
Unprecedented phthalocyanine–porphyrin-fused oligomers with induced chirality nature†
Yuehong
Zhang
,
Luyang
Zhao
,
Kang
Wang
* and
Jianzhuang
Jiang
*
Beijing Key Laboratory for Science and Application of Functional Molecular and Crystalline Materials, Department of Chemistry, University of Science and Technology Beijing, Beijing 100083, China. E-mail: jianzhuang@ustb.edu.cn; kangwang@ustb.edu.cn
First published on 1st December 2016
Abstract
Optically active binaphthyl units have been introduced onto the periphery of the phthalocyanine chromophores in phthalocyanine–porphyrin-fused oligomers, resulting in the first (R)- and (S)-enantiomers of Pc–Por-fused dimer 1 and trimer 2 with induced chirality nature. This represents the largest chiral tetrapyrrole-based conjugated system, to the best of our knowledge. Electronic absorption, MCD, and CD spectroscopic results in combination with theoretical calculations disclose the effective transfer of the chiral information from the binaphthyl groups to both the central tetrapyrrole-fused chromophores, indicating the effective delocalization of the π-electron density over the fused systems due to the effective electronic communication between/among the tetrapyrrole chromophores.
Introduction
In the past few decades, for the purpose of constructing novel functional molecule materials, much research efforts have been made towards two-dimensional tetrapyrrole-fused conjugated oligomers.1–5 These explorations have resulted in quite fruitful achievements in the expansion of the phthalocyanine (Pc)/porphyrin (Por)-fused oligomer family with a quite large number of Por-fused arrays,2 Pc-fused oligomers,3 and Por–Pc-fused oligomers4 with a wide range of applications in photosynthetic antenna models, molecular sensors, NIR dyes, and PDT materials associated with the effective electronic communication between the fused tetrapyrrole chromophores.5 However, optically active tetrapyrrole-fused oligomers have been rarely investigated and significantly retarded despite their scientific importance and potential applications in a range of technologies including functional chiral materials,6 with the sole report on a doubly linked porphyrin dimer.7 As a consequence, constructing chiral tetrapyrrole-fused oligomers will not only promote research in optically active tetrapyrrole-fused oligomer systems but also lead to novel two-dimensional conjugated oligomers with application potential in chiral recognition, chiral memory, and asymmetric catalysis.In the present paper, we describe the synthesis and characterization of an unprecedented chiral Pc–Por-fused dimer and trimer, namely (R)- and (S)-enantiomers of {[H2BDAPc(OBNP)3][(H2TMP)]} (1) and {[(H2TMP)][cis-H2(BDA)2Pc(OBNP)2] [(H2TMP)]} (2) [BDAPc(OBNP)3 = dianion of tris(dinaphtho[1,2-e:1′,2′-g]-1,4-(dioxocine)[2,3-b;2′,3′-k;2′′,3′′-t]-22,25-benzodiaza-phthalocyanine), cis-(BDA)2Pc(OBNP)2 = dianion of bis(dinaphtho[1,2-e:1′,2′-g]-1,4-(dioxocine)[2,3-b;2′,3′-k]-15,18,22,25-bis(benzodiaza)phthalocyanine), TMP = dianion of 5,10,15,20-tetrakis(2,4,6-trimethylphenyl)porphyrin], Scheme 1, representing the largest chiral tetrapyrrole-based conjugated system, to the best of our knowledge. Electronic absorption, magnetic circular dichroism (MCD) and circular dichroism (CD) spectroscopic results in combination with theoretical calculations disclose the effective transfer of the chiral information from the binaphthyl groups to both the central tetrapyrrole-fused chromophores, indicative of the effective delocalization of the π-electron density over the fused systems due to the effective electronic communication between/among the tetrapyrrole chromophores.
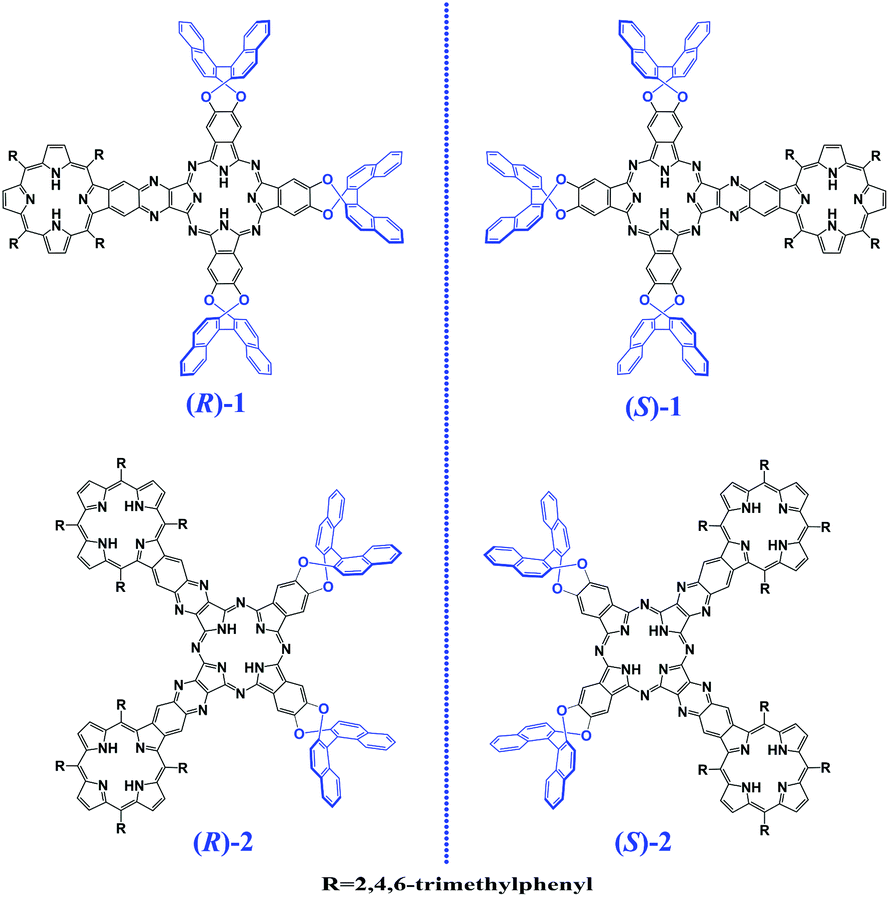 | ||
| Scheme 1 Schematic molecular structures of the (R)- and (S)-enantiomers of the Pc–Por-fused dimer 1 and trimer 2. | ||
Experimental section
General remarks
n-Pentanol was distilled from sodium. Column chromatography was carried out on silica gel (Merck, Kieselgel 60, 70–230 mesh) and biobead (BIORAD S-X1, 200–400 mesh) columns with the indicated eluents. All other reagents and solvents were used as received. The compounds (R)- and (S)-enantiomers of benzo[b]dinaphtho[2,1-e:1′,2′-g][1,4]dioxocine-2,3-dicarbonitrile8 and 5,10,15,20-tetrakis(2,4,6-trimethylphenyl)-2′,3′-di-cyanopyrazino[2,3-β]porphyrin9 were prepared according to the published procedure.1H NMR spectra were recorded on a Bruker DPX 400 spectrometer. Spectra were referenced internally using the residual solvent resonances relative to SiMe4. Electronic absorption spectra were recorded with a Hitachi U-4100 spectrophotometer. CD and MCD spectra were recorded with a JASCO J-1500 spectropolarimeter equipped with a JASCO electromagnet, which produces a magnetic field of 1.60 T with both parallel and antiparallel fields. IR spectra were recorded with KBr pellets using a Bruker Tensor 37 spectrometer with 2 cm−1 resolution. MALDI-TOF mass spectra were recorded on a Bruker BIFLEX III ultra-high resolution Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer with α-cyano-4-hydroxycinnamic acid as the matrix. Elemental analyses were performed on an Elementar Vario El III elemental analyzer.
Density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations
The DFT method of m0610 was used to simulate the CD spectra of the dimer (R)-1 and trimer (R)-2. Both the DFT and TDDFT calculations were carried out by using the 6-311 g(d) basis set. All the calculations were carried out by using the Gaussian 09 (Revision D.01) program.11Preparation of the (R)-Pc–Por-fused dimer 1 and trimer 2
A mixture of (R)-benzo[b]dinaphtho[2,1-e:1′,2′-g][1,4]dioxocine-2,3-dicarbonitrile (246.0 mg, 0.60 mmol), 5,10,15,20-tetrakis(2,4,6-trimethylphenyl)-2′,3′-dicyanopyrazino[2,3-β]porphyrin (56.2 mg, 0.060 mmol), and lithium (10.5 mg, 1.5 mmol) in n-pentanol (3.0 mL) was heated to reflux under nitrogen for 2 h. After being cooled to room temperature, the resulting green solution was poured into methanol (300 mL) containing 3.0 mL of CH3COOH. The precipitate was collected by filtration and chromatographed on a silica gel column using toluene as the eluent, giving a product containing Pc–Por-fused dimer 1, trimer 2 and symmetric phthalocyanine H2Pc(OBNP)4. This product was further purified by biobead column chromatography with CH2Cl2 as the eluent, giving the trimer (R)-2 as the first elution, which was followed by a fraction containing a mixture of the dimer (R)-1 and (R)-H2Pc(OBNP)4. The mixture was then purified by silica gel column chromatography with toluene and n-hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 v/v) as the eluent, giving (R)-1 as the first elution followed by (R)-H2Pc(OBNP)4. Repeated chromatography followed by recrystallization from CH2Cl2 and CH3OH gave (R)-1 (12.7 mg, 9.8%) as green powder and (R)-2 (4.7 mg, 5.8%) as brown powder. (R)-1, 1H NMR (CDCl3, 400 MHz): 9.88 (s, 2H), 9.62 (s, 2H), 9.30 (s, 2H), 9.00 (s, 2H), 8.74 (d, 2H, J = 4.00 Hz), 8.66 (d, 2H, J = 4.00 Hz), 8.52 (s, 2H), 8.21 (d, 2H, J = 8.00 Hz), 8.09 (t, 6H, J = 8.00 Hz), 8.03–7.86 (m, 10H), 7.80 (d, 4H, J = 8.00 Hz), 7.74 (d, 4H, J = 8.00 Hz), 7.61 (s, 4H), 7.59–7.46 (m, 10H), 7.29 (s, 4H), 3.02 (s, 6H), 2.64 (s, 6H), 2.03 (s, 6H), 1.94–1.90 (m, 18H), −0.16 (s, 2H), −2.14 (s, 2H). UV-Vis (toluene): λmax/nm (logε) = 357 (4.94), 421 (5.24), 518 (4.39), 639 (4.75), 668 (4.72), 708 (5.28), 727 (5.31). MALDI-TOF MS: an isotopic cluster peaking at m/z 2168.4, calcd for C148H99N14O6, [M + H]+ 2168.8. Anal. calcd (%) for C148H98N14O6·CH2Cl2·2C7H8: C, 80.31; H, 4.80; N, 8.04. found: C, 80.17; H, 4.78; N, 8.05. (R)-2, 1H NMR (CDCl3, 400 MHz): 9.73 (s, 2H), 9.47 (s, 2H), 9.42 (s, 2H), 9.05 (s, 2H), 8.78–8.71 (m, 6H), 8.64 (d, 2H, J = 4.00 Hz), 8.54 (s, 4H), 8.22 (d, 2H, J = 12.00 Hz), 8.11 (t, 4H, J = 8.00 Hz), 8.03 (d, 2H, J = 8.00 Hz), 7.98 (d, 2H, J = 8.00 Hz), 7.91 (d, 4H, J = 12.00 Hz), 7.82–7.78 (m, 8H), 7.66 (s, 2H), 7.61–7.32 (m, 8H), 7.31 (s, 8H), 3.31 (s, 6H), 3.06 (s, 6H), 2.67 (s, 6H), 2.65 (s, 6H), 2.17 (s, 6H), 2.07 (s, 6H), 2.02 (s, 6H), 1.98–1.94 (m, 30H), 0.54 (s, 2H), −2.05 (s, 4H). UV-Vis (toluene): λmax/nm (logε) = 428 (5.47), 526 (4.75), 565 (4.58), 680 (4.71), 719 (4.69), 761 (5.37). MALDI-TOF MS: an isotopic cluster peaking at m/z 2694.3, calcd for C184H139N20O4, [M + H]+ 2694.1. Anal. calcd (%) for C184H138N20O4·1.75CH2Cl2: C, 78.51; H, 5.02; N, 9.86. found: C, 78.57; H, 4.86; N, 9.80.
:1.5 v/v) as the eluent, giving (R)-1 as the first elution followed by (R)-H2Pc(OBNP)4. Repeated chromatography followed by recrystallization from CH2Cl2 and CH3OH gave (R)-1 (12.7 mg, 9.8%) as green powder and (R)-2 (4.7 mg, 5.8%) as brown powder. (R)-1, 1H NMR (CDCl3, 400 MHz): 9.88 (s, 2H), 9.62 (s, 2H), 9.30 (s, 2H), 9.00 (s, 2H), 8.74 (d, 2H, J = 4.00 Hz), 8.66 (d, 2H, J = 4.00 Hz), 8.52 (s, 2H), 8.21 (d, 2H, J = 8.00 Hz), 8.09 (t, 6H, J = 8.00 Hz), 8.03–7.86 (m, 10H), 7.80 (d, 4H, J = 8.00 Hz), 7.74 (d, 4H, J = 8.00 Hz), 7.61 (s, 4H), 7.59–7.46 (m, 10H), 7.29 (s, 4H), 3.02 (s, 6H), 2.64 (s, 6H), 2.03 (s, 6H), 1.94–1.90 (m, 18H), −0.16 (s, 2H), −2.14 (s, 2H). UV-Vis (toluene): λmax/nm (logε) = 357 (4.94), 421 (5.24), 518 (4.39), 639 (4.75), 668 (4.72), 708 (5.28), 727 (5.31). MALDI-TOF MS: an isotopic cluster peaking at m/z 2168.4, calcd for C148H99N14O6, [M + H]+ 2168.8. Anal. calcd (%) for C148H98N14O6·CH2Cl2·2C7H8: C, 80.31; H, 4.80; N, 8.04. found: C, 80.17; H, 4.78; N, 8.05. (R)-2, 1H NMR (CDCl3, 400 MHz): 9.73 (s, 2H), 9.47 (s, 2H), 9.42 (s, 2H), 9.05 (s, 2H), 8.78–8.71 (m, 6H), 8.64 (d, 2H, J = 4.00 Hz), 8.54 (s, 4H), 8.22 (d, 2H, J = 12.00 Hz), 8.11 (t, 4H, J = 8.00 Hz), 8.03 (d, 2H, J = 8.00 Hz), 7.98 (d, 2H, J = 8.00 Hz), 7.91 (d, 4H, J = 12.00 Hz), 7.82–7.78 (m, 8H), 7.66 (s, 2H), 7.61–7.32 (m, 8H), 7.31 (s, 8H), 3.31 (s, 6H), 3.06 (s, 6H), 2.67 (s, 6H), 2.65 (s, 6H), 2.17 (s, 6H), 2.07 (s, 6H), 2.02 (s, 6H), 1.98–1.94 (m, 30H), 0.54 (s, 2H), −2.05 (s, 4H). UV-Vis (toluene): λmax/nm (logε) = 428 (5.47), 526 (4.75), 565 (4.58), 680 (4.71), 719 (4.69), 761 (5.37). MALDI-TOF MS: an isotopic cluster peaking at m/z 2694.3, calcd for C184H139N20O4, [M + H]+ 2694.1. Anal. calcd (%) for C184H138N20O4·1.75CH2Cl2: C, 78.51; H, 5.02; N, 9.86. found: C, 78.57; H, 4.86; N, 9.80.
Preparation of the (S)-Pc–Por-fused dimer 1 and trimer 2
By employing the above-described procedure with (S)-benzo[b]dinaphtho[2,1-e:1′,2′-g][1,4]dioxocine-2,3-dicarbonitrile instead of the (R)-enantiomer precursor as the starting material, (S)-Pc–Por-fused dimer 1 and trimer 2 were also synthesized and isolated. The (S)-enantiomers of the dimer 1 and trimer 2 have identical NMR, electronic absorption, MS and elemental analysis data with the corresponding (R)-enantiomers.Results and discussion
Synthesis and spectroscopic characterization
Despite the significant achievement in the field of conjugated Por/Pc-fused oligomers, chiral tetrapyrrole-fused systems still remain undeveloped probably due to the difficulty in their synthesis and isolation, with the sole report on a porphyrin dimer with inherent chirality nature.7 Inspired with the quite lately successful synthesis of the Pc–Por-fused oligomeric compounds,12 in the present case chiral binaphthyl groups were introduced onto the phthalocyanine periphery of the Pc–Por-fused systems, resulting in chiral Pc–Por-fused oligomeric compounds 1 and 2 with induced chirality nature. The key chiral precursors, the (R)- and (S)-enantiomers of benzo[b]dinaphtho[2,1-e:1′,2′-g][1,4]dioxocine-2,3-dicarbonitrile, were synthesized following the published procedure.8 To enhance the solubility and impede the aggregation of the target oligomeric Pc–Por-fused products with a large conjugated system, bulky 2,4,6-trimethylphenyl groups were incorporated onto the porphyrin periphery in the Pc–Por-fused systems. As a consequence, mixed cyclic tetramerization of the (R)-benzo[b]dinaphtho[2,1-e:1′,2′-g][1,4]dioxocine-2,3-dicarbonitrile and 5,10,15,20-tetrakis(2,4,6-trimethylphenyl)-2′,3′-di-cyanopyrazino[2,3-β]porphyrin9 in the presence of lithium pentanolate in refluxing n-pentanol followed by treatment with acetic acid led to the isolation of the (R)-Pc–Por-fused dimer 1 (9.8%) and trimer 2 (5.8%), representing the largest chiral tetrapyrrole-based conjugated system, Scheme 1. By employing the above-described procedure with the (S)-benzo[b]dinaphtho[2,1-e:1′,2′-g][1,4]dioxocine-2,3-dicarbonitrile instead of the (R)-enantiomeric precursor as the starting material, (S)-Pc–Por-fused dimer 1 and trimer 2 were also synthesized and isolated. At the end of this paragraph, it is worth noting that despite great efforts made thus far, repeated trials still failed to afford single crystals of 1 and 2 suitable for X-ray diffraction analysis.These newly prepared Pc–Por-fused compounds 1 and 2 gave satisfactory elemental analysis data, Table S1 (ESI†). Their MALDI-TOF mass spectra clearly show intense signals for the corresponding protonated molecular ion [M + H]+. The isotopic pattern closely resembles the simulated one as exemplified by the spectra of the dimer 1 and the trimer 2, Fig. S1 and S2 (ESI†). To further clarify the corresponding molecular structures and optical properties, 1 and 2 were characterized with different spectroscopic techniques including NMR, electronic absorption, circular dichroism (CD), and magnetic CD (MCD) spectroscopy. As would normally be expected, the (R)- and (S)-enantiomers of compound 1 have identical NMR, electronic absorption, and IR spectroscopic spectra. As a consequence, only the data for (R)-1 are discussed hereafter. This is also the case for compound 2.
Satisfactory NMR spectra were obtained for the dimer 1 and trimer 2 in CDCl3, Fig. S3–S6 (ESI†) and Table S2 (ESI†). As can be found, the 1H NMR spectrum of 1 exhibits three singlets at δ = 9.88, 9.62, and 9.30 ppm due to the Pc α protons and one singlet at δ = 9.00 ppm due to the protons on the benzo[α]pyrazine moiety owing to the C2 molecular symmetry. The two doublets at δ = 8.74 and 8.66 ppm were assigned to the two kinds of β protons on the Por moiety. The other β proton signal on the Por moiety was observed at δ = 8.52 ppm. The two singlets observed at δ = 7.61 and 7.29 ppm were attributed to the protons of the meso-attached aryl groups on the Por moiety. Signals for the aromatic protons of binaphthyl on the Pc moiety exhibited several doublets and multiplets ranging from δ 8.21 to 7.46 ppm. The signals attributed to the methyl protons on the Por moiety appear as three singlets and one multiplet at δ = 3.02, 2.64, 2.03, and 1.94–1.90 ppm, respectively. In addition, the two singlets at δ = −0.16 and −2.14 ppm were assigned to the inner isoindole and pyrrole protons, respectively.
In the 1H NMR spectrum of the trimer 2, Fig. S5 (ESI†), observation of two Pc α proton singlets at δ = 9.73 and 9.47 ppm, and two singlets for protons on benzo[α]pyrazine rings at δ = 9.42 and 9.05 ppm clearly reveals the C2 molecular symmetry of this trimer with a cis-configuration rather than D2-symmetry with a trans-configuration. This was further supported by the observation of four signals due to the β protons on the Por moieties at δ = 8.77, 8.72, 8.64, and 8.54 ppm. Signals for the protons in the meso-attached aryl moieties on the Por moieties appear at δ 7.82–7.78, 7.66, and 7.31 ppm, while the aromatic protons of binaphthyl on the Pc moiety exhibit several doublets and multiplets in the range from δ 8.22 to 7.32 ppm. The signals attributed to the methyl protons on the Por moieties can also be unambiguously assigned in a similar way to the dimer 1 as tabulated in Table S2 (ESI†). Also, the singlets at δ = 0.54 and −2.05 ppm with an integral ratio of 1:2 can be assigned to the inner isoindole and pyrrole protons, respectively.
The IR spectra of the dimer 1 and trimer 2 are shown in Fig. S7 (ESI†). In addition to the bands associated with the aromatic Pc and Por moieties such as the C–H wagging and torsion vibrations, and the isoindole ring and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N aza group stretching vibrations,13 the bands at 2918–2921 and 2848–2854 cm−1 are assigned to the C–H stretching vibrations of the –CH3 groups on the Por moiety, while those at 1263 and 1088–1090 cm−1 are ascribed to the C–O–C stretching vibrations of the binaphthyl groups on the Pc moiety.14 In addition, one weak band due to the asymmetrical N–H stretching vibration of the isoindole and pyrrole moieties is observed at ca. 3300 cm−1.15
N aza group stretching vibrations,13 the bands at 2918–2921 and 2848–2854 cm−1 are assigned to the C–H stretching vibrations of the –CH3 groups on the Por moiety, while those at 1263 and 1088–1090 cm−1 are ascribed to the C–O–C stretching vibrations of the binaphthyl groups on the Pc moiety.14 In addition, one weak band due to the asymmetrical N–H stretching vibration of the isoindole and pyrrole moieties is observed at ca. 3300 cm−1.15
Optical properties
The optical properties of the (R)- and (S)-enantiomers of dimer 1 and trimer 2 in toluene are thoroughly investigated via electronic absorption, MCD, and CD spectroscopy studies. Two Soret-like bands at 357 and 421 nm and split Q-like bands at 708 and 727 nm together with two shoulders at 639 and 668 nm were observed for the dimer 1, Fig. 1a. This is also true for the trimer 2, which exhibits one pronounced Soret-like band and one intensified Q-like band at 428 and 761 nm, respectively, Fig. 1b. In the MCD spectrum of 1, Fig. 1a, one set of coupled oppositely-signed Faraday B terms were observed at 731 and 711 nm with the −ve/+ve sequence in ascending energy, indicating the absence of three-fold or higher rotation symmetry element of this system.16 This is also true for the trimer 2, with the oppositely-signed Faraday B terms observed at 776 and 755 nm. It is worth noting that in contrast to the increase in the absorption intensity, the MCD intensity of the lowest energy absorption band for the trimer gets decreased compared to that of the dimer, probably due to the smaller energy gap between the two Faraday B terms of 2 than that of 1. This in turn leads to the partly offset for the two Faraday B terms intensities of 2.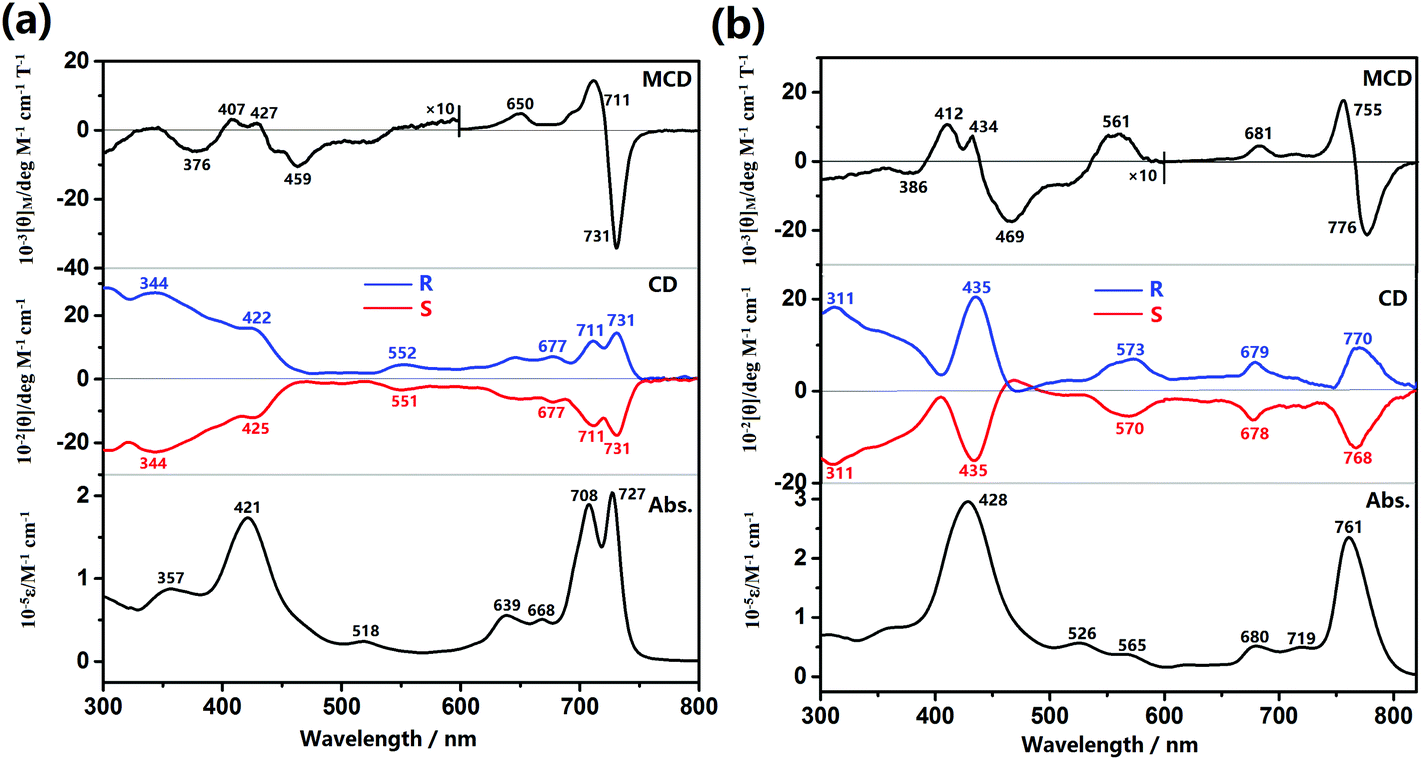 | ||
| Fig. 1 MCD, CD, and electronic absorption spectra for the (R)- and (S)-enantiomers of dimer 1 (a) and trimer 2 (b) in toluene. | ||
The CD spectra of (R)- and (S)-enantiomers of 1 and 2 are also shown in Fig. 1, in which the spectral mirror image for the two enantiomers of both 1 and 2 is clearly exhibited in the range of 300–800 nm. Nevertheless, along with the increased energy, (R)-1 presents two positive CD signals at 731 and 711 nm in the Q-band region and two positive discernible signals at 422 and 344 nm ascribed mainly to the Soret band of Por and Pc moieties, respectively. In a similar way, reverse signals at the same region are observed for (S)-1. As for the trimer, (R)-2 presents three positive CD signals at 770, 679, and 573 nm in the Q-band region and two positive signals at 435 and 311 nm in the Soret band region, and reverse signals at the same region are also observed for (S)-2. Since the binaphthyl moiety is only optically active at wavelengths shorter than ca. 260 nm, all the CD signals observed from 300–800 nm with significant intensity are actually induced from the chiral binaphthyl units to the achiral Pc/Por chromophores and can therefore be interpreted by the Kuhn–Kirkwood coupled oscillator and “CD stealing” mechanisms.17 In other words, the chiral information has been effectively transferred from the binaphthyl groups to both the Pc and Por chromophores due to the effective delocalization of the π-electrons of the fused systems and effective electronic communication between/among these chromophores. In addition, since the induced CD intensity is proportional to νNνCh/(νN2 − νCh2) (νN and νCh are the frequencies of the absorption of naphthalene and tetrapyrrole chromophore, respectively)17 and positively correlated with the absorption intensity for each transition according to the quantum theory of CD,17c the Soret band CD signals are therefore reasonably stronger than those of the Q-band CD signals for both the dimer and trimer. Due to the same reason, the intensity of the Por Soret-band CD signal for the trimer with a larger molar extinction coefficient is stronger than that of the dimer with a smaller molar extinction coefficient for its Por Soret band, Table S3 (ESI†). To quantitatively evaluate the chiral dissymmetry of these novel optically active tetrapyrrole-fused oligomers, the anisotropic factor g for the corresponding Q-bands and Soret bands is calculated based on the equation g = θ/(33 × ε)18 and tabulated in Table S3 (ESI†). In accordance with the literature result,17b the anisotropic factor g values for these absorptions are all in the magnitude of 10−4, disclosing the considerable chirality-inducing ability of the binaphthyl units in these systems. Nevertheless, the g values for the dimer amount to ca. 2.0 × 10−4, which are significantly larger than those for the trimer, ca. 1.6 × 10−4, revealing the effect of the number of chiral binaphthyl units relative to the number of the central fused tetrapyrrole chromophores.
The fluorescence spectra of compounds 1 and 2 were also recorded in toluene at room temperature, Fig. S8 (ESI†). The maxima for the fluorescence bands were observed at 734 and 780 nm for 1 and 2, respectively. The fluorescence quantum yields were estimated as 0.51 for 1 and 0.31 for 2, respectively.
Electronic structures
In order to gain further insight into the absorption and CD spectra, TD-DFT molecular orbital calculations were carried out at the level of m06/6-311 g(d).10 Partial molecular orbital (MO) energy diagrams of (R)-1 and (R)-2 are shown in Fig. 2. In line with the previous result,12 both the optically active Pc–Por-fused dimer and trimer behave as single, totally π-conjugated molecular systems due to the delocalized molecular orbital over the entire molecular skeletons in the corresponding HOMOs. The calculated absorption and CD spectra are shown in Fig. S9 (ESI†), and the calculated transition energies, oscillator strengths (f), and configurations are summarized in Tables S4 and S5 (ESI†). Despite the slightly overestimated vertical transition energies, the simulated electronic absorption spectra of (R)-1 and (R)-2 are in good agreement with the experimental ones. As can be seen for the dimer, the intense absorptions at 670 and 664 nm mainly originating from the transitions from the HOMO to the LUMO/LUMO+1 correspond nicely to the experimental Q-bands observed at 727 and 708 nm, while the absorptions in the range of 300–450 nm associated with the transitions from the HOMO−9/HOMO−12/HOMO−13/HOMO−19/HOMO−22 to the LUMO contribute to the observed Soret bands. In addition, by using the (R)-enantiomer, the calculated CD spectrum with positive signals almost reproduces the experimental spectrum with well-matched relative intensity for all the Q band and Soret band signals. Similar features can also be observed for the trimer (R)-2.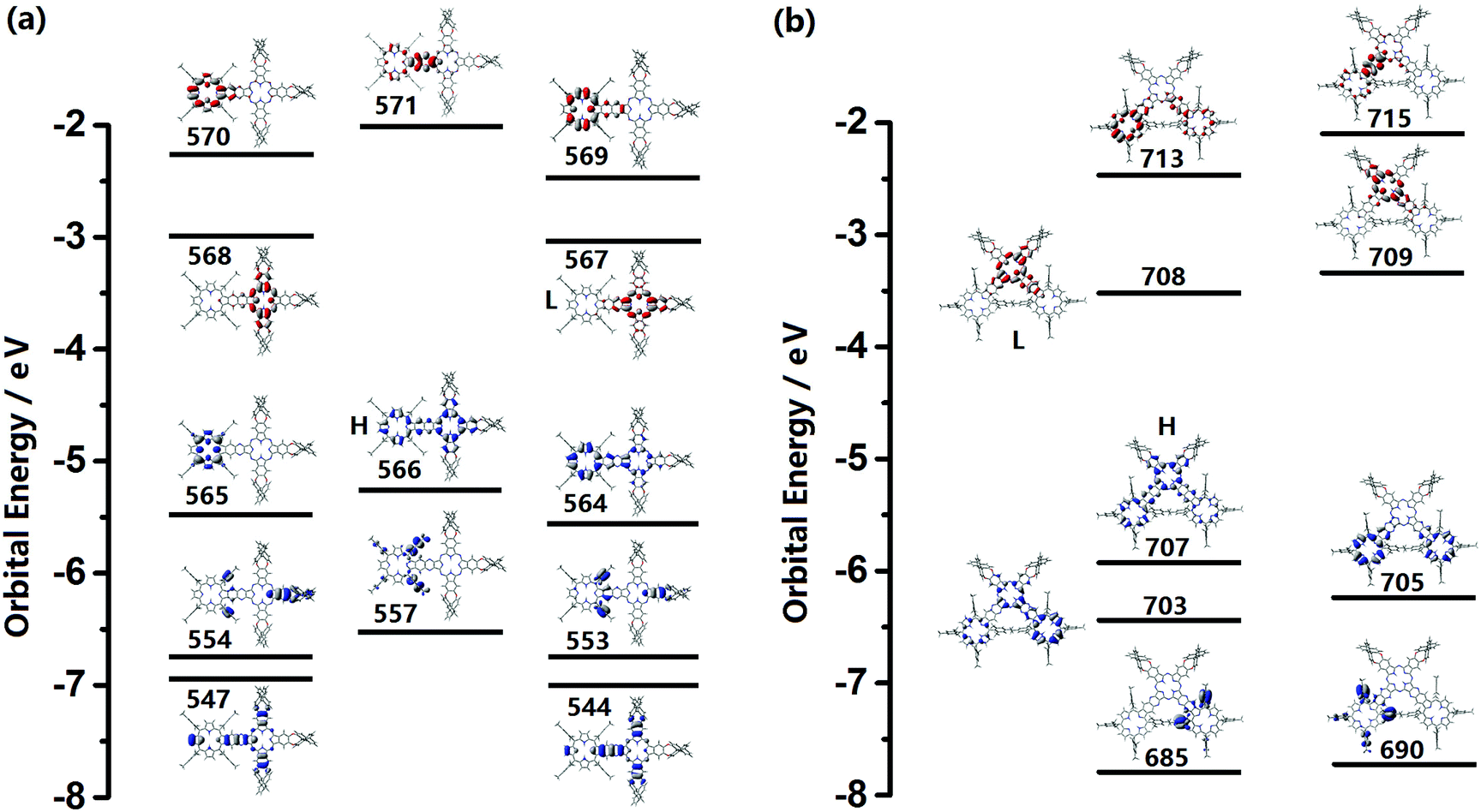 | ||
| Fig. 2 Partial molecular energy diagrams and orbitals for (R)-1 (a) and (R)-2 (b) (HOMO and LUMO are represented as H and L). Blue and red plots indicate the occupied and unoccupied MOs, respectively. | ||
Conclusions
Briefly summarizing above, an optically active Pc–Por-fused dimer and trimer, representing the largest chiral tetrapyrrole-based conjugated systems, have been synthesized and characterized for the first time through incorporating chiral binaphthyl units at the phthalocyanine periphery. This result is surely helpful for the design, synthesis, and understanding of the optical activity of chiral systems with large two-dimensional conjugated electronic structures.Acknowledgements
Financial support from the National Key Basic Research Program of China (Grant No. 2013CB933402), the Natural Science Foundation of China (no. 21290174, 21401009, and 21631003), the Beijing Municipal Commission of Education, and the University of Science and Technology Beijing is gratefully acknowledged.References
- (a) T. Tanaka and A. Osuka, Chem. Soc. Rev., 2015, 44, 943 RSC; (b) N. Kobayashi, H. Lam, W. A. Nevin, P. Janda, C. C. Leznoff, T. Koyama, A. Monden and H. Shirai, J. Am. Chem. Soc., 1994, 116, 879 CrossRef CAS; (c) C. C. Leznoff, H. Lam, S. M. Marcuccio, W. A. Nevin, P. Janda, N. Kobayashi and A. B. P. Lever, J. Chem. Soc., Chem. Commun., 1987, 699 RSC.
- (a) Y. Nakamura, S. Y. Jang, T. Tanaka, N. Aratani, J. M. Lim, K. S. Kim, D. Kim and A. Osuka, Chem. – Eur. J., 2008, 14, 8279 CrossRef CAS PubMed; (b) H. Uoyama, K. S. Kim, K. Kuroki, J.-Y. Shin, T. Nagata, T. Okujima, H. Yamada, N. Ono, D. Kim and H. Uno, Chem. – Eur. J., 2010, 16, 4063 CrossRef CAS PubMed; (c) H. Uno, K. Nakamoto, K. Kuroki, A. Fujimoto and N. Ono, Chem. – Eur. J., 2007, 13, 5773 CrossRef CAS PubMed; (d) M. J. Crossley and P. Thordarson, Angew. Chem., Int. Ed., 2002, 41, 1709 CrossRef CAS; (e) K. Sendt, L. A. Johnston, W. A. Hough, M. J. Crossley, N. S. Hush and J. R. Reimers, J. Am. Chem. Soc., 2002, 124, 9299 CrossRef CAS PubMed; (f) A. Tsuda and A. Osuka, Science, 2001, 293, 79 CrossRef CAS PubMed; (g) S. Hiroto, K. Furukawa, H. Shinokubo and A. Osuka, J. Am. Chem. Soc., 2006, 128, 12380 CrossRef CAS PubMed; (h) H. S. Cho, D. H. Jeong, S. Cho, D. Kim, Y. Matsuzaki, K. Tanaka, A. Tsuda and A. Osuka, J. Am. Chem. Soc., 2002, 124, 14642 CrossRef CAS PubMed.
- (a) C. Huang, K. Wang, J. Sun and J. Jiang, Dyes Pigm., 2014, 109, 163 CrossRef CAS; (b) K. Wang, D. Qi, H. Wang, W. Cao, W. Li, T. Liu, C. Duan and J. Jiang, Chem. – Eur. J., 2013, 19, 11162 CrossRef CAS PubMed; (c) S. G. Makarov, A. V. Piskunov, O. N. Suvorova, G. Schnurpfeil, G. A. Domrachev and D. Wöhrle, Chem. – Eur. J., 2007, 13, 3227 CrossRef CAS PubMed; (d) K. Wang, C. Huang, H. Pan, N. Kobayashi and J. Jiang, Inorg. Chem. Front., 2016 10.1039/c6qi00408c.
- (a) A. M. V. M. Pereira, A. R. M. Soares, A. Hausmann, M. G. P. M. S. Neves, A. C. Tomé, A. M. S. Silva, J. A. S. Cavaleiro, D. M. Guldi and T. Torres, Phys. Chem. Chem. Phys., 2011, 13, 11858 RSC; (b) K. Wang, D. Qi, J. Mack, H. Wang, W. Li, Y. Bian, N. Kobayashi and J. Jiang, Chin. J. Inorg. Chem., 2012, 28, 1779 CAS; (c) M. J. F. Calvete, J. P. C. Tomé and J. A. S. Cavaleiro, Curr. Org. Chem., 2013, 17, 1103 CrossRef CAS.
- (a) S. Makarov, C. Litwinski, E. A. Ermilov, O. Suvorova, B. Röder and D. Wöhrle, Chem. – Eur. J., 2006, 12, 1468 CrossRef CAS PubMed; (b) M.-C. Yoon, S. B. Noh, A. Tsuda, Y. Nakamura, A. Osuka and D. Kim, J. Am. Chem. Soc., 2007, 129, 10080 CrossRef CAS PubMed; (c) Y. Shi and X. Li, Org. Electron., 2014, 15, 286 CrossRef CAS.
- (a) N. Kobayashi, in Handbook of Porphyrin Science, ed. K. Kadish, K. Smith and R. Guilard, World Scientific, Singapore, 2012, vol. 23, ch. 110, pp. 373–440 Search PubMed; (b) H. Lu and N. Kobayashi, Chem. Rev., 2016, 116, 6184 CrossRef CAS PubMed; (c) N. Kobayashi, Coord. Chem. Rev., 2001, 219–221, 99 CrossRef CAS; (d) N. Kobayashi, in Handbook of Porphyrin Science, ed. K. Kadish, K. Smith and R. Guilard, World Scientific, Singapore, 2010, vol. 7, ch. 33, pp. 147–240 Search PubMed; (e) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40 CrossRef CAS PubMed; (f) D. Holten, D. F. Bocian and J. S. Lindsey, Acc. Chem. Res., 2002, 35, 57 CrossRef CAS PubMed; (g) K. Gopalaiah, Chem. Rev., 2013, 113, 3248 CrossRef CAS PubMed; (h) K. Konishi, K. Yabara, H. Toshishige, T. Aida and S. Inoue, J. Am. Chem. Soc., 1994, 116, 1337 CrossRef CAS; (i) K. Wang, D. Qi, H. Wang, W. Cao, W. Li and J. Jiang, Chem. – Eur. J., 2012, 18, 15948 CrossRef CAS PubMed.
- (a) A. Tsuda, H. Furuta and A. Osuka, J. Am. Chem. Soc., 2001, 123, 10304 CrossRef CAS PubMed; (b) A. Muranaka, Y. Asano, A. Tsuda, A. Osuka and N. Kobayashi, ChemPhysChem, 2006, 7, 1235 CrossRef CAS PubMed.
- K. Wang, H. Wang, J. Mack, W. Li, N. Kobayashi and J. Jiang, Acta Chim. Sin., 2012, 70, 1791 CrossRef CAS.
- S. Zhao, M. G. P. M. S. Neves, A. C. Tomé, A. M. S. Silva, J. A. S. Cavaleiro, M. R. M. Domingues and A. J. F. Correia, Tetrahedron Lett., 2005, 46, 2189 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- Y. Zhang, J. Oh, K. Wang, C. Chen, W. Cao, K. H. Park, D. Kim and J. Jiang, Chem. – Eur. J., 2016, 22, 4492 CrossRef CAS PubMed.
- (a) J. Jiang, M. Bao, L. Rintoul and D. P. Arnold, Coord. Chem. Rev., 2006, 250, 424 CrossRef CAS and references therein; (b) S. Dong, D. Qi, Y. Zhang, J. Jiang and Y. Bian, Vib. Spectrosc., 2011, 56, 245 CrossRef CAS.
- (a) H. Shang, H. Wang, W. Li and J. Jiang, Vib. Spectrosc., 2013, 69, 8 CrossRef CAS; (b) Y. Zhang, W. Cao, K. Wang and J. Jiang, Dalton Trans., 2014, 43, 9152 RSC.
- (a) X. Zhang, Y. Zhang and J. Jiang, Vib. Spectrosc., 2003, 33, 153 CrossRef CAS; (b) Y. Zhang, P. Ma, P. Zhu, X. Zhang, Y. Gao, D. Qi, Y. Bian, N. Kobayashi and J. Jiang, J. Mater. Chem., 2011, 21, 6515 RSC.
- (a) J. Mack, M. J. Stillman and N. Kobayashi, Coord. Chem. Rev., 2007, 251, 429 CrossRef CAS; (b) J. Michl, J. Am. Chem. Soc., 1978, 100, 6801 CrossRef CAS.
- (a) N. Kobayashi, R. Higashi, B. C. Titeca, F. Lamote and A. Ceulemans, J. Am. Chem. Soc., 1999, 121, 12018 CrossRef CAS; (b) L. Zhao, K. Wang, T. Furuyama, J. Jiang and N. Kobayashi, Chem. Commun., 2014, 50, 7663 RSC; (c) J. A. Schellman, Chem. Rev., 1975, 75, 323 CrossRef CAS.
- (a) Y. Zhou, Z. Zhu, W. Huang, W. Liu, S. Wu, X. Liu, Y. Gao, W. Zhang and Z. Tang, Angew. Chem., Int. Ed., 2011, 50, 11456 CrossRef CAS PubMed; (b) B. Han, Z. Zhu, Z. Li, W. Zhang and Z. Tang, J. Am. Chem. Soc., 2014, 136, 16104 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: MS spectra; NMR and IR spectra for 1 and 2; simulated electronic absorption and CD spectra for 1 and 2; elemental analytical and mass spectroscopic data, NMR data and electronic absorption data for 1 and 2; electron density difference plots of electron transitions for 1 and 2. See DOI: 10.1039/c6qi00496b |
| This journal is © the Partner Organisations 2017 |