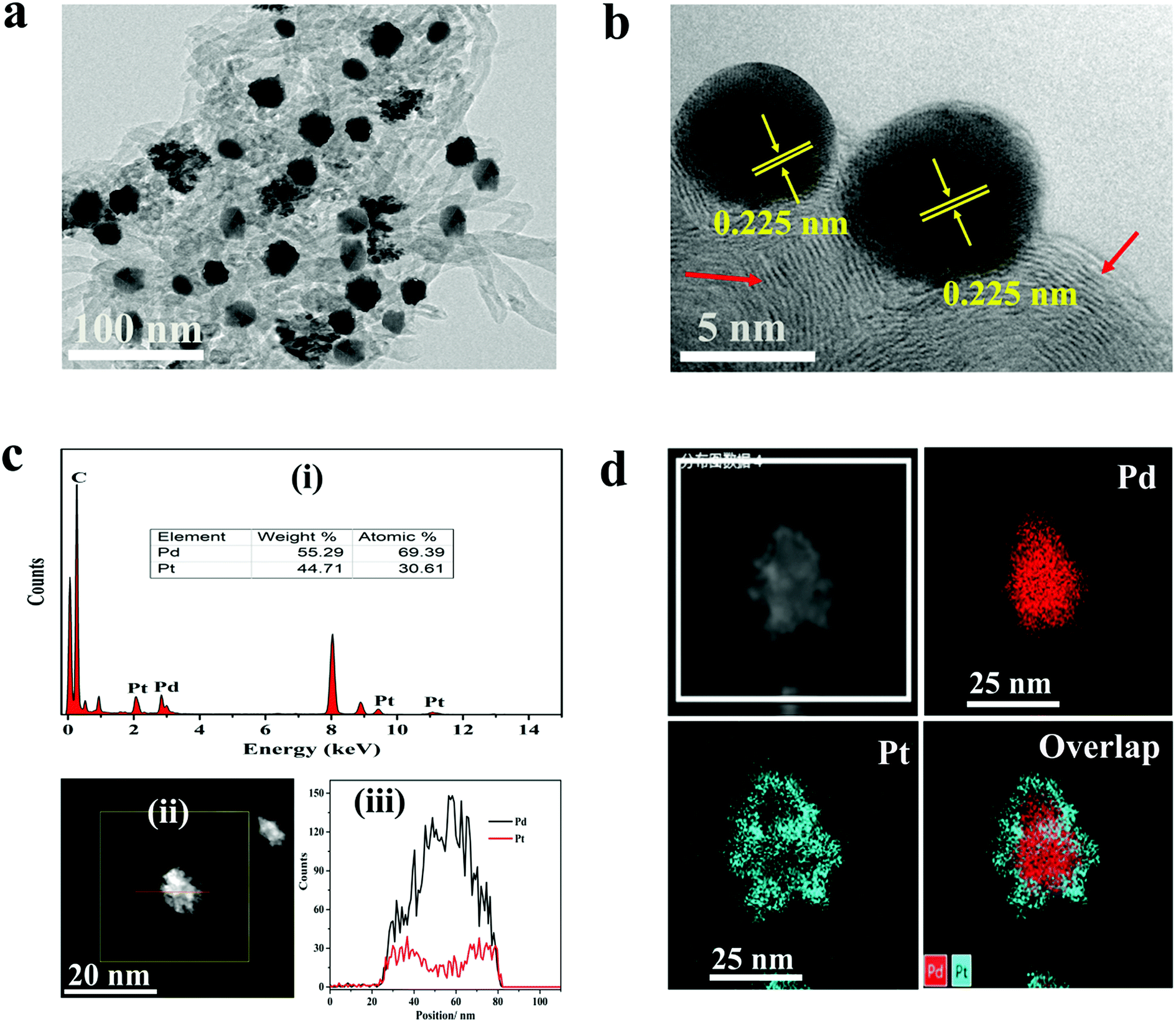 | ||
| Fig. 1 (a) TEM and (b) HRTEM images of Pd@Pt/MWCNT (the red arrow shows the MWCNT surface). (c) (i) EDS spectrum, (ii) STEM image and (iii) elemental line profile of a Pd@Pt nanoparticle. (d) HAADF-STEM-EDS mapping images of Pd, Pt and Pd@Pt. | ||
The crystal structure of the materials was characterised by XRD analysis (Fig. 2a). The Pd@Pt/MWCNT shows characteristic diffraction peaks at 39.9°, 46.4°, 67.9°, 81.8° and 86.2°, which are mainly ascribed to the fcc structure of Pd (JCPDS no. 46–1043) and Pt (JCPDS no. 04–0802). Note that the diffraction peak at 26.2° corresponds to the (002) crystal plane of MWCNT. The diffraction peaks obtained are sharp and intense, confirming high purity and better crystallinity of the Pd@Pt/MWCNT. It is difficult to find any difference in the XRD patterns of Pd and Pt due to a high lattice match.36 The mesoporous structure of Pd@Pt is confirmed from the N2 adsorption–desorption isotherm and the average pore size was found to be 13.62 nm (Fig. 2b). The BET specific surface area of the Pd@Pt/MWCNT is 142.87 m2 g−1.
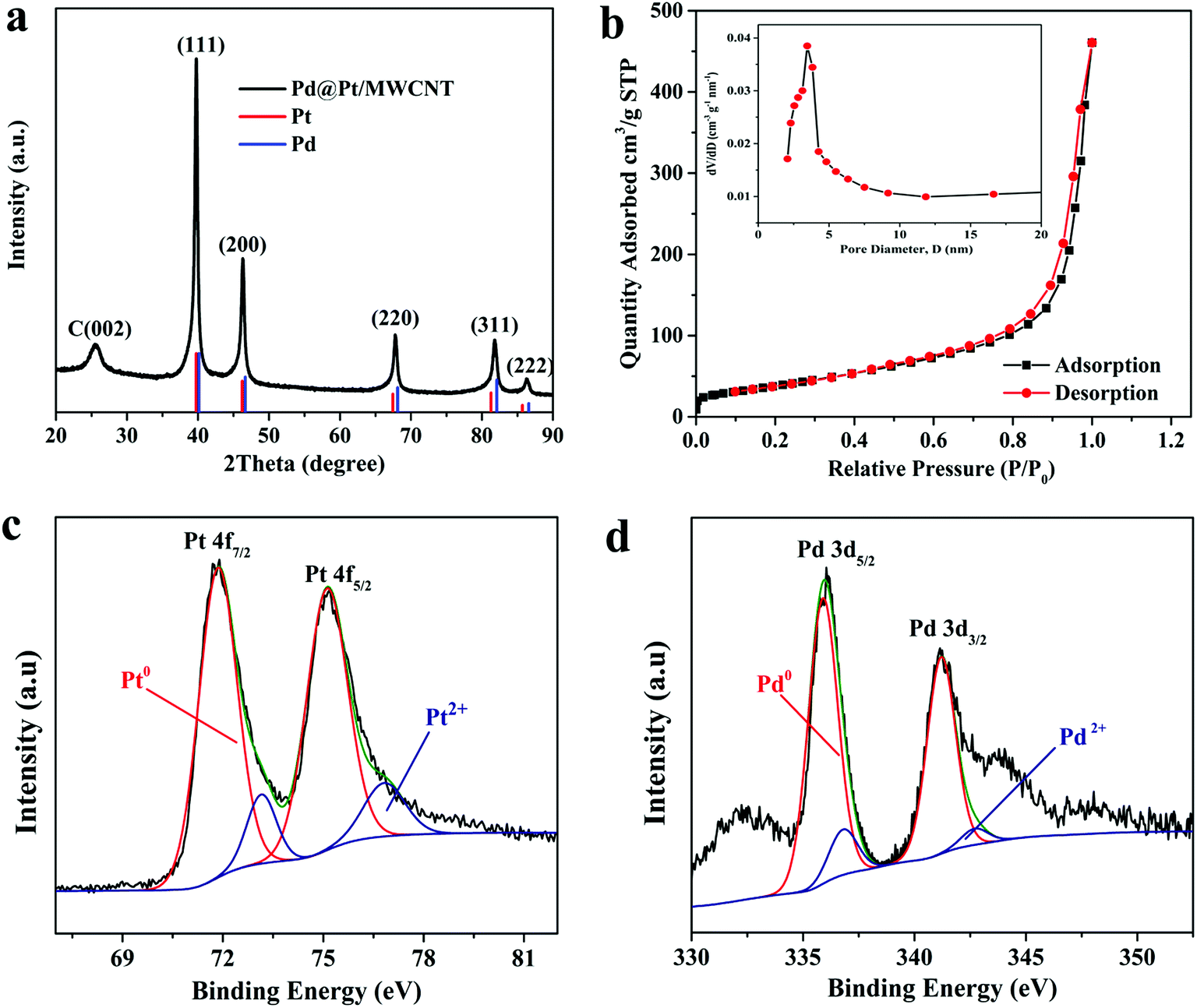 | ||
| Fig. 2 (a) XRD pattern and (b) N2 adsorption–desorption isotherm of Pd@Pt/MWCNT (inset: BJH pore size distribution curve). High-resolution XPS spectra of (c) Pt 4f and (d) Pd 3d. | ||
X-ray photoelectron spectroscopy (XPS) was performed to study the surface chemical compositions of the materials. The Pt 4f spectrum (Fig. 2c) shows two pairs of peaks. The strong peaks at 71.51 and 75.02 eV correspond to Pd0 and the weak peaks at 73.01 and 76.71 eV are indexed to Pt2+.37 Similarly, the Pd 3d spectrum (Fig. 2d) shows strong peaks at 336.02 and 341.01 and weak peaks at 337.22 and 342.71 eV, corresponding to Pd0 and Pd2+, respectively.37 From this study it is concluded that metallic Pt0 is the major species of the electrode system. However, the presence of Pt2+ is either due to incomplete reduction of metal precursors or surface oxidation of materials under ambient conditions.
3.2. Electrochemical characterization of Pd@Pt/MWCNT
The active surface area of the electrodes was calculated by employing the Randles–Sevcik equation using 10 mM [Fe (CN)6]3–4− in 0.5 M KCl as a redox probe. Fig. 3a shows CVs of the [Fe(CN)6]3–4− redox couple at GCE, MWCNT, Pd/MWCNT, Pt/MWCNT and Pd@Pt/MWCNT electrodes. In this respect, the largest peak current was obtained for Pd@Pt/MWCNT among the five electrodes due to the excellent properties of the material such as high surface area, superior electrocatalytic activity and excellent conductivity. Additionally, the peak separations between GCE and Pd@Pt/MWCNT changed from 79 mV to 69 mV with a shift in peak potential towards less positive potential. The surface areas for GCE, MWCNT, Pd/MWCNT, Pt/MWCNT and Pd@Pt/MWCNT were found to be 0.018, 0.025, 0.029, 0.032 and 0.038 cm2, respectively.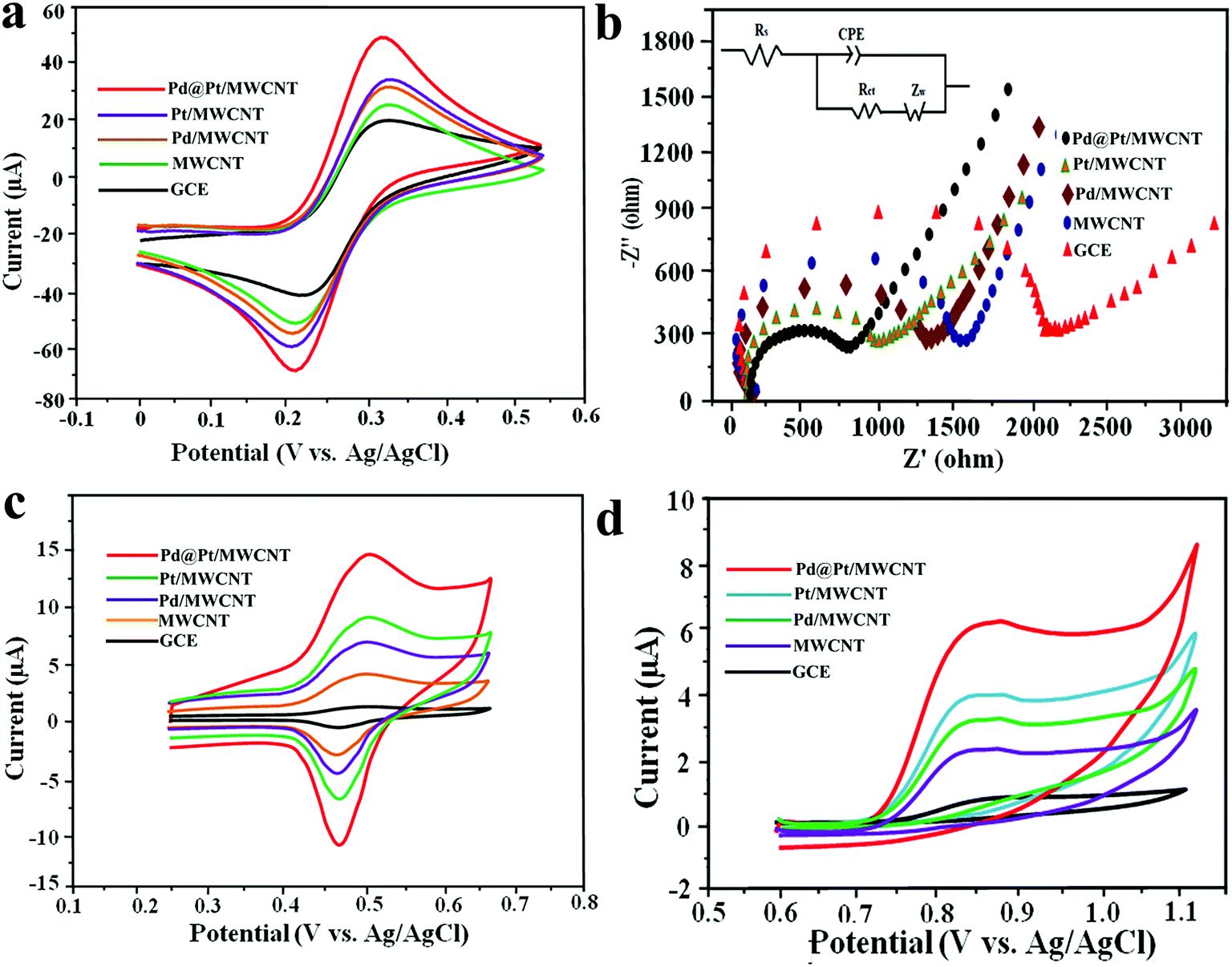 | ||
| Fig. 3 (a) Cyclic voltammetric curves and (b) Nyquist plot of 10 mM [Fe(CN)6]3−/4− in 0.5 M KCl at the bare GCE, MWCNT, Pd/MWCNT, Pt/MWCNT and Pd@Pt/MWCNT [inset: equivalent circuit used for data fitting]. Cyclic voltammetric curves of (c) 8.0 μM DOX and (d) 4.95 μM DAS at the bare GCE, MWCNT, Pd/MWCNT, Pt/MWCNT and Pd@Pt/MWCNT in PBS (0.1 M, pH 6.0) with a scan rate of 50 mV s−1. | ||
Electrochemical impedance spectroscopy (EIS) is widely used to obtain information on the impedance changes after electrode modification. Fig. 3b depicts Nyquist plots for GCE, MWCNT, Pd/MWCNT, Pt/MWCNT and Pd@Pt/MWCNT electrodes employing 10 mM [Fe(CN)6]3–4− in 0.5 M KCl. It is observed that as we move from GCE to Pd@Pt/MWCNT, the diameter of the semicircle is decreased drastically, indicating low charge transfer resistance (Rct). Moreover, the Rct values obtained for GCE, MWCNT, Pd/MWCNT, Pt/MWCNT and Pd@Pt/MWCNT were 1.866, 1.435, 1.225, 0.897, and 0.712 kΩ, respectively. The lowest Rct for Pd@Pt/MWCNT suggested a high electron transfer rate at the electrode surface and outstanding electrical conductivity of the material.
3.3. Electrochemical behaviour of DOX and DAS at Pd@Pt/MWCNT
The electrocatalytic behaviour of 8.0 μM DOX and 4.95 μM DAS was investigated by CV at GCE, MWCNT, Pd/MWCNT, Pt/MWCNT and Pd@Pt/MWCNT. For example, Fig. 3c and d illustrate CVs recorded for anticancer agents in the potential ranges of 0.25–0.65 and 0.6–1.1 V at a scan rate of 50 mV s−1. DOX and DAS exhibit strong anodic peaks at ≈0.5 V and ≈0.84 V, respectively. Additionally, DOX showed a reduction peak at 0.46 V (quasi-reversible behaviour). Moreover, the electrochemical response of both drugs was significantly enhanced at the Pd@Pt/MWCNT electrode as compared to the bare GCE due to the high active surface area, synergistic effect of Pd and Pt, unique mesoporous core–shell structure and good electrical conductivity of the materials. The oxidation of DAS is found to be completely irreversible due to the absence of reduction peak signals in the reverse scan. After modification of the GCE, a negative shift in the peak potential was observed, suggesting a higher electron transfer rate at the electrode surface and superior electrocatalytic behaviour of the Pd@Pt/MWCNT towards DOX and DAS. Due to the high electron transfer rate at the electrode surface, enhanced electrochemical response and superior electrocatalytic activity, we chose Pd@Pt/MWCNT as a highly sensitive sensor for DOX and DAS.The influence of the pH of the supporting electrolyte on the electrochemical behaviour of 2.1 μM DOX and 3.1 μM DAS was investigated in Britton–Robinson (BR) buffer over a wide range of pH, viz., 2.0–9.0, through differential pulse voltammetry (DPV, n = 3). It was observed that with raising the pH, the peak potentials shifted to less positive values, demonstrating participation of protons in the oxidation reactions (Fig. 4a and b). The peak current of DAS gradually increases from pH 2.0 to 6.0 and decreases afterwards due to peak broadening. DOX gives well-defined oxidation peaks at all pH, except at higher pH 9.0, where peak broadening was observed. The peak current of DOX increases from pH 2.0 to 6.0 and there was a high loss in current response between pH 7.0 and 9.0. Hence, pH 6.0 of the supporting electrolyte was chosen as the optimum medium for simultaneous determination of both drugs at which the highest current magnitude and well-defined peaks were obtained.
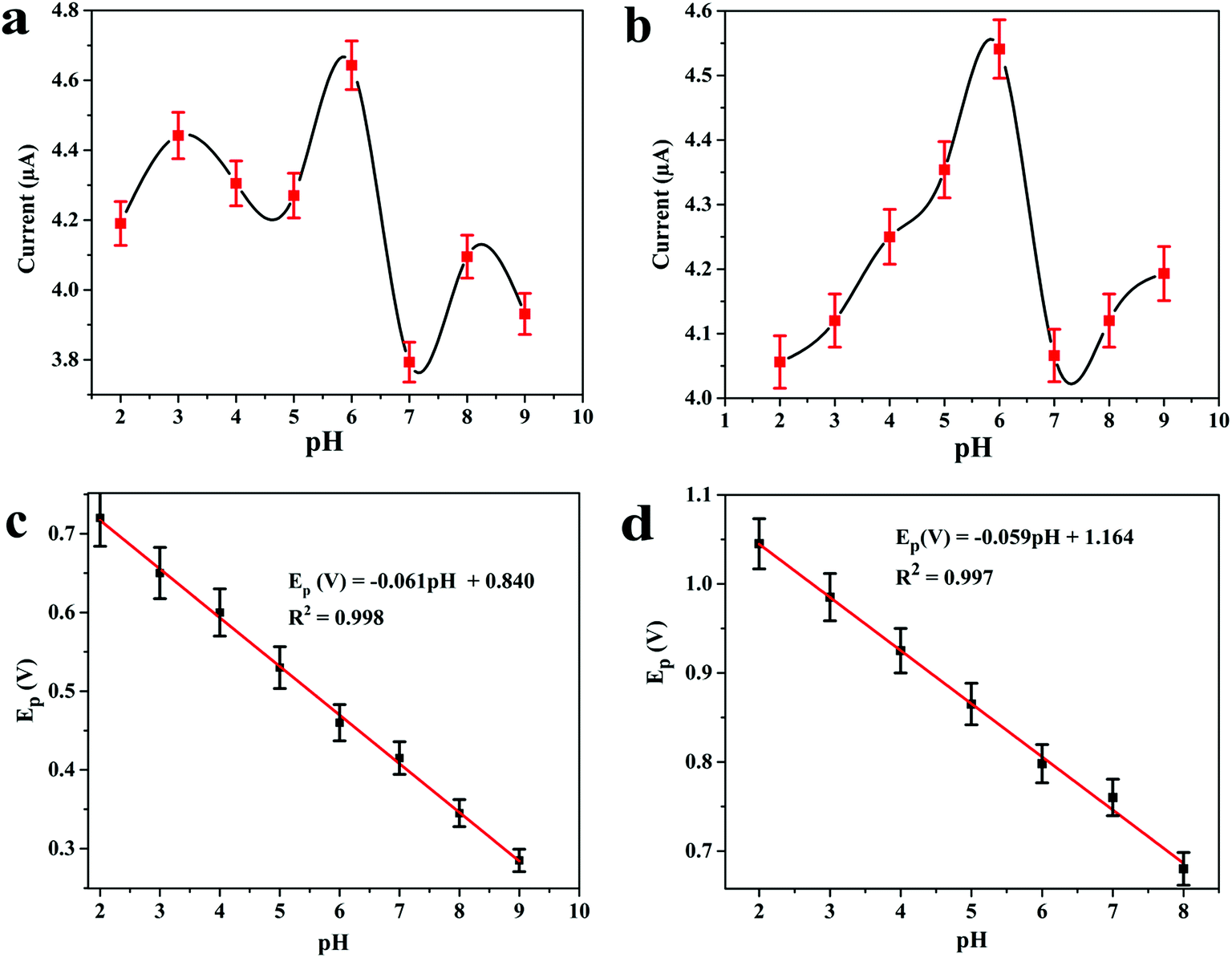 | ||
| Fig. 4 Effect of pH on differential pulse voltammograms of (a) 2.1 μM DOX and (b) 3.1 μM DAS in 0.04 M B.R. buffer and variation in the peak potentials of (c) DOX and (d) DAS with pH of the solution at Pd@Pt/MWCNT; step potential = 5 mV and modulation amplitude = 50 mV. | ||
Additionally, the regression equations of the plot Epvs. pH for DOX and DAS can be presented as Ep(V) = −0.061 pH + 0.840 (R2 = 0.998) and Ep(V) = −0.059 pH + 1.164 (R2 = 0.997), respectively. The slopes of the Epvs. pH plots were found to be −0.061 and −0.059 V per pH (close to the Nernstian slope of 0.059 V per pH), confirming that an equal number of protons and electrons are involved in the electrochemical reaction of both drug (Fig. 4c and d). In addition to this, the effect of other buffers such as phosphate and citrate phosphate was also investigated at pH 6.0. It is important to note that the peak current was further enhanced when 0.1 M phosphate buffer was employed (data not shown). Hence, for further measurements, 0.1 M phosphate (pH 6.0) buffer was employed.
In the next step, the effect of scan rate (ν) on oxidation peak current of 3.1 μM DOX and 0.25 μM DAS was investigated by CV employing various scan rates ranging from 10 to 170 mV s−1. The peak currents were found to increase with increasing scan rate, with a shift in peak potential towards a more positive window, confirming the irreversible nature of the electrode processes (Fig. 5a and b). The dependence of the peak current on the scan rate (ν) and square root (ν1/2) of the scan rate was plotted to find the nature of the electrode processes. The relationship between the scan rate (ν) and the peak current (Ip) can be expressed by the regression equations Ip (μA) = 0.181ν (mV s−1) + 4.418 (R2 = 0.995, DOX) and Ip (μA) = 0.052ν (mV s−1) + 0.540 (R2 = 0.996, DAS). Based on the above equations, a linear plot of peak current vs. scan rate was obtained, indicating adsorption controlled processes of DOX and DAS (Fig. 5c and d).
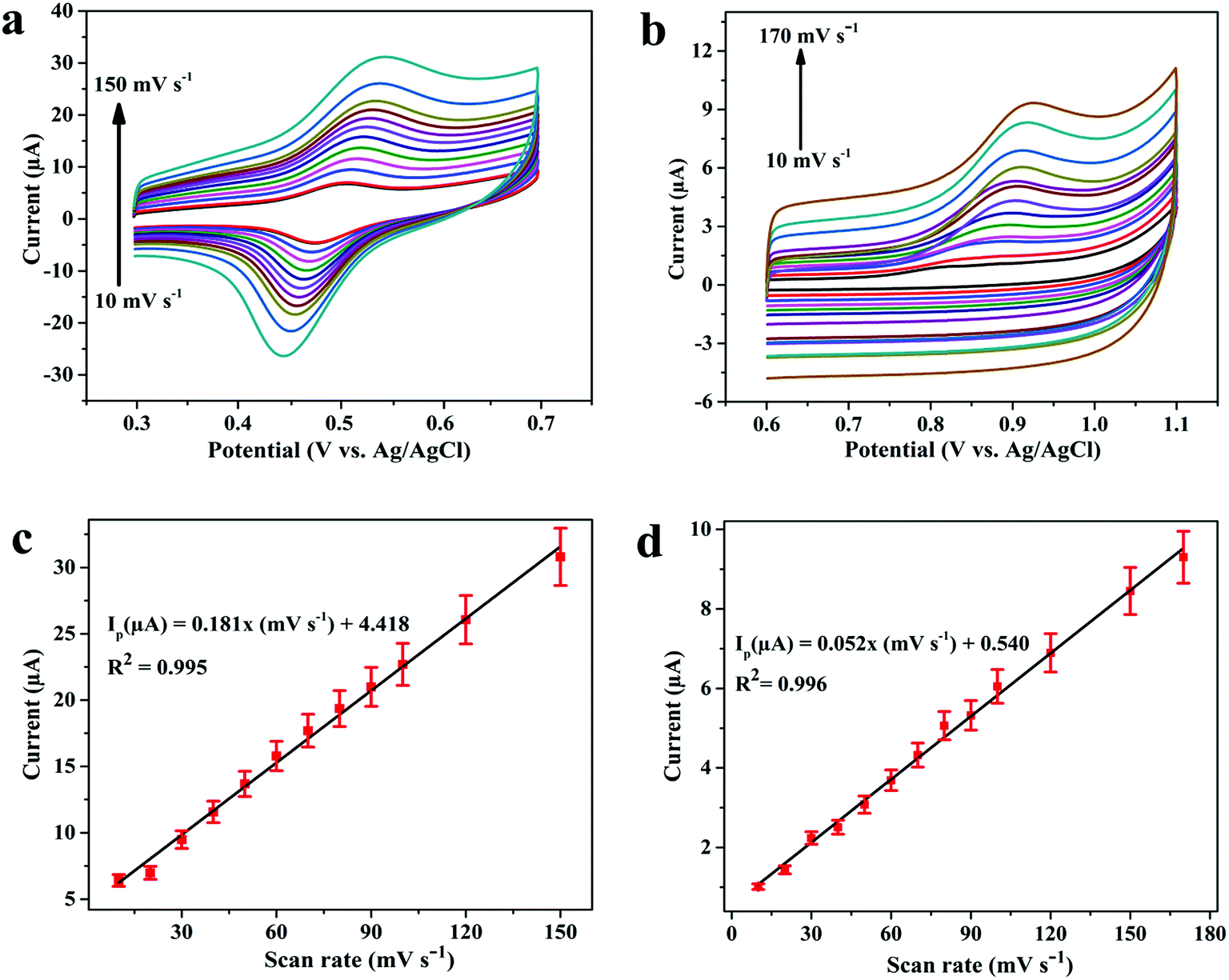 | ||
| Fig. 5 Cyclic voltammograms for (a) 3.1 μM DOX and (b) 0.25 μM DAS at Pd@Pt/MWCNT in PBS (0.1 M, pH 6.0) employing varying scan rates of 10–170 mV s−1. A linear relation of peak current vs. scan rate for (c) DOX and (d) DAS, respectively. | ||
To calculate the number of electrons involved in the electrochemical reaction of DOX, Laviron theory was used, and is given by the following equation:38
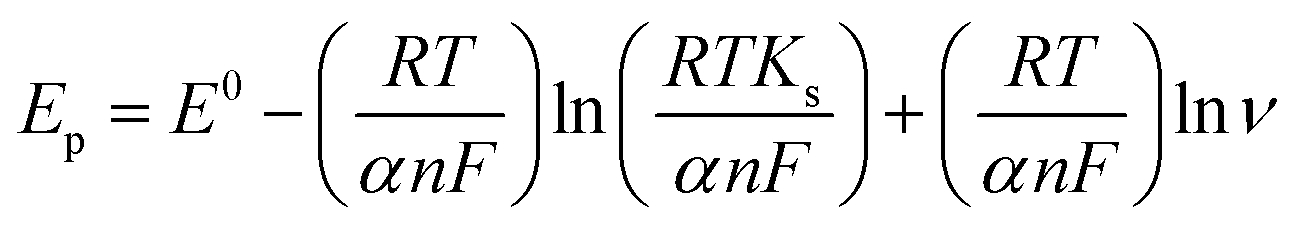 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 480 C mol−1, T = 298 K and R = 8.314 J K−1. The slope of the linear plot of Epvs. lnν was used to calculate the number of electrons using eqn (1). By using α = 0.5 and the slope value (Ep = 0.023 lnν + 0.592) (Fig. S3†), the number of electrons transferred in the electrochemical reaction of DOX was found to be approximately 2. We know from the slope of the Epvs. pH plot that an equal number of electrons and protons are involved in the electrochemical reaction of DOX and DAS. The oxidation of DOX is based on the transfer of two electrons and two protons with formation of quinone (Scheme 1a).39,40 On the other hand, oxidation of DAS is a two-step process (Scheme 1b). The process involves transfer of two electrons and two protons from a sulphur atom (in the presence of H2O) yielding the sulphoxide, followed by an electrochemical reaction with water to give sulphone.28
480 C mol−1, T = 298 K and R = 8.314 J K−1. The slope of the linear plot of Epvs. lnν was used to calculate the number of electrons using eqn (1). By using α = 0.5 and the slope value (Ep = 0.023 lnν + 0.592) (Fig. S3†), the number of electrons transferred in the electrochemical reaction of DOX was found to be approximately 2. We know from the slope of the Epvs. pH plot that an equal number of electrons and protons are involved in the electrochemical reaction of DOX and DAS. The oxidation of DOX is based on the transfer of two electrons and two protons with formation of quinone (Scheme 1a).39,40 On the other hand, oxidation of DAS is a two-step process (Scheme 1b). The process involves transfer of two electrons and two protons from a sulphur atom (in the presence of H2O) yielding the sulphoxide, followed by an electrochemical reaction with water to give sulphone.28
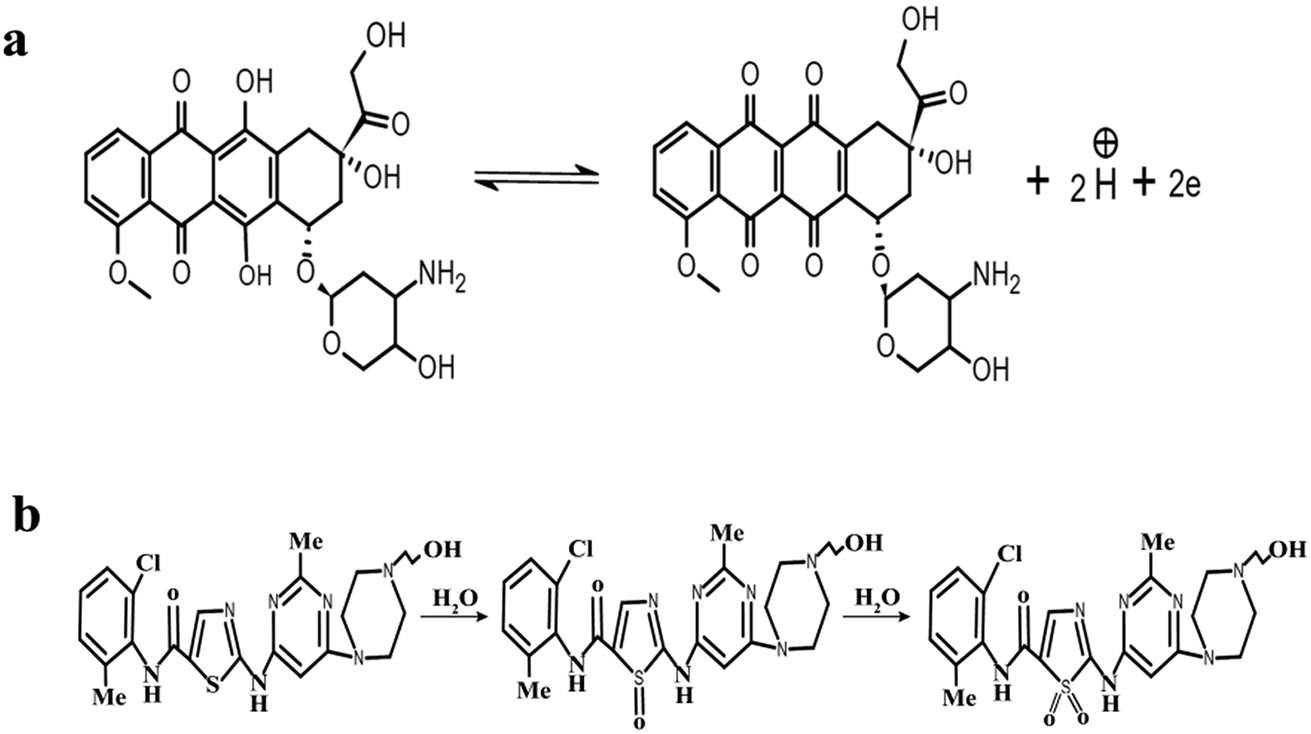 | ||
| Scheme 1 The electrochemical reaction mechanisms of (a) DOX and (b) DAS. | ||
3.4. Analytical performance of Pd@Pt/MWCNT
The optimization of SWV parameters is explained in the ESI (2).† The main focus of the present study is to determine DOX and DAS at low concentration using AdSSWV. In this respect, the calibration plots were constructed and analytical performance was accessed under the optimized conditions (n = 5). In case 1, the concentration of DOX was increased linearly in the presence of a fixed concentration of DAS and vice versa (Fig. 6a and b). Two well-resolved oxidation peaks at ≈0.468 and ≈0.792 V were obtained for DOX and DAS with a potential separation of ≈ 0.324 V. In case 2, the concentrations of both drugs were increased simultaneously (Fig. 6c). In both the cases, the peak current increased proportionally with the concentration (C) of drugs. Additionally, individual determination was also performed separately by the same method. The analytical results for all above cases are shown in Table S1.† The comparison of simultaneous determination with their individual determinations proved that the simultaneous determination is as efficient as their individual determinations. The reasons for enhanced sensitivity of Pd@Pt/MWCNT towards DOX and DAS can be explained on the basis of the following factors: (a) a unique mesoporous core–shell structure displays attractive characteristics such as larger specific surface area, superior electrocatalytic activity, high electrical conductivity and excellence adsorptive ability; (b) highly conducting MWCNT prevents agglomeration of Pd@Pt nanoparticles and also acts as a signal amplifier. Thus, the synergistic effect of Pd@Pt and MWCNT leads to facile oxidation of DOX and DAS on Pd@Pt/MWCNT. The calibration plots presented in Fig. 6d are highly linear in the investigated concentration ranges (4.4–8580 nM for DOX and 38–9880 nM for DAS). The corresponding linear regression equations obtained for DOX and DAS can be presented as Ip (μA) = 0.019C (10−8 M) + 1.501 (R2 = 0.991) and Ip (μA) = 0.013C (10−8 M) + 1.228 (R2 = 0.991), respectively.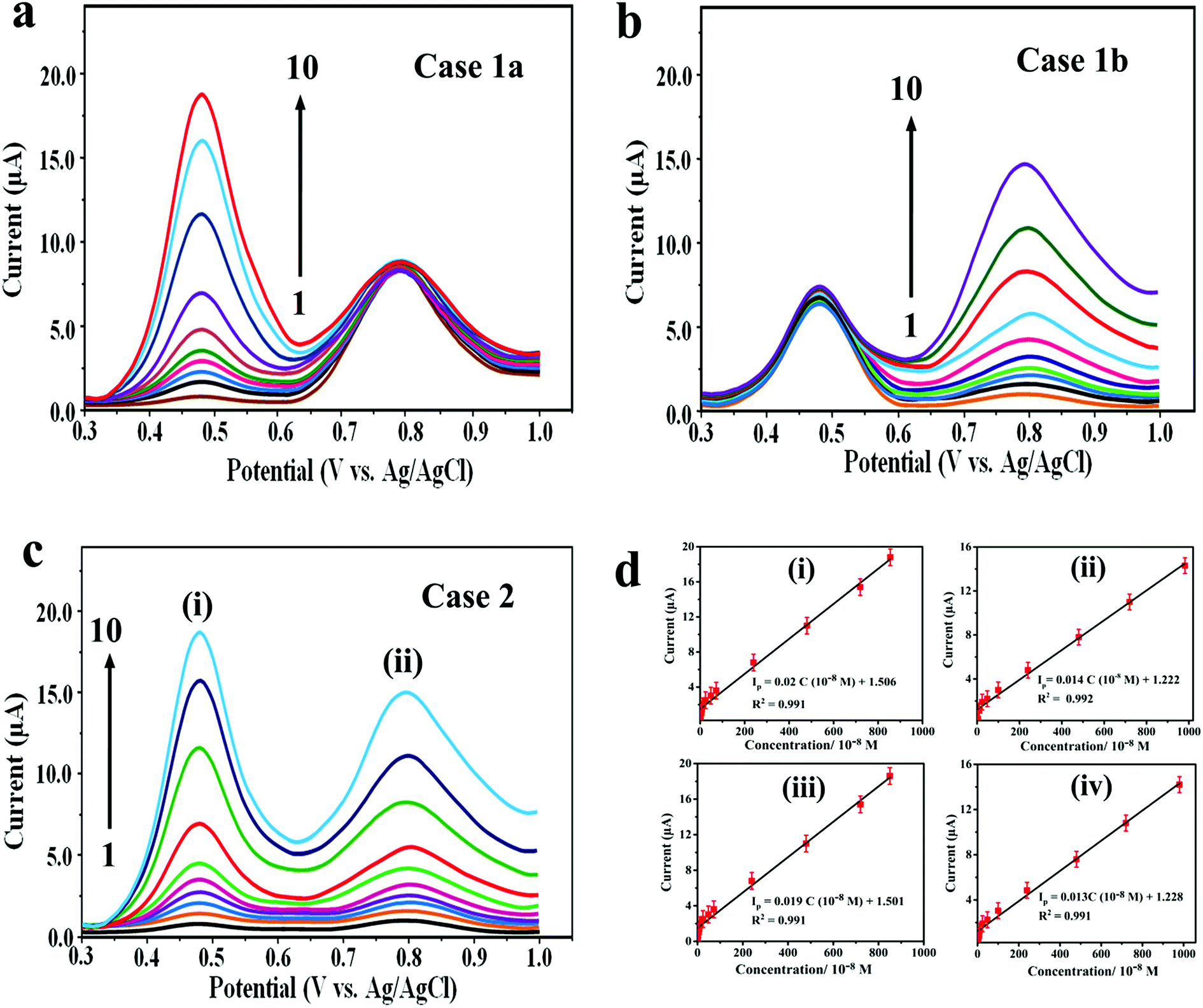 | ||
| Fig. 6 AdSSWV curves for different concentrations of (a) DOX (0.42 × 10−8 to 852 × 10−8 M) in the presence of a fixed concentration of 4.1 μM DAS and (b) DAS (3.75 × 10−8 to 982 × 10−8 M) in the presence of a fixed concentration of 2.4 μM DOX. (c) AdSSWV curves for the mixture containing (i) DOX (0.44 × 10−8 to 858 × 10−8 M) and (ii) DAS (3.8 × 10−8 to 988 × 10−8 M) at the Pd@Pt/MWCNT electrode. (d) The corresponding calibration plots for (i) DOX (case 1a), (ii) DAS (case 1b), (iii) DOX (case 2) and (iv) DAS (case 2). | ||
Based on the calibration plots, the LODs obtained for the simultaneous determination of DOX and DAS were 0.86 and 6.72 nM, respectively. Similarly, LODs and LWRs obtained for individual determination of DOX and DAS were 0.73 and 5.83 in the linear concentration range of 3.8–8410 and 37–9720 nM, respectively (Table S1†). The LOQs obtained for simultaneous determination of DOX and DAS were 2.87 and 22.4 nM, respectively. Similarly, the LOQs obtained for individual determination of DOX and DAS were 2.43 and 19.4 nM, respectively. Additionally, the Pd@Pt/MWCNT electrode displayed a high sensitivity of 1.9 μA μM−1 and 1.3 μA μM−1 for simultaneous determination of DOX and DAS, respectively.
Table S2† illustrates a comparison of the proposed study with literature methods reported by some research groups. It was exciting to discover that the developed platform offers many advantages over previously reported procedures in term of unique simultaneous determination of these drugs, wider linear ranges, efficiency, operational simplicity and high stability. Although the DNA sensor based on ds–DNA/Au [12] could be used for sensitive determination of DOX with an LOD of 1 pM, the fabrication process is too complicated and time consuming with a limited working range. Additionally, the stability of the sensor is a big issue due to instability of DNA. The proposed sensing platform exhibited the lowest limit of detection for DAS.
3.5. Interference, stability, reproducibility and repeatability studies
The selectivity of the proposed electrochemical platform was assessed in the presence of potential interfering compounds found in biological fluids. For this purpose, the influence of some inorganic, organic and biological compounds on the electrochemical response of 0.7 μM DOX and 1.0 μM DAS was investigated under the optimum conditions (Fig. 7a). Firstly, the peak current intensities of DOX and DAS were measured and fixed as the 100% value. In the next step, the potential interferents such as BSA, glucose, sucrose, lactose, ascorbic acid, urea, aspartic acid, KCl and NaCl (1 mM each) were added into a solution containing DOX and DAS. Secondly, the peak current intensities of DOX and DAS in the presence of interferents were measured at the Pd@Pt/MWCNT electrode. Interestingly, it was found that these interfering compounds did not affect the response of DOX and DAS considering the change of 5% (tolerance limit). These results proved that the proposed platform is highly selective towards anticancer drugs as compared to the reported sensor.15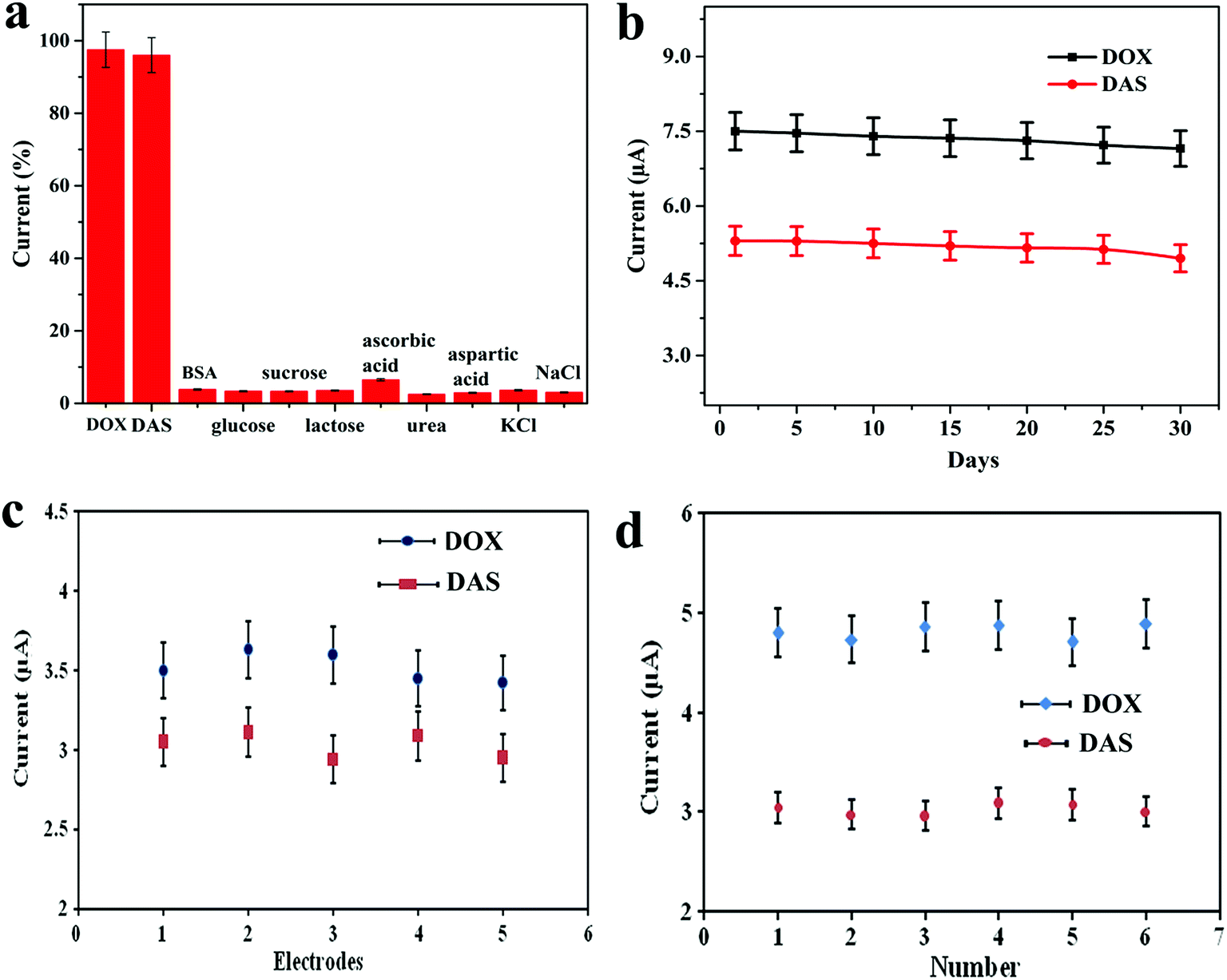 | ||
| Fig. 7 (a) The oxidation peak current intensities of Pd@Pt/MWCNT in 0.1 M PBS (pH 6.0) containing various interferents. (b) Storage stability of the Pd@Pt/MWCNT electrode over 30 days for 3.5 μM DOX and 2.9 μM DAS. Voltammetric conditions: Eacc = OCP, tacc = 90 s, f = 5 Hz, a = 30 mV, ΔEs = 4 mV and equilibrium time = 10 s. (c) Reproducibility of the five Pd@Pt/MWCNT electrodes for 0.7 μM DOX and 1 μM DAS. (d) Repeatability of the Pd@Pt/MWCNT for six measurements of DOX and DAS (1 μM each). | ||
The long-term stability of the biosensor was tested by keeping Pd@Pt/MWCNT at ambient temperature for 30 days. The sensor retained ≈95.5% of its initial response (on an average for both drugs) even after 30 days, confirming that our platform has an appreciable stability (Fig. 7b). To test the reproducibility of the Pd@Pt/MWCNT, five electrodes were fabricated using the same method and they were tested for 0.7 μM DOX and 1.0 μM DAS. Employing AdSSWV, the electrodes showed a low RSD value of 3.1%, proving the acceptable reproducibility of the platform (Fig. 7c). After that, the repeatability of the Pd@Pt/MWCNT was checked by examining DOX and DAS (1.0 μM each) six times. The RSD of peak current measurements was found to be 2.2%, indicating good repeatability of the platform (Fig. 7d). Subsequently, intra-day and inter-day (over a period of 5 days) repeatability of the platform was checked and the results are showcased in Table S3.† As can be seen, the method provides good repeatability and precision under the optimized conditions. These outcomes indicated that the Pd@Pt/MWCNT electrode is reusable and durable as compared to reported sensors; thus the established method is efficient for DOX and DAS determination.
3.6. Analysis of complex biological samples
The practical applicability of the platform was validated by determination of both drugs in untreated human blood serum and urine samples under optimal conditions. Next, varying amounts of known DOX and DAS were spiked in drug free urine and blood serum samples. The voltammograms were recorded for simultaneous and individual determination. The recoveries and RSD obtained using the standard addition method are in the range of 98.39–100.57% and 1.86–2.23%, respectively (Table 1 and Table S4†). According to the above results, Pd@Pt/MWCNT can be used effectively for simultaneous and individual detection of both drugs without any important matrix interference, proving the high feasibility and reliability of the platform. Thus, we can declare that the platform is useful for these drugs in samples with complex matrices.| Sample | DOX | DAS | ||||
|---|---|---|---|---|---|---|
| Spiked (10−7 M) | Detected (10−7 M) | % R ± RSD | Spiked (10−7 M) | Detected (10−7 M) | % R ± RSD | |
| Urine sample 1 | Not spiked | Not found | — | Not spiked | Not found | — |
| 0.45 | 0.443 | 98.44 ± 1.92 | 0.38 | 0.378 | 99.47 ± 1.96 | |
| 0.82 | 0.815 | 99.39 ± 1.98 | 0.78 | 0.774 | 99.23 ± 2.02 | |
| 1.34 | 1.33 | 99.25 ± 2.16 | 1.3 | 1.3 | 100.0 ± 2.06 | |
| Urine sample 2 | Not spiked | Not found | — | Not spiked | Not found | — |
| 0.31 | 0.305 | 98.39 ± 1.86 | 0.48 | 0.474 | 98.75 ± 2.08 | |
| 0.68 | 0.679 | 99.85 ± 2.18 | 0.83 | 0.825 | 99.39 ± 1.92 | |
| 1.1 | 1.102 | 100.18 ± 1.97 | 1.35 | 1.343 | 99.48 ± 2.1 | |
| Blood serum sample 1 | Not spiked | Not found | — | Not spiked | Not found | — |
| 0.43 | 0.426 | 99.06 ± 2.03 | 0.52 | 0.523 | 100.57 ± 1.9 | |
| 0.76 | 0.758 | 99.73 ± 2.2 | 0.83 | 0.828 | 99.76 ± 2.11 | |
| 1.3 | 1.28 | 98.5 ± 2.12 | 1.4 | 1.394 | 99.57 ± 2.05 | |
| Blood serum sample 2 | Not spiked | Not found | — | Not spiked | Not found | — |
| 0.48 | 0.479 | 99.79 ± 2.23 | 0.36 | 0.361 | 100.28 ± 2.06 | |
| 0.78 | 0.783 | 100.38 ± 2.18 | 0.65 | 0.648 | 99.69 ± 2.13 | |
| 1.3 | 1.3 | 100.0 ± 2.2 | 1.2 | 1.19 | 99.17 ± 1.96 | |
4. Conclusion
Herein, highly sensitive individual and simultaneous electrochemical determination of DOX and DAS is introduced on a Pd@Pt/MWCNT platform. The Pd@Pt/MWCNT is prepared by a simple one-pot synthesis method and characterized by XRD, XPS, TEM, BET and electrochemical methods including CV, DPV, EIS and AdSSWV. The experimental parameters were carefully optimized in order to get a better electrochemical response. The developed platform demonstrated excellent electrochemical response towards DOX and DAS with LODs of 0.86 nM and 6.72 nM, respectively. The established sensor is superior to reported sensors in terms of low detection limit, wide linear working range and sensitivity. It should be highlighted that the outstanding performance of the Pd@Pt/MWCNT was associated with the excellent properties of the material such as high electrical conductivity, synergistic effect between Pd, Pt and MWCNT, large effective surface area and unique mesoporous core–shell structure, which facilitate fast electron and mass transfer. Additionally, the proposed platform has advantages of wide linear working ranges, long-term stability, high reproducibility and good selectivity in the presence of potential interfering compounds. The practical utility was successfully demonstrated in complex biological samples including human serum and urine samples with acceptable recoveries. The influence of commonly found compounds in body fluids such as ascorbic acid, BSA, glucose, sucrose, lactose, aspartic acid, NaCl and KCl was negligible. Overall, the obtained results demonstrated that our sensor could detect low concentrations of these drugs in biological samples without any complex pre-treatment of samples. We believe that the developed sensor may be applied for quality control of drug formulations composed of these drugs and for the optimization of chemotherapeutic treatment.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 51632001). The authors thank the Analytical and Testing Centre and State Key Laboratory of Materials Processing and Die & Mould Technology of HUST for providing TEM, BET and XRD measurements.References
- S. E. Lipshultz, T. R. Cochran, V. I Franco and T. L. Miller, Nat. Rev. Clin. Oncol., 2013, 10, 697–710 CrossRef CAS PubMed.
- P. K. Singal and N. Iliskovic, N. Engl. J. Med., 1998, 339, 900–905 CrossRef CAS PubMed.
- Y. Pommier, E. Leo, H. Zhang and C. Marchand, Chem. Biol., 2010, 17, 421–433 CrossRef CAS PubMed.
- P. Ma and R. J. Mumper, Nano Today, 2013, 8, 313–331 CrossRef CAS PubMed.
- G. N. Hortobagyi, J. U. Gutterman, G. R. Blumenschein, C. K. Tashima, M. A. Burgess and L. Einhorn, Cancer, 1979, 44, 1955–1962 CrossRef CAS PubMed.
- J. M. Nabholtz, J. R. Mackey, M. Smylie, A. Paterson, D. R. Noel and T. Al-Tweigeri, J. Clin. Oncol., 2001, 19, 314–321 CrossRef CAS PubMed.
- H. Kantarjian, N. P. Shah, A. Hochhaus, J. Cortes, S. Shah, M. Ayala, B. Moiraghi, Z. Shen, J. Mayer and R. Pasquini, N. Engl. J. Med., 2010, 362, 2260–2270 CrossRef CAS PubMed.
- E. Weisberg, P. W. Manley, S. W. Cowan-Jacob, A. Hochhaus and J. D. Griffin, Nat. Rev. Cancer, 2007, 7, 345–356 CrossRef CAS PubMed.
- J. Cortes, E. Jabbour, H. Kantarjian, C. C. Yin, J. Shan, S. O'Brien, G. Garcia-Manero, F. Giles, M. Breeden, N. Reeves, W. G. Wierda and D. Jones, Blood, 2007, 110, 4005–4011 CrossRef CAS PubMed.
- P. Ceppi, M. Papotti, V. Monica, M. L. Iacono, S. Saviozzi, M. Pautasso, S. Novello, S. Mussino, E. Bracco and M. Volante, Mol. Cancer Ther., 2009, 8, 3066–3074 CrossRef CAS PubMed.
- G. Konecny, R. Glas, J. Dering, K. Manivong, J. Qi, R. Finn, G. Yang, K. Hong, C. Ginther and B. Winterhoff, Br. J. Cancer, 2009, 101, 1699–1708 CrossRef CAS PubMed.
- C. S. Pichot, S. M. Hartig, L. Xia, C. Arvanitis, D. Monisvais and F. Y. Lee, Br. J. Cancer, 2009, 101, 38–47 CrossRef CAS PubMed.
- P. Zhang, J. Li, M. Ghazwani, W. Zhao, Y. Huang, X. Zhang, R. Venkataramanan and S. Li, Biomaterials, 2015, 67, 104–114 CrossRef CAS PubMed.
- N. Aggerholm-Pedersen, C. Demuth, A. Safwat, P. Meldgaard, M. Kassem and B. S. Sorensen, Stem Cells Int., 2016, 2016, 1–8 CrossRef PubMed.
- S. A. R. Alavi-Tabari, M. A. Khalilzadeh and H. Karimi-Maleh, J. Electroanal. Chem., 2018, 811, 84–88 CrossRef CAS.
- K. Alhareth, C. Vauthier, C. Gueutin, G. Ponchel and F. Moussa, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 887, 128–132 CrossRef PubMed.
- J. S. Perez-Blanco, M. M. F. De-Gatta, J. M. Hernandez-Rivas, M. J. Garcia Sanchez, M. L. Sayalero Marinero and F. G. Lopez, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 955–956, 93–97 CrossRef CAS PubMed.
- A. B. Anderson, C. M. Ciriacks, K. M. Fuller and E. A. Arriaga, Anal. Chem., 2003, 75, 8–15 CrossRef CAS PubMed.
- A. B. Anderson, J. Gergen and E. A. Arriaga, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2002, 769, 97–106 CrossRef CAS.
- Z. Chen, S. Qian, G. Liu, X. Chen and J. Chen, Microchim. Acta, 2011, 175, 217–223 CrossRef CAS.
- M. L. Soriano, C. Carrillo-Carrion, C. Ruiz-Palomero and M. Valcarcel, Microchim. Acta, 2018, 185, 115 CrossRef PubMed.
- M. G. Kassem, E. Ezzeldin, H. M. Korashy and G. A. E. Mostafa, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2013, 939, 73–79 CrossRef CAS PubMed.
- E. Kralj, J. Trontelj, T. Pajic and A. Kristl, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 903, 150–156 CrossRef CAS PubMed.
- H. W. Darwish and A. H. Bakheit, RSC Adv., 2015, 5(67), 54471–54480 RSC.
- M. Taei, F. Hasanpour, H. Salavati and S. Mohammadian, Microchim. Acta, 2016, 183, 49–56 CrossRef CAS.
- H. Guo, H. Jin, R. Gui, Z. Wang, J. Xia and F. Zhang, Sens. Actuators, B, 2017, 253, 50–57 CrossRef CAS.
- H. Karimi-Maleh, A. F. Shojaei, K. Tabatabaeian, F. Karimi, S. Shakeri and R. Moradi, Biosens. Bioelectron., 2016, 86, 879–884 CrossRef CAS PubMed.
- C. S. H. Jesus and V. C. Diculescu, J. Electroanal. Chem., 2015, 752, 47–53 CrossRef CAS.
- P. K. Kalambate and A. K. Srivastava, Sens. Actuators, B, 2016, 233, 237–248 CrossRef CAS.
- P. K. Kalambate, C. R. Rawool and A. K. Srivastava, New J. Chem., 2017, 41, 7061–7072 RSC.
- P. K. Kalambate, B. J. Sanghavi, S. P. Karna and A. K. Srivastava, Sens. Actuators, B, 2015, 213, 285–294 CrossRef CAS.
- L. Farzin, M. Shamsipur, L. Samandari and S. Sheibani, Microchim. Acta, 2018, 185, 276 CrossRef PubMed.
- R. Mandal, A. Baranwal, A. Srivastava and P. Chandra, Biosens. Bioelectron., 2018, 117, 546–561 CrossRef CAS PubMed.
- Q. Liu, Y.-R. Xu, A. J. Wang and J. J. Feng, J. Power Sources, 2016, 302, 394–401 CrossRef CAS.
- P. Song, L. Liu, A. J. Wang, X. Zhang, S. Y. Zhou and J. J. Feng, Electrochim. Acta, 2015, 164, 323–329 CrossRef CAS.
- J.-S. Ye, B.-D. Hong, Y.-S. Wu, H.-R. Chen and C.-L. Lee, Microchim. Acta, 2016, 183, 3311–3320 CrossRef CAS.
- G. Fu, K. Wu, J. Lin, Y. Tang, Y. Chen, Y. Zhou and T. Lu, J. Phys. Chem. C, 2013, 117, 9826–9834 CrossRef CAS.
- E. Laviron, J. Electroanal. Chem. Interfacial Electrochem., 1972, 39, 1–23 CrossRef CAS.
- M. Taei, F. Hasanpour and E. Dehghani, J. Taiwan Inst. Chem. Eng., 2015, 54, 183–190 CrossRef CAS.
- E. Haghshenas, T. Madrakian and A. Afkhami, Anal. Bioanal. Chem., 2016, 408, 2577–2586 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ay02381f |
| This journal is © The Royal Society of Chemistry 2019 |