
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral analysis of β-methylamino alanine (BMAA) enantiomers after (+)-1-(9-fluorenyl)-ethyl chloroformate (FLEC) derivatization and LC-MS/MS†
Javier
Zurita‡
 a,
Nadezda
Zguna‡
a,
Rudolf
Andrýs
b,
Anna
Strzelczak
a,
Liying
Jiang
c,
Gunnar
Thorsen
*d and
Leopold L.
Ilag
*a
a,
Nadezda
Zguna‡
a,
Rudolf
Andrýs
b,
Anna
Strzelczak
a,
Liying
Jiang
c,
Gunnar
Thorsen
*d and
Leopold L.
Ilag
*a
aDepartment of Environmental Science and Analytical Chemistry, Stockholm University, Stockholm, Sweden. E-mail: Leopold.Ilag@aces.su.se
bDepartment of Chemistry, Faculty of Science, University of Hradec Králové, Hradec Králové, Czech Republic
cDrug Control Centre, Department of Analytical, Environmental & Forensic Sciences, King's College London, UK
dIVL Swedish Environmental Research Institute, Stockholm, Sweden. E-mail: Gunnar.Thorsen@ivl.se
First published on 12th December 2018
Abstract
β-Methylamino-L-alanine, a neurotoxin first isolated from the seeds of cycad tree Cycas circinalis, is widely spread in a variety of environments. New sensitive techniques and robust methodologies are needed to detect its presence in complex biological samples and to further understand its biochemical properties. In this context, the determination of the enantiomeric composition of natural BMAA is of great importance. In this study, a simple and easily implemented LC-ESI-MS/MS method was developed to determine the presence of both D- and L-BMAA enantiomers in samples of cycad seed (Cycas micronesica). The samples were subjected to enzymatic hydrolysis to avoid racemization that occurs during strong acid hydrolysis. Derivatization with (+)-1-(9-fluorenyl)-ethyl chloroformate (FLEC) was performed prior to LC-ESI-MS/MS to produce chromatographically separable derivatives of D- and L-BMAA. Together with the retention time, two MRM transitions and their peak area ratios were used to identify the compounds. The LOQ obtained was 0.3 μg BMAA per g wet weight for each enantiomer. Method repeatability was within 3 RSD% both intraday and interday and accuracy was 98–108%. An accurate enantiomeric composition was obtained from the samples of cycad seed, where L- and D-BMAA were detected at 50.13 ± 0.05 and 4.08 ± 0.04 μg BMAA per g wet weight respectively (n = 3).
Introduction
β-Methylamino-L-alanine (BMAA) is a non-protein amino acid suggested to have a link to amyotrophic lateral sclerosis (ALS) and other neurodegenerative disorders recently reviewed by Nunn.1 Originally found in the tropical island of Guam, it was first isolated from the seeds of cycad tree Cycas circinalis in 1967. Produced by ubiquitous organisms such as cyanobacteria, as well as diatoms and dinoflagellates, BMAA occurs in many different locations globally encompassing diverse terrestrial and aquatic environments. However, it should be pointed out that the occurrence of BMAA in many matrices has been questioned, not least in aquatic environments.2The neurotoxin has been found both as a free amino acid and in a protein bound form in the brain tissue from Chamorro people that suffered from ALS/Parkinson's disease (PD) complex. Interestingly, the protein bound form has been discovered in much higher concentration than the free fraction.3–5 Other studies have shown the occurrence of BMAA in Canadians with Alzheimer's disease,5,6 although others have questioned these findings.7 It has been hypothesized that BMAA could act as an endogenous neurotoxic reservoir when it binds to proteins which in turn release and accumulate the neurotoxin into cerebral tissue.4 Artificial peptides have been used to demonstrate the incorporation of BMAA into proteins and the study has consequently indicated the possibility of BMAA mis-incorporation.8 However, other research studies performed recently have questioned this possibility and suggested instead that BMAA toxicity may be caused primarily by excitotoxicity9,10
A fraction known as soluble bound BMAA has been discovered recently.11,12 It was reported that hydrolysis of free BMAA extracts resulted in the increase of BMAA concentration, implying the presence of trichloroacetic acid (TCA)-soluble BMAA-containing compounds, which release BMAA even under mild hydrolysis conditions (i.e. weaker acidic environment, lower temperature, and shorter time). Possible soluble-bound forms of BMAA are still being studied; however, from the previous studies of BMAA, several forms have been suggested, namely, metal complexes,13 carbamates14 and low-molecular weight peptides.1,15
It is known that BMAA is readily taken up by animals and plants. Possible mechanisms and routes of exposure have been addressed. Animal experiments have shown that BMAA administrated to rats tends to be deposited in their brain and liver tissues.16 It was also found that BMAA can be transferred via the breast milk from an exposed mother to an offspring.17 Renewed interest in BMAA came with the report on BMAA presence in aquatic food webs in the Baltic Sea.18 Although that study analyzed samples from geographically distant food webs in the Baltic Sea, thus making it not suitable for the assessment of bioaccumulation, it stimulated further scientific interest and the fact that BMAA was found, for instance, in common seafood from Swedish supermarkets, mainly in mollusks19 brings the reality of urban exposure closer to home.
Not surprisingly, BMAA research has overall raised public health concerns, although a recent study by Downing who used hair as an indicator of human exposure to BMAA has put this into perspective.20 While a positive correlation was observed between seafood consumption and hair BMAA content among local residents, no correlation could be found for other environmental variables such as proximity to cyanobacterial blooms. In general, a direct relationship between exposure to an environmental toxin and the development of neurodegenerative diseases is not easy to establish, given the long latencies of these diseases and the fact that people's lifestyle (eating habits, living environment, etc.) changes during their lifetime. However, the strengthening body of evidence for BMAA neurotoxicity and its worldwide natural distribution demand continued exploration and identification of possible human exposure routes and their impact. Understanding the chemistry of toxicity is just as important as assessing exposure to the toxin. One aspect rather poorly studied is the effect of chirality.
It is well known that living organisms predominantly use L-isomers of amino acids to build proteins while the D-isomers of monosaccharides are preferred to build complex carbohydrates.21 In general, D-enantiomers of amino acids have been less studied and their specific functions in living organisms remain to be fully elucidated. However their importance in biochemical processes is supported by different studies, many of which have been recently reviewed by Genchi.22 Moreover, in recent years there has been growing knowledge regarding the presence of D-amino acids in plants and increasing evidence that supports a more active role than what was commonly believed in the past. Evidence has been provided concerning their utilization by plants as a nitrogen source, and there are indications of their participation in signaling processes, such as D-Ser binding to glutamate receptors which may influence pathogen response.23
Similar to protein amino acids, BMAA is an optically active biomolecule existing as L- and D-BMAA. According to a study by Andersson et al.17 performed in rats, the uptake of L-BMAA was much higher than the uptake of the D-enantiomer. Furthermore, up to now it has been commonly believed that the L-isoform was exclusively responsible for the neurotoxic effect of BMAA while D-BMAA had little or no harmful effect.3,24 However Metcalf et al.25 in a recent study assessed the in vitro toxicity of D-BMAA towards mixed cortical cell cultures from mice and found it to be comparable to that of L-BMAA. Another finding reported in the same article showed that D-BMAA was detected in the central nervous system of mice and vervets orally dosed with L-BMAA, while only L-BMAA was found in the peripheral nervous system. Moreover, D-BMAA was also present in the liver of mice, suggesting that racemization occurs during digestion. These findings raised the relevance of further D-BMAA investigation and posed many questions about BMAA metabolism, since the process of amino acid incorporation into proteins is apparently specific for L-forms.1
The biological role of BMAA as well as its defined origin is not still fully elucidated and may require novel analytical methodologies to be developed. Chiral analysis may become an invaluable tool to resolve those issues, and thus it is critical to determine the biological distribution of both enantiomers. Many strategies have been employed to resolve chiral amino acids in complex matrices. Direct chiral separation using enantioselective stationary phases has been used over the years.26–28 Derivatization has often been used to simplify the method development and to reduce the time of analysis due to the possibility to separate and detect several compounds in a single run.29–31 Enantiomers of BMAA have previously been separated by high performance liquid chromatography (HPLC) using pre-derivatization with o-phthaldialdehyde and detection by fluorimetry32 and recently, Metcalf et al. have developed a liquid chromatography-tandem mass spectrometry (LC-MS/MS) methodology including a fractionation step using chiral chromatography.25
The study reported here describes a simple LC-MS/MS method in order to determine the presence of both D- and L-isomers of BMAA in complex samples. The sample preparation procedure includes enzymatic hydrolysis using Pronase followed by derivatization with (+)-1-(9-fluorenyl)-ethyl chloroformate (FLEC). The original studies on derivatization of amino acids with FLEC and a similar reagent 1-(9-fluorenyl)-methyl chloroformate (FMOC) were carried out in the 1980s by Einarsson and co-workers, where they derivatized 20 essential amino acids prior to separation and detection on a reversed phase HPLC-FLD (fluorescence detector). Also, the separation of enantiomers of optically active amino acids was achieved.33,34 Later, Kisby and co-workers successfully used FMOC to derivatize BMAA and other amino acids in cycad seeds, which, however, was not suitable for resolving optical isomers.35 Derivatization of amino acids with FLEC is still being used by different groups, as an effective method for simultaneous determination of D- and L-amino acids,36 although, to the best of our knowledge, it has not been previously used for chiral analysis of BMAA.
Although it is common to use strong acid hydrolysis to break amide bonds in order to release individual amino acids from proteins, it is known that during this process optical inversion may occur thus leading to racemization.37 Our method essentially overcomes racemization artefacts in order to accurately determine the presence of enantiomers of BMAA and separate them from its structural isomers. Furthermore, derivatization of enantiomers of BMAA with FLEC results in the formation of diastereomers that can be separated by reversed phase HPLC. In this work, we applied the new methodology to determine the enantiomeric profile of cycad seed, allowing for the first time the detection of naturally occurring D-BMAA in this biological material. The methodology applied here can be further extended to the analysis of environmental samples as well as toxicological studies in animals.
Experimental
Chemicals and reagents
The L-BMAA standard (B107, Germany), L-2,4-diaminobutyric acid (DAB) standard (D8376, Switzerland), Pronase (P5147, Japan) and (+)-1-(9-fluorenyl)-ethyl chloroformate (FLEC) (23182, Switzerland) were obtained from Sigma Aldrich (St. Louis, MO, USA). The N-(2-aminoethyl)glycine (AEG) standard (A1153, Japan) was purchased from TCI (Japan). β-Amino-N-methyl-alanine (BAMA) and deuterium labelled BMAA (d3-BMAA) standards were synthesized as previously described.38,39 The D-BMAA standard was synthesized by Peter Wyatt (Queen Mary University of London, UK). Solvents were purchased from WVR international (Radnor, Pennsylvania, USA) and were of HPLC grade. Ammonium bicarbonate and calcium chloride were purchased from WVR international (Radnor, Pennsylvania, USA). Water was purified in-house to Milli-Q grade with a resistivity of 18.2 MΩ cm using a water purification system (Millipore Synergy 185; Molsheim, France). Cycad seeds were given as a gift by Johan Rosén (National Food Administration, SE-75126 Uppsala, Sweden).Preparation of cycad seeds (Cycas micronesica)
Cycad seeds were treated according to the method of Jiang et al.19 An equivalent to 10 mg of endosperm of cycad seeds was incubated with 600 μL of 6 M HCl at 110 °C for 20 h. Alternatively the same amounts were incubated with Pronase (final concentration of 2 mg mL−1) in 50 mM NH4HCO3 at pH 8 with 1 mM CaCl2 as well as in the same buffer without Pronase. The reaction mixture was left at 37 °C for 48 hours. To observe the effect that the different hydrolysis treatments have on optically pure isomers, standards of L- and D-BMAA (20 ng) were treated under the same conditions.Hydrolysates were then filtered through vial spin filters (0.2 μm cut-off; Thermo Scientific, USA). Solid phase extraction (SPE) was performed on the filtered acid hydrolysates using Isolute HCX-3 columns (100 mg; sorbent AB, Sweden) according to the method of Jiang et al.40 Both the eluates from SPE and the Pronase hydrolysates were then dried under a nitrogen flow and reconstituted in 30 μl of 20 mM HCl before derivatization.
Derivatization with (+)-1-(9-fluorenyl)-ethyl chloroformate (FLEC)
A derivatization procedure of BMAA with the FLEC chiral reagent was developed in this work, based on previous reports33–35 and improved considerably. Briefly, samples prepared as indicated above or pure standards (20 μl 1 mg L−1) were incubated with 60 μl of borate buffer (5% sodium tetraborate in water, pH 8.8) from a commercial AccQ·TagReagent Kit (Waters, Milford, MA, USA), 80 μl of acetone and 15 μl of FLEC derivatization reagent (final concentration in reaction 1.54 nmol L−1). Different reaction times between 15 and 60 min were compared. After 30 minutes, the yield was not significantly improved. The use of gentle heating (temperature ≤40 °C) was also tested and the reaction yield was improved compared with using the derivatization at room temperature. Finally, optimal conditions for incubation were set to 30 minutes of reaction at 40 °C. After incubation, the excess of FLEC tag was extracted using 200 μl of hexane:ethylacetate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solvent and vortexing for 30 s. The organic part was discarded and the aqueous part was dried under nitrogen and reconstituted in 10:90:0.075 of acetonitrile:water:acetic acid.
:1) solvent and vortexing for 30 s. The organic part was discarded and the aqueous part was dried under nitrogen and reconstituted in 10:90:0.075 of acetonitrile:water:acetic acid.
Liquid chromatography coupled to tandem mass spectrometry
In order to be sure of the presence of detectable BMAA enantiomers in the samples that are to be chirally resolved, various specimens were screened. For the screening, the usual 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AQC) derivatization protocol coupled to LC-MS/MS was implemented. The detection of BMAA in fish (char, trout, Baltic herring, perch and plaice), squid and blue mussel samples derivatized with the AQC tag was performed on a TSQ Vantage triple-quadrupole mass analyser. The column, gradient, mobile phase, flow rate and MS/MS parameters were set according to the method of Jiang et al.40For the preliminary evaluation of the FLEC-derivatized BMAA fragmentation pattern, a method for linear ion trap LC-ESI-MS/MS was implemented on a liquid chromatography system (Agilent 1100 series) consisting of a degasser (G1322A), a binary pump (G1312A) and an autoinjector (G1313A) coupled to an ion trap mass spectrometer (Finnigan LTQ, Thermo, USA). BMAA enantiomers and isomers were separated on a reversed phase C18 column (2.1 × 150 mm, 3.5 μm; Sunfire columns, Waters). The gradient was run for 25 min (mobile phase A was 0.075% acetic acid in water and mobile phase B was 0.075% acetic acid in acetonitrile) at a flow rate of 0.3 mL min−1. MS/MS detection was performed by multiple reaction monitoring (MRM) of general product ions (m/z 355.17) and diagnostic product ions (m/z 268.13) for BMAA. For the isomers only the general product ions were chosen except for BAMA where the diagnostic product ion at m/z 268.13 was also chosen. The precursor ion mass for BMAA and its isomers was m/z 591.25. The precursor ion at m/z 594.27 and the diagnostic product ion at m/z 358.17 were chosen for the derivatised internal standard (d3-DL-BMAA).
The method of choice for the quantification of L- and D-BMAA used a Xevo TQ-S micro-mass spectrometer (Waters, Milford, MA, USA) system with ESI+ in MRM mode. To separate the enantiomers, ultra-high performance liquid chromatography (UHPLC) runs were performed using a Hypersil Gold C18 column (150 × 2.1 mm, 1.9 μm particle size; Thermo Scientific) with a binary mobile phase (solvent A, water with 0.075% acetic acid; solvent B, acetonitrile with 0.075% acetic acid) at a flow rate of 300 μl min−1. The gradient used was: 0.0 min, 60% B; 10.0 min, 65% B; 10.01 min, 90% B; 12.0 min, 90% B; 12.01 min, 60% B; and 15.0 min, 60% B. In MS/MS the following m/z transitions were monitored in MRM mode: 591.0 > 355.0, CE = 15 eV; and 591.0 > 193.0, CE = 30 eV. The ion source parameters were optimized as follows: capillary voltage (3000 V), source temperature (150 °C), desolvation temperature (400 °C), cone gas flow (50 L hour−1) and desolvation gas flow (700 L hour−1).
Method evaluation
d3-DL-BMAA, synthesized as described in Jiang et al.,39 was added as a surrogate internal standard to all standards and samples (c = 10 μg L−1 in standards and 4 μg L−1 in samples). A calibration curve reflecting the peak area ratio (standard:IS) vs. standard concentration was constructed for the 0.1 to 12 mg L−1 range. The limit of quantification (LOQ) was estimated and accuracy and both intraday and interday repeatability were calculated from quality control (QC) samples. The developed method was applied to quantify L- and D-BMAA contents in cycad seed.
Results & discussion
Screening of BMAA in cycad and seafood samples
Samples analysed for corresponding BMAA content were measured using the standard AQC-derivatization strategy coupled to LC-MS/MS.39 The analysis was performed and only cycad and blue mussel samples were positive in both the pellet and supernatant fractions that respectively distinguish between bound and free BMAA. Unfortunately the levels detected in the blue mussel sample were below the limit of detection (LOD) of the FLEC developed method presented here; therefore only cycad results are presented.Method development for chiral analysis of BMAA
Our initial work started with LC runs over 20 min but this was easily reduced to about 10 min giving reliable information on the chiral composition of the biological sample. Complete and clear separation of enantiomers of BMAA from its structural isomers was achieved (Fig. 1). BMAA and its diastereoisomers after derivatization with the optically pure (+)-FLEC reagent have [M + H]+ at m/z 591. It can be seen that (+)FLEC-L-BMAA and (+)-FLEC-D-BMAA are completely separated and therefore can be easily identified and quantified, satisfying one of the main objectives of this paper. The presence of a small contaminant in all the chromatograms at around 7.5 min has been observed; however this does not interfere with the peaks of interest. Additionally, structural isomers of BMAA have been separated and the fact that BMAA isomers can be determined simultaneously using the developed method constitutes an advantage over other methodologies that may not take into account all possible interference. A detailed study on the presence of AEG, DAB or BAMA is beyond the scope of this paper, so it was sufficient to have the separation of BMAA from the rest of its structural isomers.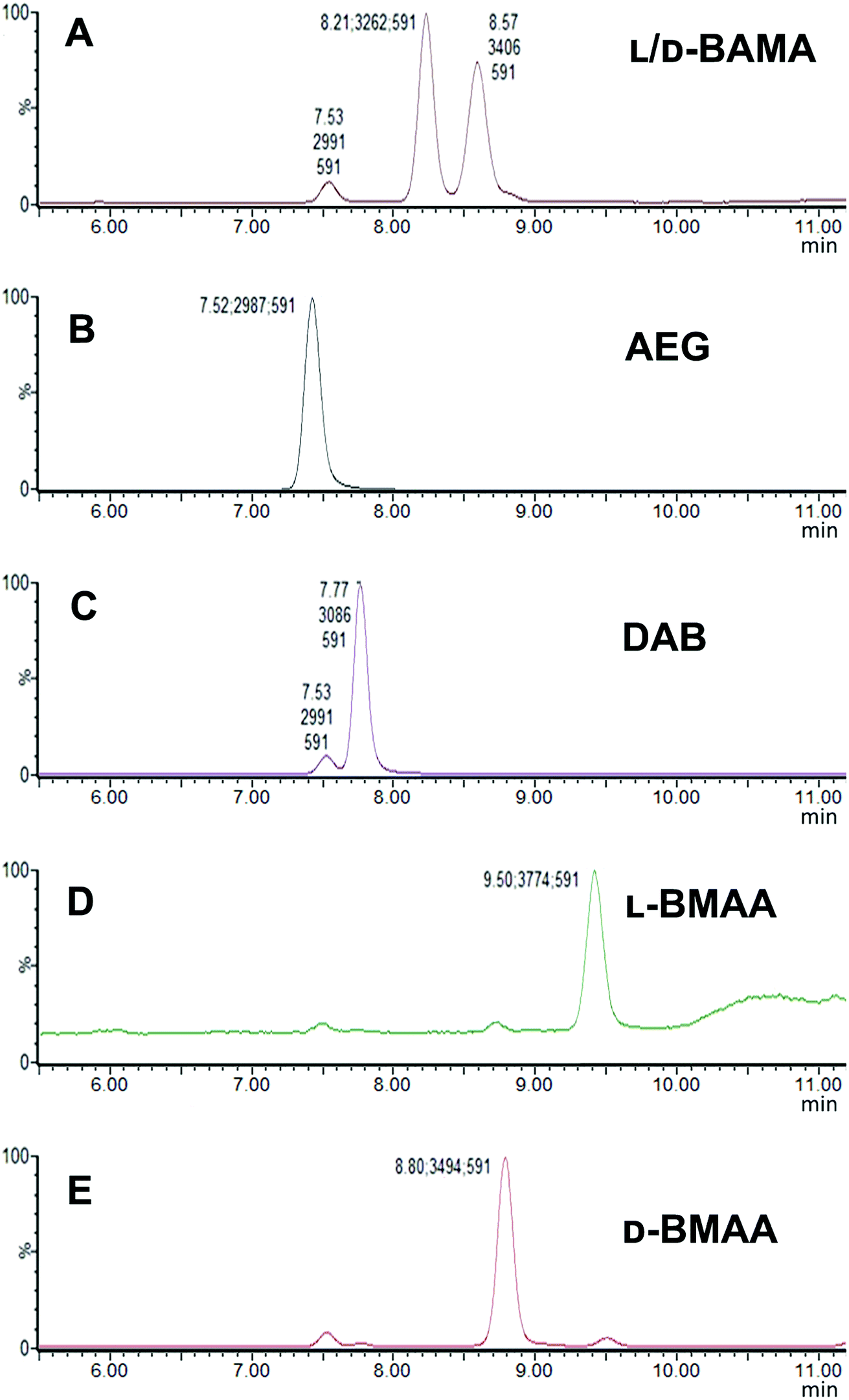 | ||
| Fig. 1 SIM (m/z 591) chromatograms of standards of BMAA and its structural isomers obtained with a Xevo TQ-S micro-mass spectrometer showing characteristic retention times: (A) L- and D-BAMA (8.21 min, 8.57 min), (B) AEG (7.52 min), (C) L-DAB (7.77 min) (D) L-BMAA (9.50 min) and (E) D-BMAA (8.80 min). | ||
MS/MS was used to further confirm the identities of L and D-BMAA. The initial method development employed tandem MS using a linear ion trap but later tested two triple quadrupole instruments, API 2000 and Xevo, with the latter becoming the instrument of choice for the final method. Depending on the kind of MS instrumentation used, the fragmentation patterns vary (shown in ESI Fig. 1†) and slightly different intensities of fragment ions were observed. However, the strongest signals were in common for all instruments and it was not possible to get appreciable signals by using only diagnostic ions, so general ions were used for quantification in this study. For the Xevo mass spectrometer, which exhibited the best sensitivity, product ions at m/z 193 and 355 were selected for identification (Fig. 2). Moreover, MRM mode resulted in the contaminant at 7.5 min disappearing from the chromatograms. A chromatogram of the pure compounds using MRM is shown in Fig. 3a.
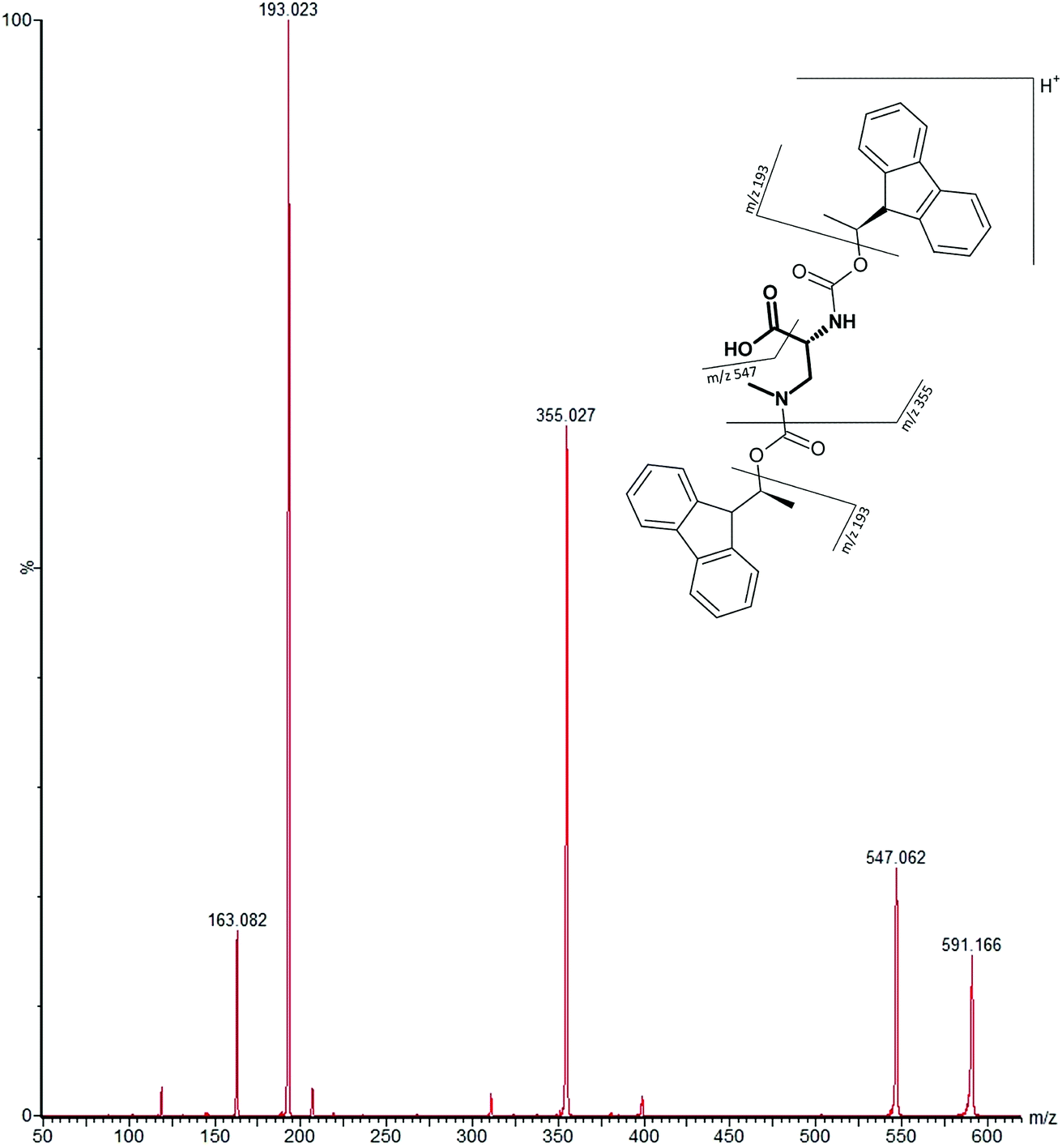 | ||
| Fig. 2 MS/MS spectrum of FLEC-(D)-BMAA showing the proposed fragmentation of the m/z 591 ion corresponding to the molecular ion (most intense product ions are m/z 193 and m/z 355). | ||
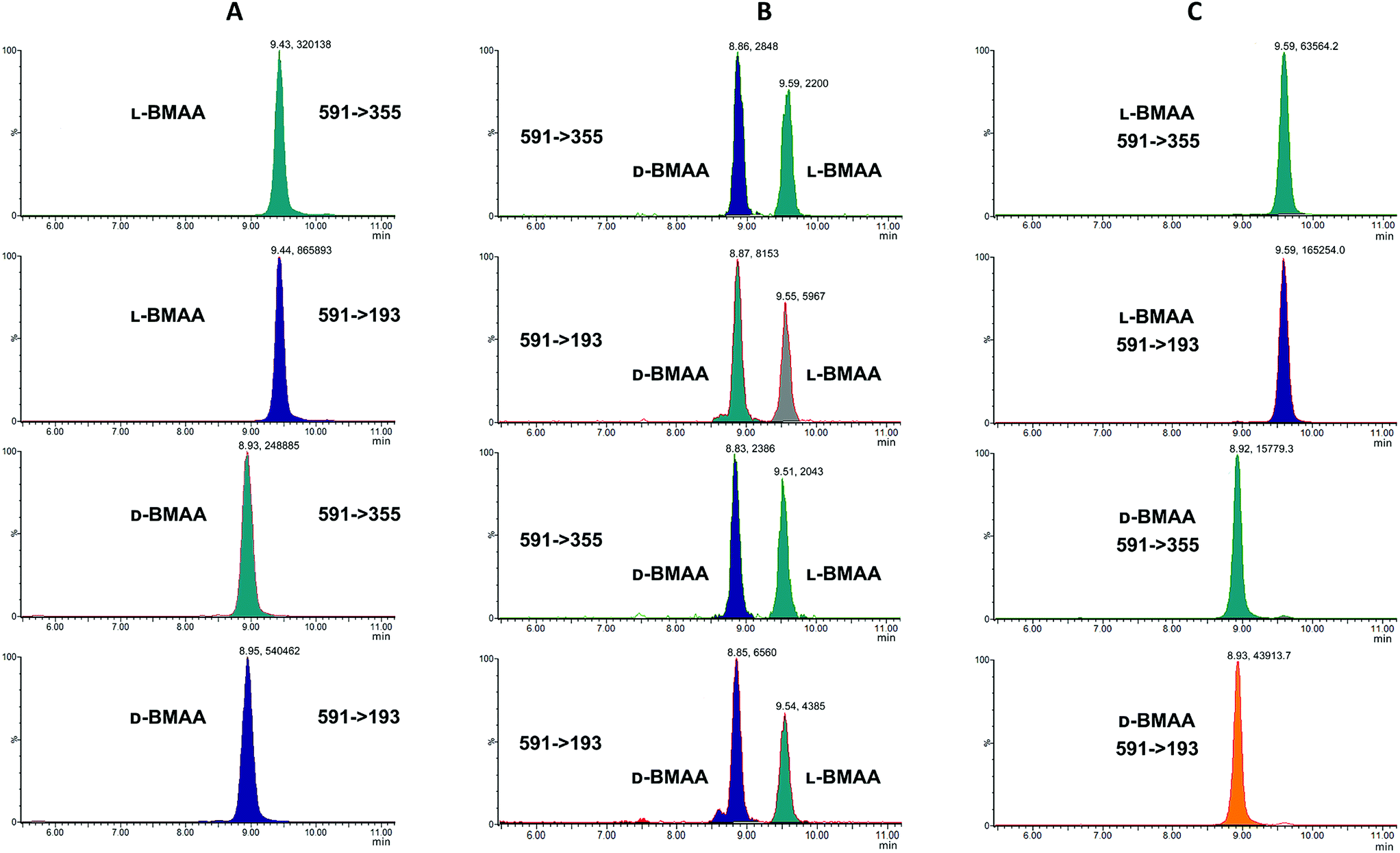 | ||
| Fig. 3 MRM chromatogram of L and D standard compounds (1 mg L−1). Chromatograms for specific transitions corresponding to: (A) L-BMAA and D-BMAA standard solutions; (B) L-BMAA and D-BMAA standard solutions after acid hydrolysis show that racemization of L- and D-BMAA pure enantiomers occurs; but in (C) L-BMAA and D-BMAA standard solutions after enzymatic hydrolysis do not racemize, and stereochemistry of L- and D-BMAA pure enantiomers is preserved. | ||
Acid vs. enzymatic hydrolysis
Studies on the presence of BMAA in food have often used strong acid hydrolysis to release protein-associated BMAA.19,41,42 However, because they can lead to racemization, strong acid conditions may not be appropriate to determine the enantiomeric composition of BMAA in biological matrices. Thus in this analysis commercial standards of optically pure enantiomers were exposed to conditions typical of strong acid hydrolysis in order to determine whether racemization of BMAA occurs, and the results were compared with those obtained after gentle enzymatic treatment.As can be seen in Fig. 3b, after treatment with HCl (6 M) for 20 hours at 110 °C the chromatogram shows two intense peaks at the retention times corresponding to the BMAA enantiomers. This shows the inadequacy of using strong acid hydrolysis for chiral BMAA analysis, as this will lead to an ambiguity regarding how much of the L-isomer is actually present in a sample and can lead to false positives for D-BMAA. In contrast, gentle enzymatic treatment with Pronase for 48 hours at 37 °C resulted in a very low percentage of L-BMAA after incubation of the D-BMAA standard (<2%) and even lower of the D-enantiomer after incubation of the L-BMAA standard (<0.3%) (Fig. 3c).
Cycad seed enantiomeric composition
Because L-BMAA was the only enantiomer initially isolated from cycad seeds,24,43 this has been the only enantiomer considered for studies on toxicity. However, the study by Metcalf et al.25 has shown that exposure to D-BMAA could also have toxic effects. In addition, it is important to point out that with the methods used in the past, only free BMAA has been determined in the analysis of cycad seeds and thus certain amounts of D- and L-enantiomers bound to the matrix were overlooked.Pronase digestion, as well as strong acid hydrolysis, was applied to cycad seed samples in order to test the performance of the method in a complex biological matrix. In accordance to what was expected based on previous experiments with standards, the experiment with cycad seed samples shows that stereochemistry is maintained when the samples were enzymatically treated, while racemization occurred when the samples were hydrolyzed using hydrochloric acid. The results can be seen in Fig. 4 showing that the main isomer present in the cycad seed is the L-form but there is D-BMAA present in a smaller amount. The ion ratio was used as a criterion for BMAA identification in addition to the retention time and fragmentation pattern. Product ion ratios are shown in Table 1 and are similar (±10%), according to criteria reported by Jiang et al.40 in standards and cycad seed samples for both enantiomers (for 3 sample replicates). The presence of a small peak at 6.8 min with the same transitions as L- and D-BMAA can be observed after acid hydrolysis of cycad seed samples, not being visible after Pronase treatment. Its origin therefore can be only speculated. The fact that this peak is not present in any of the standards eliminates the possibility of it being an epimer arising from strong acid hydrolysis. The hypothesis that this can be one of the known structural isomers is easily discarded, as the retention time doesn't match those of the analyzed standards. It is possible that an unknown isomer is present in the cycad seed that is strongly linked to the seed matrix and therefore only released by strong acid hydrolysis.
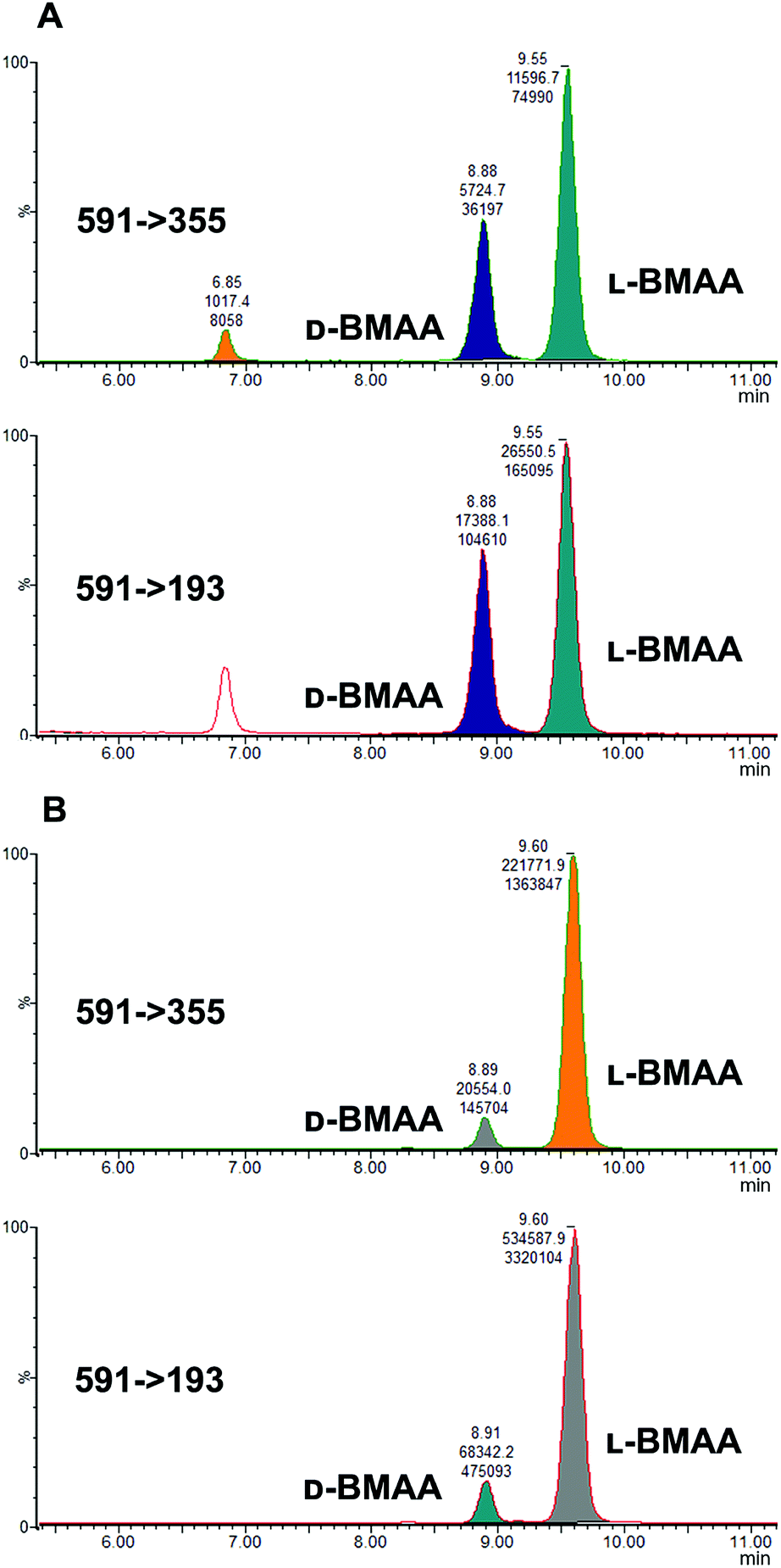 | ||
| Fig. 4 MRM chromatograms of the cycad seed sample after treatment with: (A) hydrochloric acid, and (B) Pronase. Treatment with acid resulted in an abundance of D-BMAA after racemization, while Pronase treatment showed that L-BMAA is in fact the naturally dominant enantiomer. | ||
Additionally, cycad seed samples were incubated in buffer (50 mM NH4HCO3 at pH 8 with 1 mM CaCl2) for 48 hours at 37 °C. Surprisingly, this showed similar results to the incubation with Pronase. We observed the presence of a D-BMAA peak in the chromatograms with an area approximately 10% of the L-BMAA peak. This finding suggests that BMAA in the cycad seed could be found in some conjugated form, and not only protein-bound, since apparently no peptide bond cleavage (with Pronase or strong acid) is required to release some BMAA from cycad seed samples.
For quantitative purposes, the product ion at m/z 193 was selected. Good linearity was achieved between 0.1 and 12 mg L−1 (n = 3) based on the calibration curve (Fig. 5). The LOQ was estimated to be 0.1 mg L−1 for both L- and D-BMAA. The relative standard deviation (RSD) was calculated for standards with low concentrations and it was observed that an RSD of less than 10% was found for 0.1 mg L−1 standards, while standards at lower concentrations yielded RSD > 10%. Thus, we conclude that concentrations lower than 0.1 mg L−1 are not suitable for quantitative purposes. The LOQ is at the same magnitude levels as those in previous studies performed on amino acids derivatized with FLEC that used fluorescence detection.36 In addition, the performance of the method was evaluated with QC samples at a low level (c = 0.5 mg L−1) and at a high level (c = 9 mg L−1) intraday and interday. The method evaluation data are summarized in Table 2.
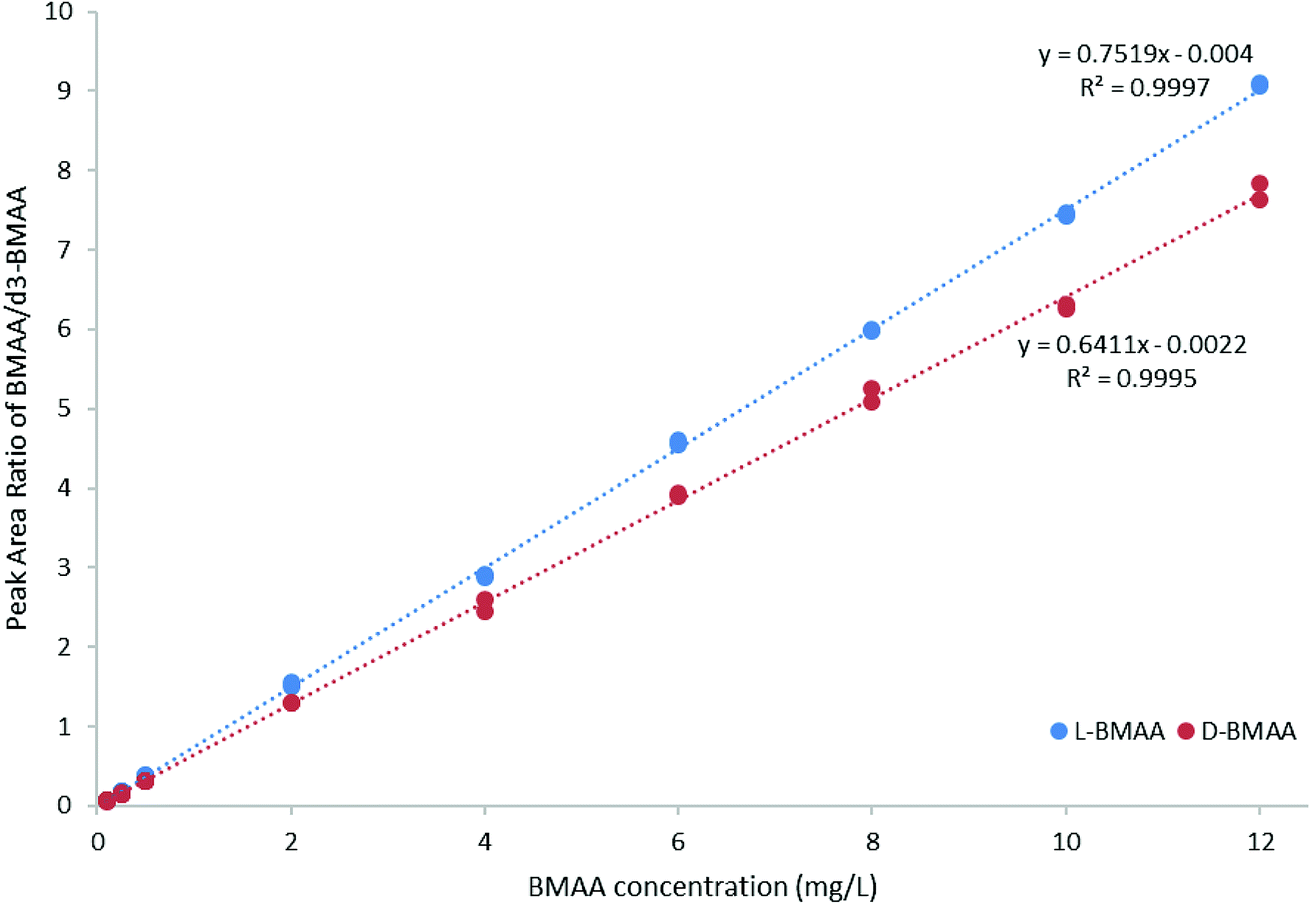 | ||
| Fig. 5 Calibration curve (0.1–12 mg L−1 BMAA, n = 3) for method evaluation and quantification of cycad seed samples. | ||
| Concentration, mg L−1 | Accuracy% | Intraday precision, RSD% | Interday precision, RSD% | |
|---|---|---|---|---|
| L-BMAA | 0.5 | 99.5 | 2.7 | 1.8 |
| 9 | 107.8 | 1.7 | 1.7 | |
| D-BMAA | 0.5 | 97.9 | 1.0 | 1.3 |
| 9 | 107.9 | 1.4 | 2.3 |
The developed method was used to quantify L- and D-BMAA in cycad seed samples (n = 3) (Fig. 6). The calculated concentrations were 50.13 ± 0.05 and 4.08 ± 0.04 μg BMAA per g (Cycas micronesica, wet weight) for L- and D-BMAA respectively. This values are in accordance with previous studies performed on the total BMAA content in cycad seeds.44,45 Calculations were also made for the acid hydrolysis and incubation with buffer; the obtained concentrations were as follows: 58.6 ± 3.0 and 74.5 ± 3.8 μg BMAA per g wet weight for L- and D-BMAA respectively in samples after acidic hydrolysis; 45.9 ± 3.5 and 4.9 ± 0.5 μg BMAA per g wet weight for L- and D-BMAA respectively in samples after incubation with buffer.
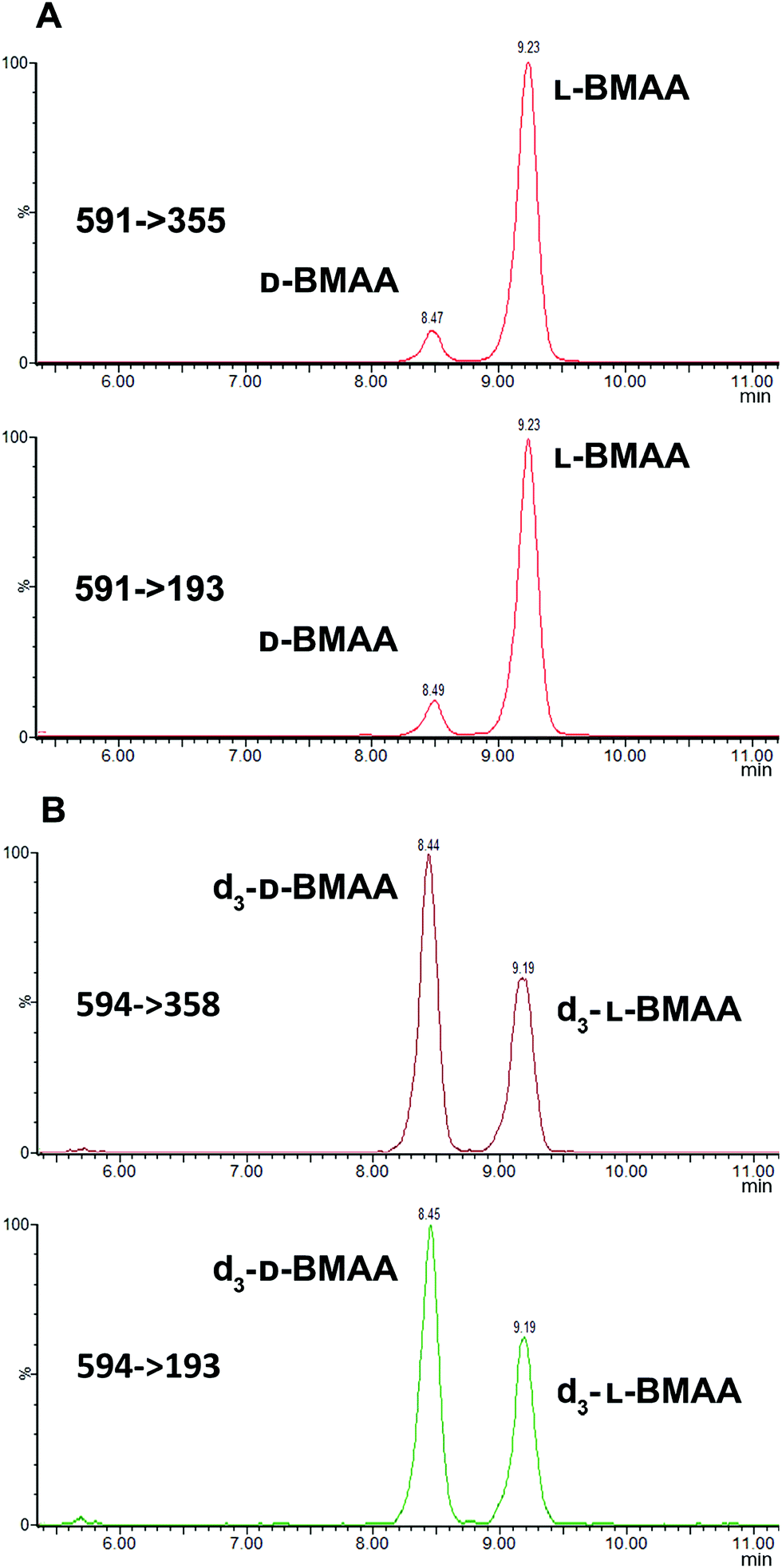 | ||
| Fig. 6 Cycad seed sample after treatment with Pronase reveals the presence of both D- and L-BMAA. Confirmatory transitions shown in the MRM chromatograms correspond to (A) D- and L-BMAA present in the cycad sample and (B) d3-DL-BMAA used as the internal standard for quantification. | ||
The total amount of detected L- and D-BMAA in cycad seed was highest after acidic hydrolysis (133.1 ± 3.4 μg BMAA per g wet weight), and then after Pronase hydrolysis (54.2 ± 5.9 μg BMAA per g wet weight) and lowest after buffer incubation (50.8 ± 3.4 μg BMAA per g wet weight). However, the difference between the latter two is quite small and can simply result from quantification variation. The results strongly suggest that BMAA is present in cycad seeds as some conjugated form, which gets released even only after mild hydrolysis or incubation with just buffer solution. However, Pronase hydrolysis is not as efficient as strong acid hydrolysis and that fact should be taken into consideration for absolute quantification. It should be noted that the form by which BMAA exists (free or bound) is sample-dependent. In this specific case (cycad seed), there is an apparent predominance of conjugated-BMAA species given the increased detection of BMAA after mere incubation in buffer. This may give the impression that the use of Pronase is negligible. However, it should be noted that the intended method is for general use; thus, it is important to incorporate the use of enzymatic hydrolysis for cases when it is necessary to deal with e.g. protein-bound BMAA.
Previous studies by Metcalf et al.25 did not find D-BMAA in cycad seeds which is consistent with the initial studies where only L-BMAA was crystalized from an alcohol extract of these plant seeds. It is crucial to note that in these studies the samples were not hydrolyzed and therefore they could not report information on any bound form. In the same publication, Metcalf and co-workers proposed that L-BMAA is metabolized to D-BMAA within the brain and some neurotoxicity may be derived from that enantiomer. An important fact, that should not be ignored, is that racemization of amino acids occurs with aging of samples. However it requires a long time for amino acids to racemize, e.g. it takes 2000 years for histidine to reach a 10% D/L ratio.46 The rate may be faster for BMAA due to it being prone to racemize even under acidic hydrolysis (racemic mixture was obtained in 20 h of 6 M HCl hydrolysis), while histidine forms only negligible amounts of the D-isomer during 24 h of 6 M HCl hydrolysis.46 Therefore, the presence of D-BMAA bound to the matrix of cycad seeds could indeed be a finding of significant importance in understanding the role of each enantiomer in the neurotoxicity of BMAA. The enantiomeric composition of cyanobacterial blooms and food as well as the mechanism of possible enzymatic inversion in vivo both need to be studied in detail to assess the real impact on health. Studies on BMAA present in food should also consider the chirality and include simple methodologies able to distinguish the different enantiomers together with structural isomers, i.e. AEG, BAMA and DAB.
Conclusions
Overall, we established a simple and accurate method for BMAA enantiomer analysis and discovered D-BMAA presence in cycad seed, which is an important finding and will most certainly encourage further studies regarding its implications.Method development towards chiral analyses of BMAA without the need for chiral columns was not a straightforward endeavor. This required establishing quick but clean separation of isomers; determination of useful fragment ions for confident identification; avoiding artefacts due to work-up of the samples and reasonable LOD for relevant monitoring purposes. The method described in this paper extends strategies regarding the analysis of BMAA. Many studies have been conducted showing the presence of BMAA in certain foods and environments. However, the existing methodologies almost never address resolving enantiomers to determine accurately the true amount of either L- or D-BMAA. Chromatographic resolution together with tandem mass spectrometry allows accurate identification and quantification of both L- and D-enantiomers of BMAA. The proposed methodology can be extended to other biological matrices and studies on food, as well as environmental and clinical samples. It will be valuable in the detection of L and D-BMAA in a variety of matrices and improve the understanding of the contributions of enantiomers to toxicity. Furthermore, the methodology proposed in this work could provide the tools to assist in unraveling the yet unknown activities of BMAA and the role that chirality plays in neurodegeneration.
A key aspect in this study is the recognition that racemization of BMAA occurs as a consequence of strong acid hydrolysis; a widely used step for measuring bound BMAA from complex matrices. The problem could be avoided by using enzymatic digestion instead and D-BMAA could be quantified using an efficient facile LC-MS/MS method. According to our quantification results, D-BMAA is apparently present in the soluble bound form in cycad seed and thus point to a need to reconsider the contribution of D-BMAA in the context of toxicity of cycad derived materials.1
Author contributions
L. L. I., G. T., and L. J. were responsible for producing the ideas regarding the strategies. N. Z., J. Z. and R. A. designed the experiments. A. S. (MSc student) and L. J. performed preliminary experiments. N. Z and J. Z. performed the main part of the experiments and analysed the data. N. Z and J. Z. wrote the manuscript, and L. L. I., G. T., L. J. and R. A. contributed with comments.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge the Baltic Ecosystem Adaptive Management (BEAM) program (funded by the Swedish government) for their financial support of this study. The authors would also like to thank Peter Wyatt for providing the D-BMAA standard and Johan Rosén for the cycad seed samples.References
- P. B. Nunn, 50 Years of Research on α-Amino-β-Methylaminopropionic Acid (β-Methylaminoalanine), Phytochemistry, 2017, 144, 271–281 CrossRef CAS PubMed.
- E. J. Faassen, Presence of the neurotoxin BMAA in aquatic ecosystems: What do we really know?, Toxins, 2014, 6(3), 1109–1138 CrossRef PubMed.
- F. I. Polsky, P. B. Nunn, E. A. Bell, Distribution and toxicity of alpha-amino-beta-methylaminopropionic acid, Fed. Proc., 1972, 31(5), 1473–1475 Search PubMed.
- S. J. Murch, P. A. Cox and S. A. Banack, A mechanism for slow release of biomagnified cyanobacterial neurotoxins and neurodegenerative disease in Guam, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(33), 12228–12231 CrossRef CAS PubMed.
- S. J. Murch, P. A. Cox, S. A. Banack, J. C. Steele and O. W. Sacks, Occurrence of β-methylamino-L-alanine (BMAA) in. ALS/PDC patients from Guam, Acta Neurol. Scand., 2004, 110(4), 267–269 CrossRef CAS.
- P. A. Cox, S. A. Banack and S. J. Murch, Biomagnification of cyanobacterial neurotoxins and neurodegenerative disease among the Chamorro people of Guam, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(23), 13380–13383 CrossRef CAS PubMed.
- T. J. Montine, K. Li, D. P. Perl and D. Galasko, Lack of b-methylamino-L-alanine in brain from controls, AD, or Chamorros with PDC, Neurology, 2005, 65(5), 768 CrossRef CAS PubMed.
- D. Seebach, A. Stude, E. Pfammatter and H. Widmer, Synthesis of Tri-, Penta-, and Heptapeptides containing and (R)-2-alkyl-2-amino-3-(methylamino)-propionic acid residue in the central position, Helvetica Chimica Acta, 1994, 77(7), 2035–2050 CrossRef CAS.
- R. van Onselen, L. Venables, M. van de Venter and T. G. Downing, β-N-Methylamino-L-Alanine Toxicity in PC12: Excitotoxicity vs. Misincorporation, Neurotoxic. Res., 2017, 33(1), 15–23 CrossRef PubMed.
- R. Van Onselen, N. A. Cook, R. R. Phelan and T. G. Downing, Bacteria do not incorporate β-N-methylamino-l-alanine into their proteins, Toxicon, 2015, 102, 55–61 CrossRef CAS.
- E. J. Faassen, M. G. Antoniou, W. Beekman-Lukassen, L. Blahova, E. Chernova and C. Christophoridis, et al., A collaborative evaluation of LC-MS/MS based methods for BMAA analysis: Soluble bound BMAA found to be an important fraction, Mar. Drugs, 2016, 14(3), 45 CrossRef.
- J. Rosén, E. Westerberg, S. Schmiedt and K. E. Hellenäs, BMAA detected as neither free nor protein bound amino acid in blue mussels, Toxicon, 2016, 109, 45–50 CrossRef PubMed.
- P. B. Nunn, P. O'Brien, L. D. Pettit and S. I. Pyburn, Complexes of zinc, copper, and nickel with the nonprotein amino acid L-alpha-amino-beta-methylaminopropionic acid: a naturally occurring neurotoxin, J. Inorg. Biochem., 1989, 37(2), 175–183 CrossRef CAS.
- P. B. Nunn and P. O'Brien, The interaction of β-N-methylamino-L-alanine with bicarbonate: an 1H-NMR study, FEBS Lett., 1989, 251(1–2), 31–35 CrossRef CAS.
- R. Cheng and S. A. Banack, Previous studies underestimate BMAA concentrations in cycad flour, Amyotrophic Lateral Scler., 2009, 10(suppl. 2), 41–43 CrossRef CAS.
- O. Karlsson, L. Jiang, M. Andersson, L. L. Ilag and E. B. Brittebo, Protein association of the neurotoxin and non-protein amino acid BMAA (β-N-methylamino-l-alanine) in the liver and brain following neonatal administration in rats, Toxicol Lett, 2014, 226(1), 1–5, DOI:10.1016/j.toxlet.2014.01.027.
- M. Andersson, L. Ersson, I. Brandt and U. Bergstr�m, Potential transfer of neurotoxic amino acid β-N-methylamino-alanine (BMAA) from mother to infant during breast-feeding: predictions from human cell lines, Toxicol Appl Pharmacol, 2017, 320, 40–50, DOI:10.1016/j.taap.2017.02.004.
- S. Jonasson, J. Eriksson, L. Berntzon, Z. Spácil, L. L. Ilag and L.-O. Ronnevi, et al., Transfer of a cyanobacterial neurotoxin within a temperate aquatic ecosystem suggests pathways for human exposure, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(20), 9252–9257 CrossRef CAS PubMed.
- L. Jiang, N. Kiselova, J. Rosén and L. L. Ilag, Quantification of neurotoxin BMAA (β-N-methylamino-L-alanine) in seafood from Swedish markets, Sci. Rep., 2014, 4, 1–7 Search PubMed.
- S. Downing, L. L. Scott, N. Zguna and T. G. Downing, Human scalp hair as an indicator of exposure to the environmental toxin β-N-methylamino-L-alanine, Toxins, 2018, 10(1), 14 CrossRef.
- L. Morozov, Mirror symmetry breaking in biochemical evolution, Origins Life, 1979, 9(3), 187–217 CrossRef CAS.
- G. Genchi, An overview on d-amino acids, Amino Acids, 2017, 49(9), 1521–1533 CrossRef CAS.
- U. Koluksaoglu and J. Suarez. D-Amino Acids in Plants: New insights and Aspects, but also More Open Questions, in Amino Acids – New Insights and Roles in Plant and Animal, Intech, 2017, pp. 155–69 Search PubMed.
- A. Vega, E. A. Bell and P. B. Nunn, Preparation of L- and D-α-amino-β-methylaminopropionic acids and the identification of the compound isolated from Cycas circinalis as the L-isomer, Phytochemistry, 1968, 7, 1885–1887 CrossRef CAS.
- J. S. Metcalf, D. Lobner, S. A. Banack, G. A. Cox, P. B. Nunn and P. B. Wyatt, et al., Analysis of BMAA enantiomers in cycads, cyanobacteria, and mammals: in vivo formation and toxicity of d-BMAA, Amino Acids, 2017, 49(8), 1427–1439 CrossRef CAS.
- C. Fontanarosa, F. Pane, N. Sepe, G. Pinto, M. Trifuoggi and M. Squillace, et al., Quantitative determination of free D-Asp, LAsp and N-methyl-D-aspartate in mouse brain tissues by chiral separation and Multiple Reaction Monitoring tandem mass spectrometry, PLoS One, 2017, 12(6), 1–18 CrossRef PubMed.
- T. Zhang, E. Holder, P. Franco and W. Lindner, Zwitterionic chiral stationary phases based on cinchona and chiral sulfonic acids for the direct stereoselective separation of amino acids and other amphoteric compounds, J. Sep. Sci., 2014, 37(11), 1237–1247 CrossRef CAS PubMed.
- H. H. Myung, C. H. Sang, B. H. Lipshutz, Y. J. Shin and C. J. Welch, Liquid chromatographic resolution of racemic amines, amino alcohols and related compounds on a chiral crown ether stationary phase, J. Chromatogr. A, 2002, 959(1–2), 75–83 Search PubMed.
- A. F. Kotthaus and H. J. Altenbach, A new chiral derivatizing agent for the HPLC separation of α-amino acids on a standard reverse-phase column, Amino Acids, 2011, 40(2), 527–532 CrossRef CAS.
- R. Kühnreich and U. Holzgrabe, High-performance liquid chromatography evaluation of the enantiomeric purity of amino acids by means of automated precolumn derivatization with ortho-phthalaldehyde and chiral thiols, Chirality, 2016, 28(12), 795–804 CrossRef PubMed.
- R. Bhushan and H. Nagar, Indirect enantio separation of selenomethionine by reversed-phase high-performance liquid chromatography using a newly synthesized chiral derivatizing reagent based on (S)-naproxen moiety, Biomed. Chromatogr., 2014, 28(1), 106–111 CrossRef CAS PubMed.
- R. Euerby, Z. Partridge and B. Nunn, Resolution of neuroactive non-protein amino acid enantiomers by high-performance liquid chromatography utilising pre-column derivatisation with o-phthaldialdehyde-chiral thiols: Application to 2-amino-ω-phosphonoalkanoic acid homologues and α-amino-β-N-m, J. Chromatogr. A, 1989, 469, 412–419 CrossRef.
- S. Einarsson, B. Josefsson and S. Lagerkvist, Determination of Amino Acid With 9-Fluorenylmethyl Chloroformate and Reversed phase High Performance Liquid Chromatography, J. Chromatogr. A, 1983,(282), 609–618 CrossRef CAS.
- B. Josefsson, S. Einarsson, P. Moller and D. Sanchez, Separation of Amino Acid Enantiomers and Chiral Amines Using Precolumn Derivatization with (+)-1-(9-Fluorenyl)ethyl Chloroformate and Reversed-Phase Liquid Chromatography, Anal. Chem., 1987, 59(8), 1191–1195 CrossRef.
- G. E. Kisby, D. N. Roy and P. S. Spencer, Determination of β-N-methylamino-l-alanine (BMAA) in plant (Cycas circinalis L.) and animal tissue by precolumn derivatization with 9-fluorenylmethyl chloroformate (FMOC) and reversed-phase high-performance liquid chromatography, J. Neurosci. Methods, 1988, 26(1), 45–54 CrossRef CAS.
- T. Yokoyama, M. Tokuda, M. Amano and K. Mikami, Simultaneous determination of primary and secondary D- and L-amino acids by reversed-phase high-performance liquid chromatography using pre-column derivatization with two-step labelling method, Biosci., Biotechnol., Biochem., 2017, 81(9), 1681–1686, DOI:10.1080/09168451.2017.1340090.
- A. Neuberger, Stereochemistry of Amino Acids, Adv. Protein Chem., 1948, 4(C), 297–383 CrossRef CAS.
- L. Jiang, B. Aigret, W. M. De Borggraeve, Z. Spacil and L. L. Ilag, Selective LC-MS/MS method for the identification of BMAA from its isomers in biological samples, Anal. Bioanal. Chem., 2012, 403(6), 1719–1730 CrossRef CAS.
- L. Jiang, E. Johnston, K. M. Aberg, U. Nilsson and L. L. Ilag, Strategy for quantifying trace levels of BMAA in cyanobacteria by LC/MS/MS, Anal. Bioanal. Chem., 2013, 405(4), 1283–1292 CrossRef CAS PubMed.
- L. Jiang, B. Aigret, W. M. De Borggraeve, Z. Spacil and L. L. Ilag, Selective LC-MS/MS method for the identification of BMAA from its isomers in biological samples, Anal Bioanal Chem, 2012, 403(6), 1719–1730 CrossRef CAS.
- J. S. Metcalf, S. A. Banack, J. Lindsay, L. F. Morrison, P. A. Cox and G. A. Codd, Co-occurrence of β-N-methylamino-l-alanine, a neurotoxic amino acid with other cyanobacterial toxins in British waterbodies, 1990-2004, Environ. Microbiol., 2008, 10(3), 702–708 CrossRef CAS PubMed.
- L. E. Brand, J. Pablo, A. Compton, N. Hammerschlag and D. C. Mash, Cyanobacterial blooms and the occurrence of the neurotoxin, beta-N-methylamino-l-alanine (BMAA), in South Florida aquatic food webs, Harmful Algae, 2010, 9(6), 620–635, DOI:10.1016/j.hal.2010.05.002.
- A. Vega and E. A. Bell, α-Amino-β-methylaminopropionic acid, a new amino acid from seeds of Cycas circinalis, Phytochemistry, 1967, 6(5), 759–762 CrossRef CAS.
- J. Rosén and K. E. Hellenäs, Determination of the neurotoxin BMAA (â-N-methylamino-L-alanine) in cycad seed and cyanobacteria by LC-MS/MS (liquid chromatography tandem mass spectrometry), Analyst, 2008, 133(12), 1785–1789 RSC.
- S. F. Dossaji and E. A. Bell, Distribution of α-amino-β-methylaminopropionic acid in Cycas, Phytochemistry, 1973, 12(1), 143–144 CrossRef CAS.
- J. Csapo, Z. Csapo-Kiss, S. Nemethy, S. Folestad, A. Tivesten and T. G. Martin, Age determination based on amino acid racemization: a new possibility, Amino Acids, 1994, 7, 317–325 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ay02287a |
| ‡ Authors share the first authorship. |
| This journal is © The Royal Society of Chemistry 2019 |