
Towards mismatched DNA photoprobes and photoreagents: “elbow-shaped” Ru(II) complexes†
Q.
Deraedt‡
 a,
L.
Marcélis
a,
F.
Loiseau
b and
B.
Elias
*a
a,
L.
Marcélis
a,
F.
Loiseau
b and
B.
Elias
*a
aInstitute of Condensed Matter and Nanosciences, Molecules, Solids and Reactivity (IMCN/MOST), Université catholique de Louvain, Place Louis Pasteur 1/L4.01.02, 1348 Louvain-la-Neuve, Belgium. E-mail: Benjamin.Elias@uclouvain.be; Fax: +3210474168; Tel: +3210473014
bDépartement de Chimie Moléculaire, Université Grenoble-Alpes, CNRS UMR 5250, BP53 38041 Grenoble, France
First published on 2nd November 2016
Abstract
Due to their potentially harmful consequences, the detection of mismatched DNA is a subject of high interest. In order to probe DNA mismatches by means of luminescent sensitive Ru(II) complexes, “elbow-shaped” extended planar ligands based on an acridine or phenazine core have been synthesized (npp = naphtho[1,2-b]pyrido[3,2-f][1,7]phenanthroline and bdppz = benzo[h]dipyrido[3,2-a:2′,3′-c]phenazine). The photophysical studies revealed that [Ru(bpy)2npp]2+ (bpy = 2,2′-bipyridine) allows emission from a 3MLCTbpy/npp excited state, while the lowest 3MLCT for [Ru(bpy)2bdppz]2+ shifts from a bright 3MLCTbpy/![[b with combining low line]](https://www.rsc.org/images/entities/b_char_0062_0332.gif)
![[d with combining low line]](https://www.rsc.org/images/entities/b_char_0064_0332.gif)
![[p with combining low line]](https://www.rsc.org/images/entities/b_char_0070_0332.gif) pz state towards a poorly luminescent 3MLCTbdp
pz state towards a poorly luminescent 3MLCTbdp![[z with combining low line]](https://www.rsc.org/images/entities/b_char_007a_0332.gif) state, probably due to the formation of H-bonds with water molecules in the excited state. Studies of the complexes in the presence of DNA demonstrate that they strongly bind to polynucleotides. In the presence of single base mismatched hairpin oligonucleotides, while [Ru(bpy)2npp]2+ does not show remarkable sensitivity to the nature of the mismatch site, [Ru(bpy)2bdppz]2+ exhibits noticeably differential luminescence behavior correlated to the nature and stability of the mismatch compared to the well-matched sequence. Photooxidant analogues of these complexes were prepared, replacing bpy ancillary ligands by bpz (2,2′-bipyrazine). A photo-induced electron transfer from dGMP and polynucleotides to the excited states of [Ru(bpz)2npp]2+ and [Ru(bpz)2bdppz]2+ is observed. These results emphasized the role of “elbow-shaped” Ru(II) complexes as promising candidates for the development of mismatch-sensitive DNA photoprobes.
state, probably due to the formation of H-bonds with water molecules in the excited state. Studies of the complexes in the presence of DNA demonstrate that they strongly bind to polynucleotides. In the presence of single base mismatched hairpin oligonucleotides, while [Ru(bpy)2npp]2+ does not show remarkable sensitivity to the nature of the mismatch site, [Ru(bpy)2bdppz]2+ exhibits noticeably differential luminescence behavior correlated to the nature and stability of the mismatch compared to the well-matched sequence. Photooxidant analogues of these complexes were prepared, replacing bpy ancillary ligands by bpz (2,2′-bipyrazine). A photo-induced electron transfer from dGMP and polynucleotides to the excited states of [Ru(bpz)2npp]2+ and [Ru(bpz)2bdppz]2+ is observed. These results emphasized the role of “elbow-shaped” Ru(II) complexes as promising candidates for the development of mismatch-sensitive DNA photoprobes.
Introduction
DNA mutations are the substrates for evolution. However, in some cases, they are also the basis for the triggering of harmful transformations.1 To prevent these mutations, DNA has developed its own repair machinery;2–4 however, some mutations can escape this control process, and ultimately be at the origin of cancer. Moreover, deficiency in the mismatch repair machinery (MMR) can occur and is associated with several types of cancer, which are often resistant to classic chemotherapeutic strategies.2,3,5,6 The design of dyes able to specifically detect mutations and defects, like a single-base pair mismatched in the DNA structure is of great interest for therapeutic applications.In this context, acridine derivatives have been widely used since the late 19th century.7,8 These dyes have been used by histologists to stain cell and tissue sections, lying at the origin of chemotherapy. In the middle of the 20th century, a new class of DNA photoprobes emerged, i.e. transition metal complexes. These compounds offer numerous advantages with respect to their purely organic predecessors, such as the modularity of their structural, photophysical and photochemical properties. Many complexes allow continuous exposure to light, enabling the real-time monitoring of the probes by spectroscopic techniques (i.e. no photobleaching). Among them, polypyridyl Ru(II) complexes became widely used, in particular thanks to the description of the “light switch” effect observed for [Ru(bpy)2dppz]2+ (bpy = 2,2′-bipyridine, dppz = dipyrido[3,2-a:2′,3′-c]phenazine). This complex can be used as a photoprobe for DNA, since, while the complex is not emissive in water, its luminescence is turned on upon interaction with DNA. Despite the advent of transition metal complexes bearing phenazine derivatives in this field a few decades ago,9–11 their acridine counterparts have surprisingly received little attention.12–14
For several years, photoreactive Ru(II) complexes have also been developed, in order to oxidize with DNA upon irradiation. It has indeed been shown in the literature that Ru(II) complexes containing at least two π-deficient ligands such as TAP (TAP = 1,4,5,8-tetraazaphenanthrene) or bpz (bpz = 2,2′-bipyrazine) are able to trigger the oxidation of guanine moieties under irradiation.15–21
The development of metal complexes able to selectively recognize mismatched sites in DNA is of great interest and the subject of numerous studies. Even if some phenazine based complexes, like [Ru(bpy)2dppz]2+ and their analogues were shown to be able to discriminate between well-matched vs. mismatched hairpin sequences,22–24 we aimed to tune the sensitivity of such complexes to the nature of the mismatch site by modifying the shape of the intercalating ligand. We have previously shown that the Ru(II) complex bearing the acridine analogue to dppz, i.e. dpac (dpac = dipyrido[3,2-a:2′,3′-c]acridine), displays strongly different photophysical properties than the phenazine derivative, emphasizing the role of both phenazine nitrogens in the “light switch” process.25 A lack of sensitivity of the dpac-based complexes has been observed and is ascribed to the “linear” structure of the extended dpac ligand.
In this context, our work focuses on the synthesis of “elbow-shaped” extended planar aromatic ligands (npp = naphtho[1,2-b]pyrido[3,2-f][1,7]phenanthroline and bdppz = benzo[h]dipyrido[3,2-a:2′,3′-c]phenazine) based on an acridine and a phenazine core, respectively, and the photophysical studies of their respective Ru(II) complexes ([Ru(bpy)2npp]2+, 1 and [Ru(bpy)2bdppz]2+, 2) (Fig. 1a). The sensitivity of the two new “elbow-shaped” complexes to probe DNA mismatches was investigated. As our complexes show avid intercalation into the DNA base pair stack, we decided to exploit their DNA binding properties to develop new photoreactive complexes by replacing their ancillary bpy ligands by π-deficient bpz (Fig. 1). A new linear [Ru(bpz)2dpac]2+ (dpac = dipyrido[3,2-a;2′,3′-c]acridine, 3) complex has also been prepared for comparison purposes.
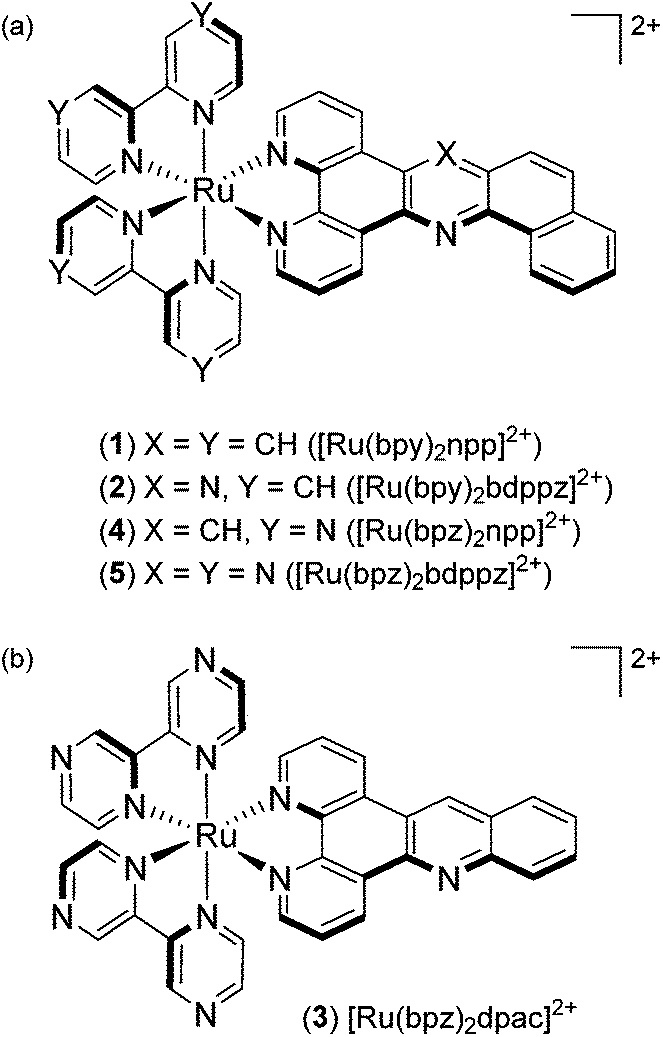 | ||
| Fig. 1 Structures of (a) [Ru(bpy)2npp]2+ (1), [Ru(bpy)2bdppz]2+ (2), [Ru(bpz)2npp]2+ (4) and [Ru(bpz)2bdppz]2+ (5); (b) [Ru(bpz)2dpac]2+ (3). | ||
In this paper, we report a comparative experimental study on “elbow-shaped” Ru(II) complexes, and analyze the influence of the acridine core, as well as bpz ancillary ligands on the photophysical properties and selective DNA interactions of the resulting Ru(II) complexes compared to the phenazine equivalents.
Results and discussion
Synthetic procedures
A Tröger's base modified synthetic scheme previously described by our group was found to be successful and allowed us to obtain npp with high yields.25 bdppz is obtained through condensation between 1,10-phenanthroline-5,6-dione and 1,2-diaminonaphthalene.The bpy-based Ru(II) complexes were synthesized by the direct chelation of the extended ligand onto a [Ru(bpy)2Cl2] precursor (Fig. S4†). While this methodology was revealed to be successful for the npp photo-oxidizing analogue, the bpz-based bdppz derivative was obtained from the condensation of a [Ru(bpz)2(1,10-phen-5,6-dione)]2+ precursor with 1,2-diaminonaphthalene (Fig. S5†). All intermediates and final compounds are protected from direct light during the synthesis and purification steps to prevent photochemical degradation. The reactions are performed under argon to avoid any oxidation of the metal center.
The complexes are characterized by HRMS and 1H NMR spectroscopy (see the Experimental section and ESI†). The 1H NMR shows unambiguously the absence of symmetry elements for all the complexes, which induces the non-equivalence of (i) the protons on the extended planar ligand and (ii) the ancillary ligands. All the multiplicity and coupling constants cannot be determined due to the superposition of several signals and/or the lack of fine resolution for some peaks in the 1H NMR spectra. The 1H NMR spectra are sensitive to concentration as some shifts occurred upon change in the concentration (data not shown). For solubility purposes, all the complexes are converted to the chloride salt for experiments conducted in water (or buffer), or to the hexafluorophosphate salt for studies in organic solvents.
Electrochemistry
The electrochemical data for the five complexes are obtained by cyclic voltammetry in dry deoxygenated MeCN or N,N-dimethylformamide (Table 1). In oxidation, complexes 1–2 display a wave at a potential close to that of the Ru2+/Ru3+ oxidation of [Ru(bpy)3]2++ (ref. 26) and [Ru(phen)2dpac]2+ (termed Ru-DPAC) (1.35 V vs. Ag/AgCl).25 This oxidation process is reversible, as ΔEp is in the range of 60–70 mV for each of the complexes (see the ESI†). A strong anodic shift is observed for complexes 3–5 (1.78–1.80 V vs. Ag/AgCl) due to the presence of the strong π-acceptor bpz ancillary ligands. Indeed, the presence of electron-withdrawing ligands such as bpz stabilizes the HOMO of these systems, which is localized on the metal cation. The nature of the extended ligand does not really affect the oxidation potential of all the complexes.| Complex |
E
1/2 ox![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a [V vs. Ag/AgCl] a [V vs. Ag/AgCl] |
E
1/2 redb [V vs. Ag/AgCl] |
|
|
||
|---|---|---|---|---|---|---|
a Measured in dry acetonitrile.
b Measured in dry N,N-dimethylformamide.
c The excited state oxidation potential was estimated from the ground state oxidation potential and the energy of emission maximum with the equation 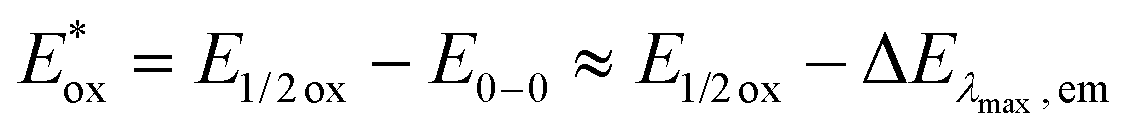 .
d The excited state reduction potential was estimated from the ground state reduction potential and the energy of emission maximum with the equation .
d The excited state reduction potential was estimated from the ground state reduction potential and the energy of emission maximum with the equation  . .
|
||||||
| [Ru(bpy)3]2+25 |
1.34 | −1.28 | −1.47 | −1.71 | −0.71 | 0.77 |
|
Ru-DPPZ27 |
1.29 | −0.97 | −1.39 | −1.62 | −0.75 | 1.07 |
| Ru-DPAC | 1.35 | −1.22 | −1.35 | −1.66 | −0.71 | 0.84 |
| [Ru(bpy)2npp]2+ (1) | 1.39 | −1.23 | −1.38 | −1.60 | −0.66 | 0.82 |
| [Ru(bpy)2bdppz]2+ (2) | 1.36 | −1.02 | −1.37 | −1.61 | −0.64 | 0.98 |
| [Ru(bpz)3]2+28 |
2.02 | −0.63 | −0.82 | −1.09 | 0.02 | 1.37 |
| [Ru(bpz)2dppz]2+29 |
1.82 | −0.72 | — | — | −0.14 | 1.24 |
| [Ru(bpz)2dpac]2+ (3) | 1.78 | −0.69 | −0.83 | — | −0.15 | 1.24 |
| [Ru(bpz)2npp]2+ (4) | 1.80 | −0.69 | −0.87 | — | −0.13 | 1.24 |
| [Ru(bpz)2bdppz]2+ (5) | 1.80 | −0.66 | −0.87 | — | −0.14 | 1.28 |

Three reduction waves are monitored for complexes 1–2, while only two for the bpz derivatives 3–5 within the investigated potential window. From the comparison with the reduction potentials of [Ru(bpy)3]2+ (−1.28 V vs. Ag/AgCl)27 and [Ru(bpy)2dppz]2+ (termed Ru-DPPZ) (−0.95 V vs. Ag/AgCl),27 the first reduction of complexes 1–2 is attributed to the addition of one electron onto the npp or bdppz ligand, respectively. The anodic shift observed for 2 (−1.02 V vs. Ag/AgCl) indicates a greater stabilization of the LUMO with respect to that of Ru-DPAC and 1 respectively, indicating that the π* orbital is centered on the bdppz ligand. The next two reduction waves for 1–2 are attributed to successive reduction of the two ancillary bpy ligands due to the similarity of the potentials with [Ru(bpy)3]2+. The two reduction waves for complexes 3–5 are attributed to the successive reduction of each of the ancillary bpz ligands by comparison with the reduction potentials of [Ru(bpz)3]2+ (−0.63 and −0.82 V vs. Ag/AgCl),28 [Ru(bpz)2dppz]2+ (−0.72 V vs. Ag/AgCl)29 and their phen/bpy counterparts.
The potentials in the excited state in water (namely, 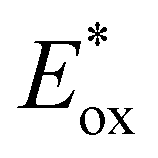 and
and  ) for all complexes have been estimated from the ground-state redox potentials and the energy of the excited state corresponding to the maximum of the emission spectrum at 298 K in water (vide infra). Not surprisingly, bpz-containing complexes 3–5 are more oxidizing in the excited state than 1–2. These three complexes should photo-oxidize DNA through guanine residues.
) for all complexes have been estimated from the ground-state redox potentials and the energy of the excited state corresponding to the maximum of the emission spectrum at 298 K in water (vide infra). Not surprisingly, bpz-containing complexes 3–5 are more oxidizing in the excited state than 1–2. These three complexes should photo-oxidize DNA through guanine residues.
Absorption spectra
The absorption data in water and acetonitrile under ambient air conditions for the five complexes, as well as for other complexes for comparison purposes, are listed in Table 2. Absorption spectra for 1–5 in acetonitrile are depicted in Fig. 2a (see the ESI† for absorption spectra in water). In our case, a comparison with the literature and absorption spectra of the free ligands (ESI†) allowed the assignment of the intense absorption (ε ≥ 105 M−1 cm−1, ca. 300 nm) bands in the UV region to ligand centered (LC) transitions and the ones in the visible region (ε ≈ 104 M−1 cm−1 around λ ≈ 400–450 nm) to metal-to-ligand-charge-transfer (MLCT) transitions Ru → L/L′, where L = bpy/bpz (1MLCTbpy/bpz) and L′ = dpac/npp/bdppz (1MLCTdpac/npp/bdppz).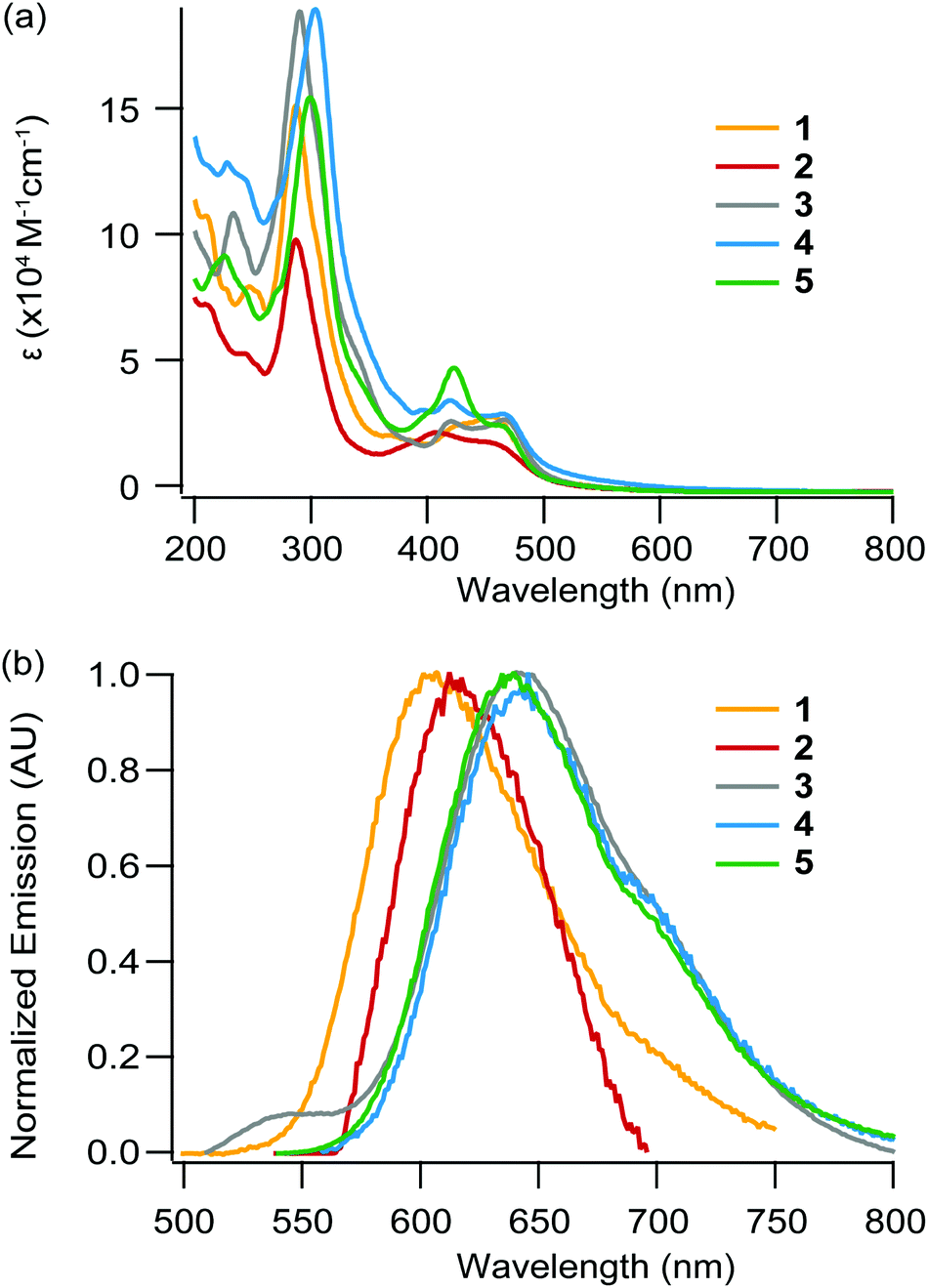 | ||
| Fig. 2 (a) Absorption and (b) emission spectra of 1 ([Ru(bpy)2npp]2+, yellow), 2 ([Ru(bpy)2npp]2+, red), 3 ([Ru(bpz)2dpac]2+, grey), 4 ([Ru(bpz)2npp]2+, blue) and 5 ([Ru(bpz)2bdppz]2+, green) in acetonitrile under air at room temperature. | ||
| Complex | Absorbance λmax [nm] (ε [104 M−1 cm−1])a | |
|---|---|---|
| CH3CN | H2O | |
| a Measurements made with solutions 1 × 10−5 mol L−1 in complex at room temperature. Extinction coefficients are reported in brackets. Absorption bands in the visible region (ε ≈ 104 M−1 cm−1 around λ ≈ 400–450 nm) are attributed to Metal-to-Ligand Charge-Transfer (MLCT) transitions. | ||
| [Ru(bpy)3]2+27 |
250 (2.51), 285 (8.71), 323 (sh), 345 (sh), 452 (1.45) | 250, 286, 322, 345, 451 |
| Ru-DPPZ 27 | 255 (4.18), 284 (9.36), 315 (sh), 342 (sh), 352 (sh), 357 (1.56), 366 (1.55), 448 (1.57) | 255, 283, 315, 341, 352, 357, 365, 448 |
| Ru-DPAC | 224 (15.1), 264 (16.3), 280 (11.6), 448 (2.62) | 224, 264 (16.2), 280, 448 (2.62) |
| [Ru(bpy)2npp]2+ (1) | 210 (11.1), 248 (9.21), 287 (15.6), 457 (2.94) | 210, 248, 287 (15.6), 457 (2.93) |
| [Ru(bpy)2bdppz]2+ (2) | 210 (7.10), 243 (5.64), 287 (9.96), 404 (2.32), 450 (1.89) | 210, 243, 287 (9.96), 404, 448 (1.87) |
| [Ru(bpz)3]2+28 |
240 (2.0), 291 (5.1), 338, 415, 440 (1.3) | — |
| [Ru(bpz)2dppz]2+29 |
290 (7.87), 420 (1.59), 445 (1.45), 456 (1.45) | — |
| [Ru(bpz)2dpac]2+ (3) | 233 (11.3), 290 (19.2), 418 (2.80), 466 (2.86) | 233, 290 (19.0), 418, 466 (2.85) |
| [Ru(bpz)2npp]2+ (4) | 229 (12.8), 245 (12.2), 303 (19.9), 395 (2.75), 419 (3.50), 460 (2.99) | 229, 245, 303 (19.8), 395, 419, 460 (2.99) |
| [Ru(bpz)2bdppz]2+ (5) | 226 (9.64), 246 (8.14), 299 (15.6), 396 (2.79), 419 (4.41), 460 (2.60) | 226, 246, 299 (15.5), 396, 419, 460 (2.58) |
The increased conjugation provided by the addition of an aromatic ring in npp and bdppz compared to the dpac and dppz structure respectively, strikingly leads to small changes in the absorption spectra. Indeed, only a small bathochromic shift (ca. 2–9 nm) of the MLCT bands is observed for 1 and 2 compared to Ru-DPAC and Ru-DPPZ respectively. The MLCT absorption bands observed for 3–5 are notably more bathochromic (shift of ca. 12–18 nm), as a result of the stabilization of the LUMO for these complexes; indeed, the lowest energy transition of 3–5 corresponds to the promotion of an electron from the metal center to one of the ancillary bpz ligand, the π* orbital of which is more stabilized, leading to a [RuIII(bpz˙−)(bpz)L′]2+ excited state. For complexes 3–5, the most bathochromic transitions 1MLCTbpz are thus slightly affected upon the change of the extended planar ligand, confirming that the latter is not involved in these electronic transitions.
Emission spectra, lifetimes, quantum yields
Complexes 1–5 display broad unstructured emission both in an organic solvent and water (Fig. 2b and ESI†), suggesting a 3MLCT-type excited state. The excited state lifetimes and luminescence quantum yields for each complex are measured in acetonitrile and water under an argon atmosphere (Table 3). The large kr values (>104 s−1), the positive solvatochromic effect (Δλ ≈ +2–11 nm when going from MeCN to water) and the hypsochromic shift of the emission band at 77 K (see Fig. S3†) confirm the formation a dipole in the excited state, consequently leading to a charge transfer excited state. A decrease in the luminescence intensity in air-equilibrated solvents with respect to argon-purged solutions is observed in all cases, indicating a photosensitization of 1O2 by the excited Ru(II) complexes (data not shown). The energy of luminescence decreases from 1 to 2, in agreement with the stabilization of the π* orbital centered on the extended planar ligand going from npp to the more electron-withdrawing bdppz core. Interestingly, complex 2 does not behave like its analogs Ru-DPPZ, as it is still emissive in water, even if its quantum yield of luminescence is weak. Ru-DPPZ, which is known as a “light switch”, is not luminescent at all in water but emits light in an organic solvent. Numerous experimental and theoretical studies have been devoted to understanding the light-switching effect.10,30–32 It is now commonly accepted that luminescence quenching in aqueous media is caused by hydrogen-bonding with two water molecules, leading to the stabilization of dark states being a 3MLCTdp and a 3LCdp, the latter being the lowest triplet state.32 The residual luminescence in water observed for 2 could likely be explained by an inhibition of the formation of the second H-bond between water and the central nitrogen atom of the bdppz ligand, cramped by its “elbow-shaped” structure.
| Complex | Emission λmaxa,b [nm] |
Emission λmaxb at 77 K [nm] |
Φ
emc,d |
τ
emc [ns] |
k r [103 s−1] | ||||
|---|---|---|---|---|---|---|---|---|---|
| CH3CN | H2O | EtOH/MeOH 4/1 | CH3CN | H2O | CH3CN | H2O | CH3CN | H2O | |
| a Measurements made with solutions 1 × 10−5 mol L−1 in complex under air. b λ exc = 448 nm. c Measurements made with solutions 1 × 10−5 mol L−1 in complex under argon. d Measured relative to [Ru(bpy)3]2+ in an aerated aqueous solution (Φairem = 0.028).28 e No luminescence is observed in H2O. | |||||||||
| [Ru(bpy)3]2+36 |
604 | 604 | 578 | 0.062 | 0.042 | 855 | 630 | 77 | 69 |
| Ru-DPPZ | 610 | —e | 581 | 0.031 | —e | 692 | —e | 44.8 | —e |
| Ru-DPAC 27 | 597 | 604 | 567 | 0.038 | 0.066 | 710 | 999 | 53.5 | 66.1 |
| [Ru(bpy)2npp]2+ (1) | 604 | 606 | 577 | 0.083 | 0.092 | 702 | 750 | 118.2 | 122.7 |
| [Ru(bpy)2bdppz]2+ (2) | 611 | 620 | 580 | 0.068 | 0.0015 | 940 (+2% 5.5) | 372 (+13% 9.4) | 72.3 | 4.1 |
| [Ru(bpz)2dpac]2+ (3) | 632 | 643 | 606 | 0.111 | 0.035 | 466 | 825 | 238.2 | 75.1 |
| [Ru(bpz)2npp]2+ (4) | 632 | 643 | 610 | 0.057 | 0.015 | 471 | 790 | 121.2 | 31.8 |
| [Ru(bpz)2bdppz]2+ (5) | 628 | 639 | 599 | 0.305 | 0.092 | 522 | 1565 (+0.5% 5.5) | 584.3 | 58.8 |
Complexes 3–5 show almost identical emission maxima, which are very similar to the ones of the reference complex [Ru(bpz)2dppz]2+ (633 nm in CH3CN).33 The chemical modification of the acridine core by addition of an aromatic ring and/or a nitrogen atom has thus only a small influence on the energy difference between the triplet excited state (3MLCT) and the ground state, as expected since the lowest 3MLCT state involves bpz ligands (noted 3MLCTbpz). Notably, the emission energy is decreased with respect to the phen/bpy analogs due to the decrease in the energy of the π* orbital involved in the radiative transition, also confirming the lowest excited state to be 3MLCTbpz. Interestingly, complex 5 displays higher quantum yields and excited state lifetimes than 3 and 4. In addition, the quantum yield of these three complexes decreases when going from acetonitrile to water, indicating a partial quenching of the luminescence by (i) H-bond formation between solvent molecules and bpz moieties, and (ii) strong stabilization of the 3MLCTbpz excited state resulting in a higher non-radiative deactivation occurrence.34,35
Our data suggest that complexes 1 and 2 follow different photophysical schemes. It has been showed in the literature that absorption of photons by Ru-DPPZ leads to the formation of a charge separated state Ru3+/dppz˙− in which an electron is transferred from the metal center either towards the bpy ligands and the phen part of the dppz ligand, or towards the phenazine part of the dppz ligand, depending on the environment of the complex.37,38 Such a behavior accounts for the unusual photophysical properties of Ru-DPPZ. We previously reported that the replacement of a nitrogen atom of the phenazine moiety of dppz by a carbon atom strongly modifies the excited state properties of the resulting Ru(II) complex.23 In the case of 1, 1MLCT states are generated upon light irradiation which rapidly undergo fast ISC to form a 3MLCT state involving either the ancillary bpy ligands ([RuIII(bpy˙−)]* – Fig. 3a) or the bpy fragment of the extended planar ligand ([RuIII(npp˙−)]*). If the “elbow-shape” of the npp ligand disfavors solvent interactions with the central nitrogen atom in the excited state, no further stabilization of the excited states towards the extended portion of npp may occur in that case. In opposition to 1, a different photophysical scheme is required to rationalize the luminescence behavior of 2. Analogously to Ru-DPPZ,23 several 3MLCT states are required to explain the experimental data. At first, after ISC39,40 from 1MLCT excited states, a luminescent 3MLCT state involving either the ancillary bpy ligands ([RuIII(bpy˙−)]* – Fig. 3b) or the bpy fragment from bdppz ([RuIII(b![[d with combining low line]](https://www.rsc.org/images/entities/char_0064_0332.gif)
![[p with combining low line]](https://www.rsc.org/images/entities/char_0070_0332.gif) pz˙−)]*) is formed. Subsequent stabilization then occurs on the phenazine moiety of the ligand, leading to the formation of a delocalized [RuIII(bdp
pz˙−)]*) is formed. Subsequent stabilization then occurs on the phenazine moiety of the ligand, leading to the formation of a delocalized [RuIII(bdp![[z with combining low line]](https://www.rsc.org/images/entities/char_007a_0332.gif) ˙−)]* excited state. Further interconversions from this state ultimately arise in aqueous media: one to a luminescent mono-hydrogen bonded excited state [RuIII(bdp˙−-H)]*, and another one to a non-luminescent bis-hydrogen bonded excited state [RuIII(bdp˙−-2H)]* (Fig. 3b).25,41 The presence of the latter is suggested by the short excited state lifetime component (see Table 3) and partial luminescence quenching of 2 in water. However, the formation of the second H-bond, responsible for the extinction of the luminescence, is disfavored due to the “elbow-shape” of the ligand. Such a hypothesis lies in agreement with the model of Lincoln and co-workers for which two H-bonds are required for the complete luminescence quenching of Ru-DPPZ in water.32 It should be noted that the analogous complex [Ru(bpy)2(dppn)]2+, where dppn (benzo[i]dipyrido[3,2-a:2′,3′-c]phenazine) is a linear equivalent of bdppz, displays a notably lower quantum yield of emission compared to dppz and bdppz complexes in organic media.38,42,43 In this case, an efficient population of a non-luminescent low-lying 3LC state located on the dppn ligand is responsible for the extremely weak emission of the complex. This low-lying LC state does not seem to play such a drastic role in the case of the elbow-shaped bdppz based complex.
˙−)]* excited state. Further interconversions from this state ultimately arise in aqueous media: one to a luminescent mono-hydrogen bonded excited state [RuIII(bdp˙−-H)]*, and another one to a non-luminescent bis-hydrogen bonded excited state [RuIII(bdp˙−-2H)]* (Fig. 3b).25,41 The presence of the latter is suggested by the short excited state lifetime component (see Table 3) and partial luminescence quenching of 2 in water. However, the formation of the second H-bond, responsible for the extinction of the luminescence, is disfavored due to the “elbow-shape” of the ligand. Such a hypothesis lies in agreement with the model of Lincoln and co-workers for which two H-bonds are required for the complete luminescence quenching of Ru-DPPZ in water.32 It should be noted that the analogous complex [Ru(bpy)2(dppn)]2+, where dppn (benzo[i]dipyrido[3,2-a:2′,3′-c]phenazine) is a linear equivalent of bdppz, displays a notably lower quantum yield of emission compared to dppz and bdppz complexes in organic media.38,42,43 In this case, an efficient population of a non-luminescent low-lying 3LC state located on the dppn ligand is responsible for the extremely weak emission of the complex. This low-lying LC state does not seem to play such a drastic role in the case of the elbow-shaped bdppz based complex.
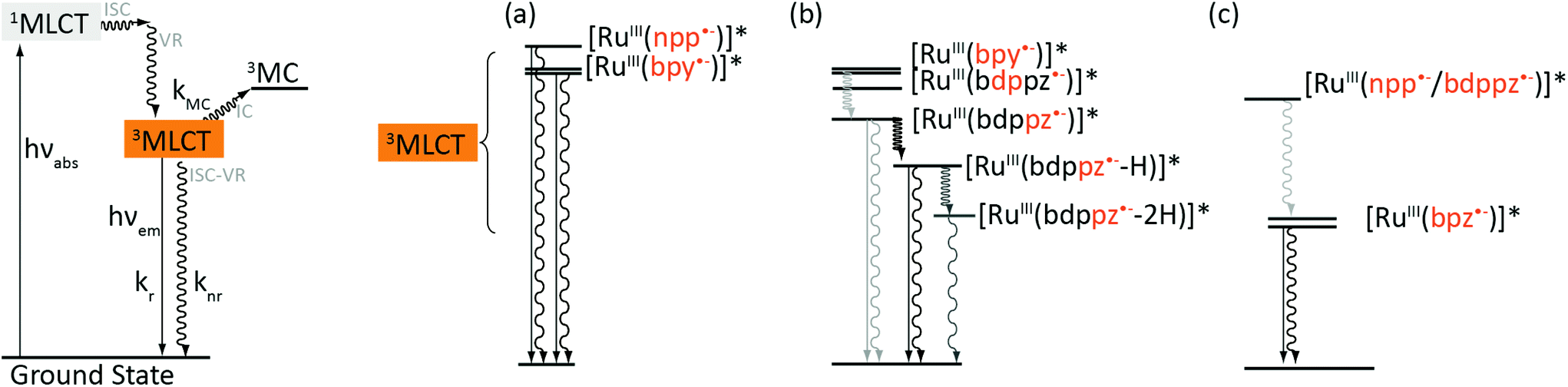 | ||
| Fig. 3 Representation of the photophysical scheme for bpy- and bpz-based complexes in water. The deactivation of 3MLCT states is emphasized on the right part of the figure for (a) [Ru(bpy)2npp]2+ (1), (b) [Ru(bpy)2bdppz]2+ (2) and (c) [Ru(bpz)2dpac]2+ (3), [Ru(bpz)2npp]2+ (4) and [Ru(bpz)2bdppz]2+ (5). Solid arrows stand for radiative transitions, wedged arrows stand for non-radiative transitions (ISC, IC or VR). | ||
Regarding the bpz-based complexes 3–5, their luminescence arises definitely from an excited state of the same nature (Fig. 3c). The similar energies of their emissive excited states suggest emission from 3MLCTbpz states, corresponding to a charge transfer from the Ru(II) center towards one of the ancillary bpz. A bathochromic shift occurs in water due to the energy decrease of these 3MLCTbpz states in a more polar solvent, caused by the stabilization of the dipole formed at the excited state. The extended planar ligand (dpac/npp/bdppz) does not seem to be involved in the lowest emissive state for 3–5.
Photophysical behavior in the presence of DNA
Ru(II) complexes have been extensively studied as DNA photoprobes and photoreagents.44,45 When used as photoprobes, the luminescence of the complexes is enhanced in the presence of DNA; this behavior can be ascribed to the interaction of the complex with DNA leading to the reduction or inhibition of non-radiative deactivation pathways. In particular, very sensitive binding to DNA could be observed for Ru(II) complexes bearing an extended planar ligand, which is intercalated between the DNA base pairs. This intercalation is usually accompanied by a strong emission increase in aqueous medium, and is characterized by a strong binding affinity (Kaff ≈ 106 L mol−1) between DNA and the complex.27,46–48 Pioneering work by Barton et al. has shown that Ru-DPPZ exhibits a luminescent-sensitive behavior in the presence of oligonucleotides.22 When used as a photoreagent (i.e. in the case of photo-oxidizing Ru-TAP/bpz complexes), the excited state generated upon irradiation is involved in electron transfer (ET) with guanine residues, the most reductive base of DNA; in this case, the luminescence of the excited complex involved in the ET is quenched. We investigate the behavior of our five new Ru(II) complexes in the presence of SS-DNA (Salmon Sperm DNA) and CT-DNA (Calf Thymus DNA).DNA titration experiments show luminescence enhancement for complexes 1–2 in the presence of increasing concentration of DNA (CT-DNA and SS-DNA) (Fig. 4). This strong emission increase suggests a high binding affinity of the complexes for these two DNAs. Binding affinity values have been estimated using a modified McGhee–von Hippel equation49 as an expression for the luminescence intensity as a function of the ratio of Ru(II) per DNA (I/I0vs. [DNA]/[complex], with I0, the intensity of luminescence in the absence of DNA and [DNA] expressed in base-pair equivalents, see Table 4 and Fig. 4). Such a behavior is ascribed to the inhibition of the non-radiative deactivation processes of the 3MLCT excited states upon binding to DNA, either by protection of the complexes which avoid luminescence quenching by oxygen, or by protection from interaction with solvent molecules. Consequently, the enhancement of luminescence is much stronger with 2 than 1, thanks to the efficient protection of the two nitrogen atoms of the bdppz ligand made by the DNA base pair stack. The DNA binding thus prevents the luminescence quenching by inhibiting the formation of the two H-bonds in the excited state. Due to the elongated planar structure of npp and bdppz and consistently with the binding values obtained, we assume our complexes intercalate into the DNA strands as other Ru-dppz analogues.
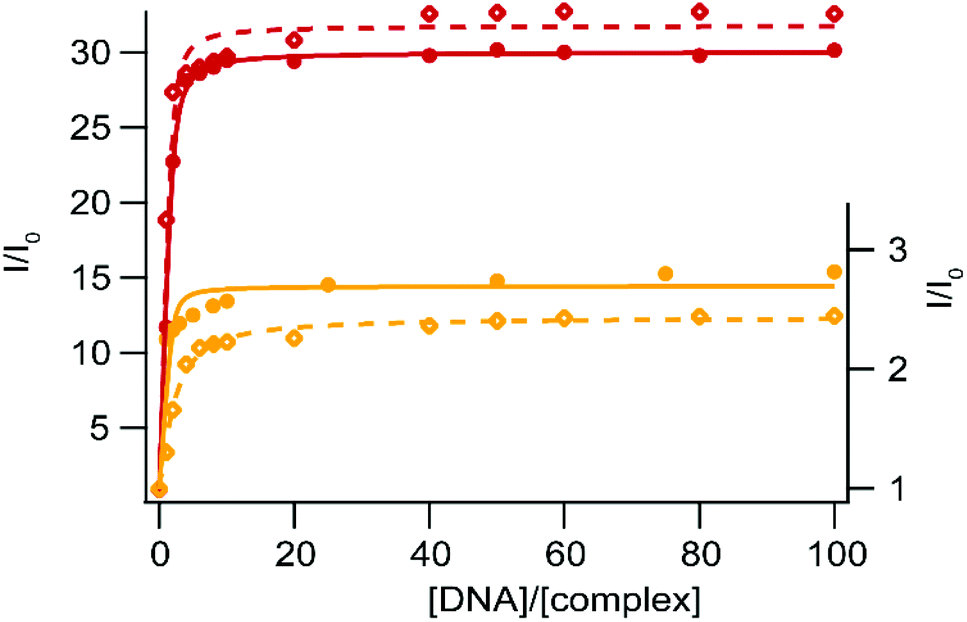 | ||
| Fig. 4 Steady-state luminescence titration of [Ru(bpy)2npp]2+ (1) (yellow – right axis) and [Ru(bpy)2bdppz]2+ (2) (red – left axis) with SS-DNA (full circles) and CT-DNA (empty diamonds). Measurements are performed using 10 μM of the complex in 50 mM Tris·HCl buffer, 50 mM NaCl at pH 7.4 under ambient air conditions. The fitted curves are obtained using a modified McGhee–Von Hippel equation and drawn as solid and dashed curves for SS- and CT-DNA respectively. | ||
| SS-DNA | CT-DNA | |||
|---|---|---|---|---|
|
K
affa (M−1) |
I/I0 max |
K
affa (M−1) |
I/I0 max | |
| a Binding constant are obtained using a McGhee–von Hippel type equation; the binding site corresponds to two base pairs per complex (best fit). Errors estimated to 5%. | ||||
| [Ru(bpy)2npp]2+ (1) | 7.95 × 105 | 2.70 | 8.31 × 104 | 2.44 |
| [Ru(bpy)2bdppz]2+ (2) | 5.06 × 105 | 30.06 | 6.86 × 105 | 31.81 |
| [Ru(bpz)2dpac]2+ (3) | 5.69 × 104 | 0.47 | 3.64 × 104 | 0.46 |
| [Ru(bpz)2npp]2+ (4) | 5.41 × 104 | 0.43 | 4.50 × 104 | 0.44 |
| [Ru(bpz)2bdppz]2+ (5) | 9.83 × 104 | 0.36 | 1.40 × 105 | 0.32 |
Due to the high binding affinity of complexes 1 and 2 for DNA, and thus their relevant application as DNA photoprobes, we investigated the behavior of the bpz “elbow-shaped” derivatives 4–5, as well as Ru-DPAC analogue 3. As stated in the Introduction, it has been shown in the literature that Ru(II) complexes bearing strong π-deficient ligands trigger oxidation of guanine bases upon irradiation due to their enhanced excited state reduction potential.15,19,50 Such properties could be exploited for phototherapies by inhibition of enzyme activity at the level of DNA,51,52 leading then to the selective silencing of the expression of the targeted gene inside cells.53,54
According to the excited state oxidation potentials of complexes 3–5 (Table 1), a photo-induced electron transfer (PIET) is thermodynamically possible between guanine and the excited complex (see eqn (1)).
| [RUIII(bpz)(bpz˙−)L]2+* + G → [RUII(bpz)(bpz˙−)L]+ + G˙+ | (1) |
Indeed, although the oxidation potential of guanine has been the subject of high debate in the literature, it is generally admitted that the EG˙+/G of isolated guanine is about +1.10 V vs. Ag/AgCl, and even less positive when stacked to other G nucleobases (EG˙+/G [poly(dG–dC)2] < +0.85 V vs. Ag/AgCl).55 Considering the excited state reduction potentials of our Ru(II) complexes (Table 1), it is expected that the PIET (eqn (1)) process is exergonic for complexes 3–5 (Table 5).
| ΔGETa (eV) |
k
qb (L mol−1 s−1) |
||
|---|---|---|---|
| dGMP | Poly(dG–dC)2c |
||
a Exergonicity of the ET between guanine and the excited complex estimated as ΔGET = −nF(ERu2+*/Ru+ − EG˙+/G), with EG˙+/G = +1.10 V for dGMP or +0.85 V for poly(dG–dC)2 and  values in Table 1.
b Quenching rate constant, kq, obtained using the Stern–Volmer equation I0/I = 1 + kqτ0[dGMP]; I0/I (where I0 is the luminescence of the complex in the absence of a quencher, here dGMP, and I is the luminescence in the presence of dGMP) as a function of the quencher concentration; τ0, the excited state lifetime of the complex in the absence of dGMP.
c ΔGET values with [poly(dG–dC)2] are given for comparison purposes in order to indicate the effect of the base pair stack. values in Table 1.
b Quenching rate constant, kq, obtained using the Stern–Volmer equation I0/I = 1 + kqτ0[dGMP]; I0/I (where I0 is the luminescence of the complex in the absence of a quencher, here dGMP, and I is the luminescence in the presence of dGMP) as a function of the quencher concentration; τ0, the excited state lifetime of the complex in the absence of dGMP.
c ΔGET values with [poly(dG–dC)2] are given for comparison purposes in order to indicate the effect of the base pair stack.
|
|||
| [Ru(bpz)2dpac]2+ (3) | −0.18 | −0.43 | 5.06 × 108 |
| [Ru(bpz)2npp]2+ (4) | −0.18 | −0.43 | 6.46 × 108 |
| [Ru(bpz)2bdppz]2+ (5) | −0.22 | −0.47 | 6.04 × 108 |
Luminescence quenching experiments have first been performed in the presence of deoxyguanosine-5′-monophosphate (dGMP). The luminescence of the three Ru(II) complexes 3–5 is progressively quenched upon using increasing amounts of dGMP (Fig. 5). A linear evolution is observed for the Stern–Volmer plot in the luminescence intensity for the three complexes, suggesting a pure dynamic quenching50 of the excited state of the three complexes by dGMP; the obtained kq values (Table 5) are close to the diffusion limit. The efficient quenching of the luminescence of complexes 3–5 by dGMP lies in agreement with a PIET process between excited complexes 3–5 and the guanine base. It should be noted that the luminescence of complexes 1 and 2 is not affected by the presence of dGMP (see the ESI†).
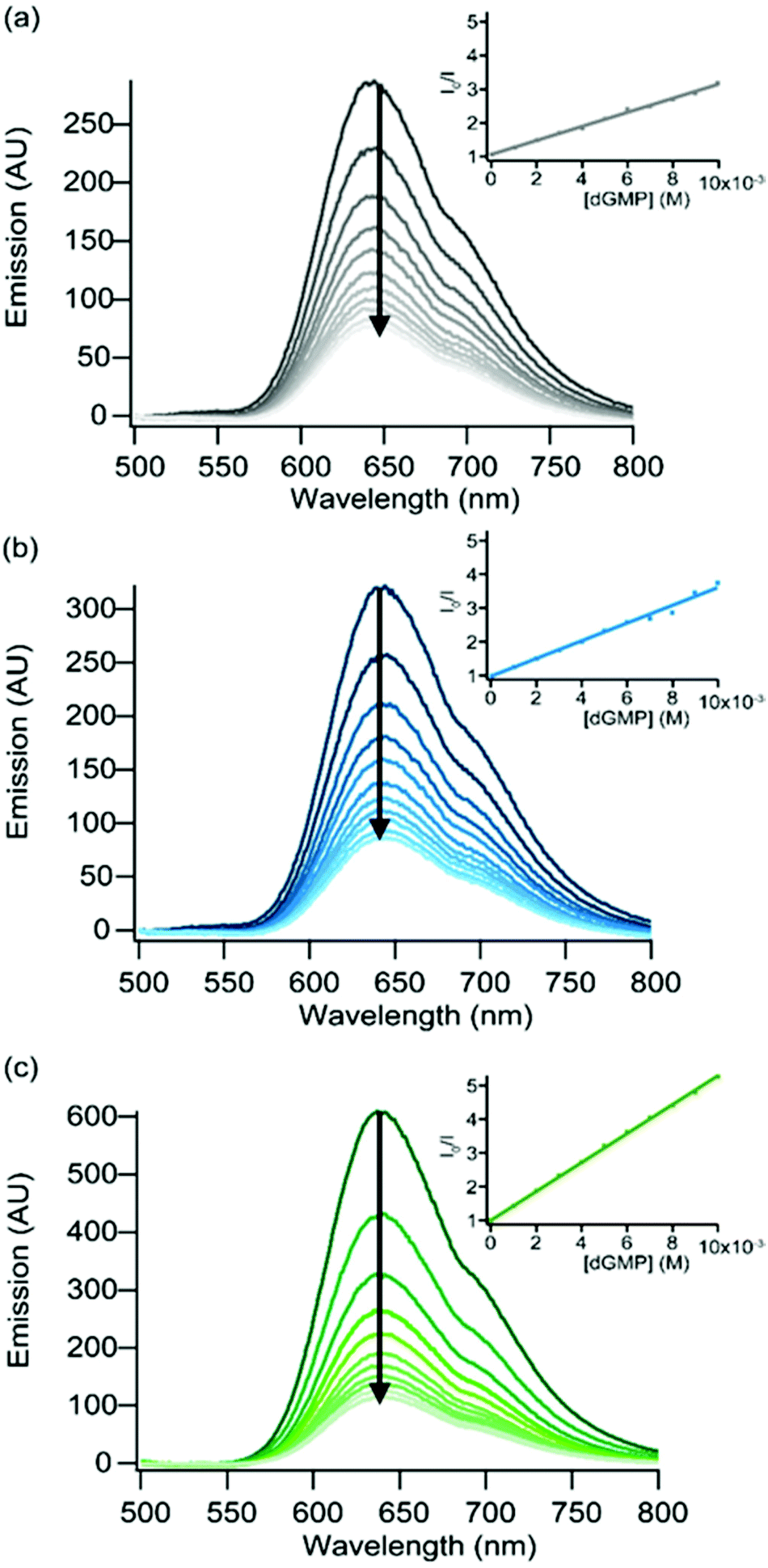 | ||
| Fig. 5 Evolution of the luminescence spectra of (a) [Ru(bpz)2dpac]2+ (3) (λexc = 466 nm), (b) [Ru(bpz)2npp]2+ (4) (λexc = 460 nm) and (c) [Ru(bpz)2bdppz]2+ (5) (λexc = 419 nm) in the presence of increasing concentrations of dGMP (dark to light). Measurements made in 5 mM Tris·HCl, 50 mM NaCl, pH = 7.4, under ambient air conditions. Insets: Stern–Volmer plots obtained upon the addition of dGMP to each complex. | ||
Similar experiments have been conducted with CT- and SS-DNA in order to probe the photoreactivity of 3–5 in the presence of nucleotides. As expected the luminescence of these complexes is quenched in the presence of increasing concentrations of DNA (Fig. 6), due to PIET with the guanine residues. The similar inhibition percentage for both types of DNA can be attributed to the similarity of the GC base-pair content for these two DNA. Binding affinity values have been calculated using a modified McGhee–von Hippel equation49 and are presented in Table 4. We can thus conclude that our complexes 4–5 can be efficiently used as photoreactive equivalents of complexes 1–2 respectively. If these latter ones were revealed to have some selectivity for specific topologies in DNA, such as single-base mismatches, the use of their bpz equivalent will allow us to target these sites, with an equivalent specificity.
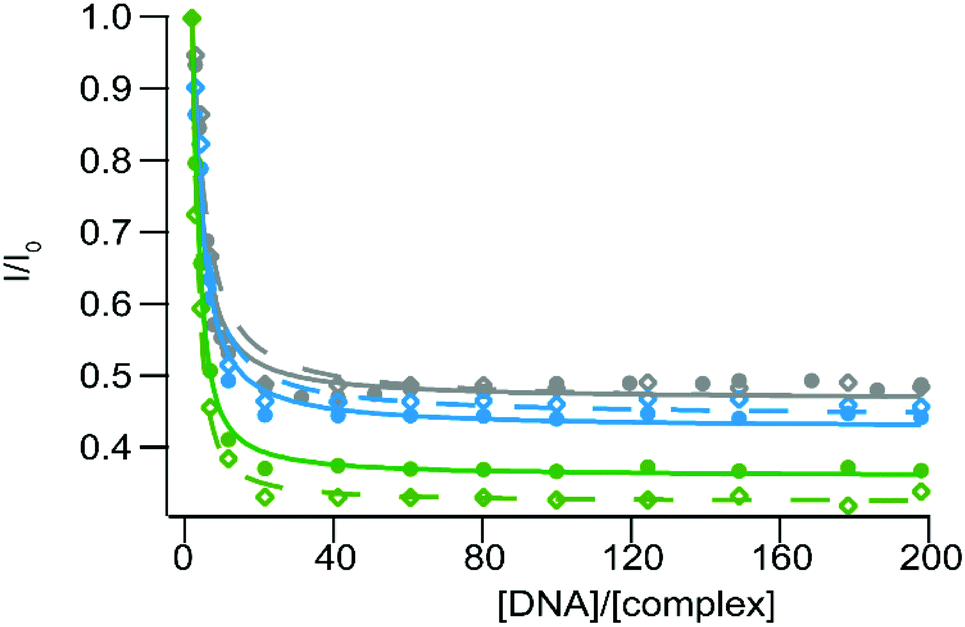 | ||
| Fig. 6 Steady-state luminescence titration of [Ru(bpz)2dpac]2+ (3) (gray), [Ru(bpz)2npp]2+ (4) (blue) and [Ru(bpz)2bdppz]2+ (5) (green) with SS-DNA (full circles) and CT-DNA (empty diamonds). Measurements are performed using 10 μM of the complex in 50 mM Tris·HCl buffer, 50 mM NaCl at pH 7.4 under ambient air conditions. The fitted curves are obtained using a modified McGhee–Von Hippel equation and drawn as solid and dashed curves for SS- and CT-DNA respectively. | ||
Photophysical behavior in the presence of mismatched DNA
As presented in the Introduction, a Ru(II) complex which would specifically associate to mismatches in DNA would be very valuable in order to detect (photoprobe) and target (photoreactant) these mutations in the genetic material. Ru-DPPZ was shown to be able to sense a single-base mismatch with a moderate specificity; the recognition of these mismatches can be correlated to their relative thermodynamic stability.20–22 Since then, other Ru(II) complexes where the intercalating ligand23,24 or the ancillary ligands51 are modified were studied. An enhanced specificity can be achieved for some of them, but these complexes were not or poorly luminescent. The best specificity for mismatched sites was obtained with rhodium(III) complexes using a bulky chrysi (5,6-chrysenequinone diimine) ligand.22,53–56 Unfortunately, these Rh(III) complexes are not luminescent and, therefore, cannot be used as photoprobes. Nevertheless, [Rh(bpy)2chrysi]3+ can photoreact at the mismatch site, leading to a strand cleavage at the level of the defect in the DNA sequence. Other Rh(III) complexes were developed and successfully used to kill (without photoactivation in this case) cancerous cells of which the mismatch repair machinery (MMR) is deficient.57,58 These cell studies strengthen the interest in the development of DNA mismatched Ru(II) photoprobes.We recently demonstrated that Ru(II) linear dpac-complexes are unfortunately unable to probe mismatched sites.25 The elbow-shaped structure of our npp and bdppz ligands is of great interest in order to obtain such a specificity for DNA mismatches; dissymmetry of the inserted ligand proved to be useful in this context for other Ru(II) complexes24 and, of course, in the case of the chrysi ligand. The ability of complexes 1–2 to detect single-base mismatches in short oligonucleotides was thus investigated by using a series of hairpin oligonucleotides containing a mismatch near the center of the duplex (see sequences in Fig. 7).
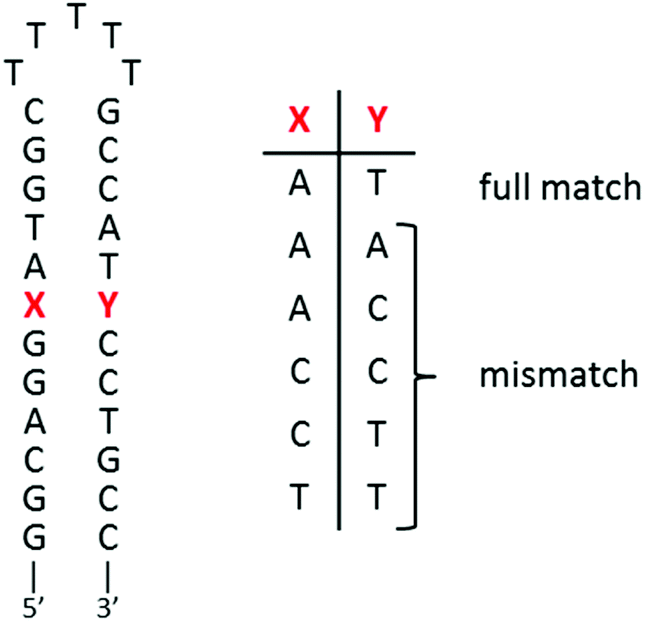 | ||
| Fig. 7 Hairpin oligonucleotide sequences used for titrations. | ||
Both complexes bearing an “elbow-shaped” intercalating ligand show distinct luminescence intensities with mismatched sequences as compared to the fully-matched one (Fig. 8). Interestingly, while the strongest luminescence enhancement is observed for the fully-matched hairpin for 1, an opposite behavior occurs for 2. The luminescence decrease observed for 1 in the presence of mismatched hairpins with respect to the well-matched one could be ascribed to a less effective interaction of this complex with the mismatched base pairs (either a weaker binding and/or less efficient protection from a non-radiative deactivation pathway), leading to an increased exposure to oxygen and solvent molecules with respect to the full matched sites. Similar behavior has been observed for linear Ru-DPAC, as a result of a looser fit of the complex within mismatched sites59 favoring non-radiative deactivations of the excited state.25
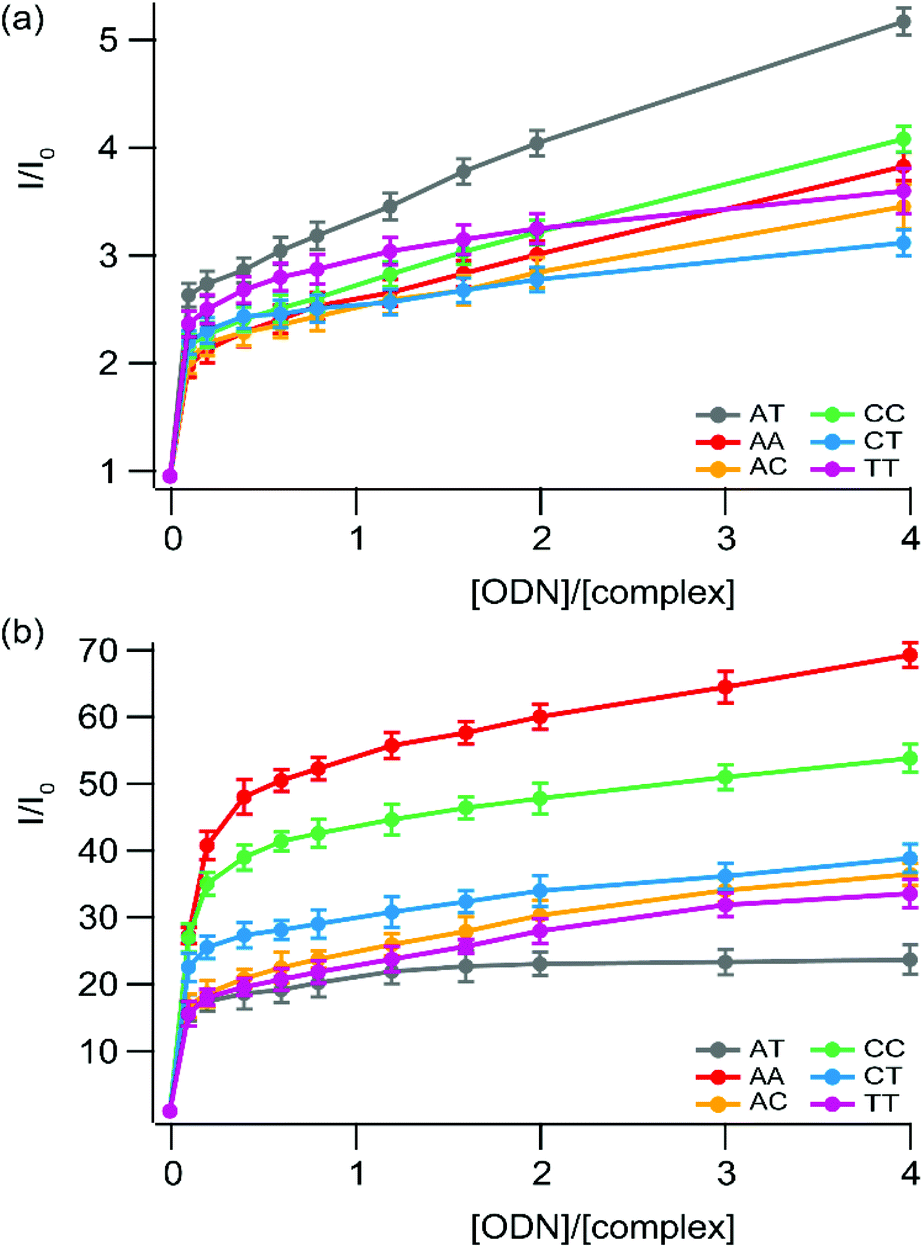 | ||
| Fig. 8 Steady-state luminescence titration of (a) [Ru(bpy)2npp]2+ (1) and (b) [Ru(bpy)2bdppz]2+ (2) with a series of small hairpin oligonucleotides which possess (or not) a mismatched base pair. Measurements are performed using 1 μM of the complex in 5 mM Tris·HCl buffer, 1 mM NaCl, pH 7.5 under ambient air conditions. Lines used as eye-guides do not correspond to the fitting process (which is depicted in the ESI† for 2 in the presence of AT and CC sequences). | ||
In contrast, the luminescence of complex 2 is greatly enhanced while the complex is in the presence of mismatched sequences, compared to the well-matched hairpin. In addition, the greater the thermodynamic instability of the mismatched site, the greater is the luminescence enhancement observed. A similar trend has previously been reported for Ru-DPPZ and [Rh(bpy)2chrysi]3+, for which binding to mismatched sites occurs via insertion with ejection of the mismatched bases towards the DNA groove.22,52–54
The enhancement of the luminescence behavior, observed for 2 and reported for Ru-DPPZ and its analogues, is due to the deeper packing allowed by insertion at the mismatched site of the hairpin, and thus better protection of the complex from the solvent with respect to the corresponding well-matched site. As a result, non-radiative relaxation pathways of the excited state of 2 are disfavored in the presence of mismatch-containing hairpins. Since this particular photophysical behavior is observed only for this complex, we suggest it binds mismatched hairpins through metallo-insertion, probably associated with favorable interactions between hairpin residues and the phenazine portion of the ligand.
The strong differential luminescence behavior of 2 in the presence of a mismatched hairpin versus the well-matched duplex corresponds to a 3.5-fold enhancement for CC mismatches compared to the well-matched ones, which is a better enhancement than the one observed using only the more favorable Δ stereoisomer of Ru-DPPZ22 under the same conditions. Using a modified McGhee–von Hippel equation to get an insight into the binding affinity of 2 for the different mismatches, we can estimate that the complex shows a one-order higher affinity for the mismatch site compared to the well-matched ones (∼107vs. 3.88 × 106 L mol−1, see the ESI†). Consequently, we can assume that the luminescence enhancement is not only due to a better protection of the excited complex inserted at the mismatched site, but also due to a greater affinity of the complex for insertion compared to intercalation within DNA.
The use of a photoreactive equivalent to induce photoreaction at these sites is thus relevant. Titrations achieved with the photoactive equivalent of 2, i.e. complex 5, demonstrate that the quenching, and thus the photoinduced electron transfer, is very efficient (see the ESI†); unfortunately, the hairpin sequence, used for comparison purposes with the literature, does not allow us to discriminate the ET process from complexes bound to the mismatched sites with complexes bound to adjacent well-matched base pairs, as the PIET can occur with distant guanine residues. A clear-cut demonstration of a preferential photoreactivity would require either the design of a specific sequence or the trapping of radical guanine, which is beyond the scope of the present study. Nevertheless, based on the similar structures of 2 and 5, and their similar behavior in binding well-matched DNA, we can safely suggest that 5 is preferentially associated with mismatched sites, and thus a photo-induced ET could preferentially occur with a guanine base in its close vicinity. Complex 5 thus represents a very interesting candidate as a photoreactant for the DNA mismatch.
Conclusions
New “elbow-shaped” Ru(II) complexes bearing acridine- and phenazine-based extended planar ligands have been prepared. The electrochemical and photophysical studies indicate that the lowest 3MLCT excited state for [Ru(bpy)2npp]2+, 1, is located on the bpy fragment of the npp ligand. The replacement of a central carbon atom in npp for a nitrogen to obtain bdppz has a strong effect on the electrochemical and photophysical properties of the resulting Ru(II) complex, as 2 is poorly luminescent in water. An excited state located on the phenazine moiety of the bdppz ligand and involved in two H-bonds is proposed to account for its partial luminescence quenching in aqueous medium. The residual luminescence observed in aqueous medium (in contrast to the non-luminescent well-known Ru-DPPZ) is rationalized by the inhibition of the second H-bond between water molecules and the second nitrogen atom of the phenazine core thanks to the “elbow-shape” of bdppz.Studies of these two complexes in the presence of DNA suggest avid binding to polynucleotides and protection of the excited complexes from non-radiative deactivation pathways upon intercalation into the base pair stack. The stronger enhancement of emission for 2 with respect to 1 is attributed to the prevention of the two H-bonds in the excited state, the second one being responsible for the complete luminescence quenching.
Photooxidizing analogs of these complexes, [Ru(bpz)2npp]2+, 4, and [Ru(bpz)2bdppz]2+, 5, have been successfully synthesized, together with their linear analogue [Ru(bpz)2dpac]2+, 3. The presence of the two bpz ancillary ligands dramatically modifies the electrochemical and photophysical properties of these new complexes compared to their bpy/phen counterparts; the excited state reduction potential is enhanced and is high enough to allow the complexes 3–5 to photo-oxidize the guanine nucleobase. Luminescence studies in the presence of dGMP and DNAs indicate that a photo-induced electron-transfer is occurring between the guanine and the excited states of 3–5; indeed, a quenching of their luminescence is observed upon addition of dGMP or DNA. In the first case, Stern–Volmer plots indicate a dynamic quenching process by dGMP, associated with a quenching rate constant close to the diffusion limit. In the presence of DNA, steady-state luminescence experiments indicate a high binding affinity of the 3–5 complexes for CT- and SS-DNA. The similar quenching percentage of the luminescence of these complexes by both types of DNAs is consistent with the similarity of the GC content in SS- and CT-DNA. Like complexes 1–2, these new complexes 3–5 are proposed to be interacting with DNA via intercalation.
The behavior of our complexes was also investigated in the presence of single-base mismatch-containing hairpins. While a small difference in the luminescence of 1 in the presence of mismatched hairpins compared to well-matched ones might be due to the poor protection of the excited state of 1 from the non-radiative relaxation pathways, a strong differential luminescence behavior in the presence of a mismatched hairpin versus the well matched duplex occurs for 2. In the presence of CC mismatches, a 3.5-fold enhancement of the luminescence is observed, compared to the intercalation with a well-matched sequence. This enhancement for the racemic mixture of 2 is more important than the ratio reported for Δ-Ru-DPPZ. Like this latter one, 2 is suggested to bind mismatched sites via metallo-insertion. This tight fit interaction leads to the better protection of the complex from the solvent compared to intercalation, and is supported by the correlation observed with the relative thermodynamic stability of the mismatch. We propose that favorable interactions between the phenazine portion of bdppz with hairpin residues are the basis for the stronger binding of 2 to mismatched hairpins, and consequently for the luminescence sensitivity of the mismatched sites. The “elbow-shaped” complex 2 might therefore represent a promising candidate for the development of DNA mismatched selective photoprobes. Moreover, thanks to the similar structure of 5, this bpz analogue could be considered as a good photoreactant candidate to efficiently photo-induce ET with guanine in close vicinity to the mismatched site.
Experimental section
Materials and instrumentation
[Ru(bpy)2Cl2],23 [Ru(bpz)2Cl2],28 (1-aminonaphthalen-2-yl)methanol,60,61 1,2-diaminonaphthalene,62 dpac25 and 1,10-phenanthroline-5,6-dione63 are synthesized according to previously described literature protocols. All solvents and reagents for the synthesis are of reagent grade and are used without any further purification. All solvents for the spectroscopic and electrochemical measurements are of spectroscopic grade. Water is purified with a Millipore Milli-Q system. Calf thymus DNA Type I (CT-DNA) and salmon sperm DNA (SS-DNA) were purchased from Sigma-Aldrich. Hairpin ODNs were purchased from Eurogentec. DNA and ODN concentrations are determined spectroscopically (λ260 nm = 6600 M−1 cm−1/bp for CT-DNA and SS-DNA64,65; λ260 nm = 260000 M−1 cm−1 for ODN-AT, 264900 M−1 cm−1 for ODN-AA, 253300 M−1 cm−1 for ODN-CC, 259100 M−1 cm−1 for ODN-AC, 254200 M−1 cm−1 for ODN-CT, 257500 M−1 cm−1 for ODN-TT). The molar extinction coefficients of hairpin oligonucleotides are values calculated based on the base content of each sequence. 1H NMR experiments are performed in CDCl3, CD3OD or CD3CN on a Bruker AC-300 Avance II (300 MHz) or on a Bruker AM-500 (500 MHz) at 20 °C. The chemical shifts (given in ppm) are measured vs. the residual peak of the solvent as the internal standard. High-resolution mass spectrometry (HRMS) spectra are recorded on a Q-Exactive orbitrap from ThermoFisher using reserpine as the internal standard. Samples are ionized by electrospray ionization (ESI; capillary temperature = 320 °C, vaporizer temperature = 320 °C, sheath gas flow rate = 5 mL min−1). UV-vis absorption spectra are recorded on a Shimadzu UV-1700. Room temperature fluorescence spectra are recorded on a Varian Cary Eclipse instrument. The luminescence intensity at 77 K is recorded on a FluoroLog3 FL3-22 from Jobin Yvon equipped with an 18 V, 450 W xenon short arc lamp and an R928P photomultiplier, using an Oxford Instrument Optistat DN nitrogen cryostat controlled by an Oxford Intelligent Temperature Controller (ITC503S) instrument. Luminescence lifetime measurements were performed after irradiation at λ = 400 nm obtained by the second harmonic of a Titanium:Sapphire laser (picosecond Tsunami laser spectra physics 3950-M1BB+39868-03 pulse picker doubler) at a 80 kHz repetition rate. The Fluotime 200 from AMS technologies was used for the decay acquisition. It consists of a GaAs microchannel plate photomultiplier tube (Hamamatsu model R3809U-50) followed by a time-correlated single photon counting system from Picoquant (PicoHarp300). The ultimate time resolution of the system is close to 30 ps. Luminescence decays were analyzed with FLUOFIT software available from Picoquant. Cyclic voltammetry is carried out in a one-compartment cell, using a glassy carbon disk working electrode (approximate area = 0.03 cm2), a platinum wire counter electrode, and an Ag/AgCl reference electrode. The potential of the working electrode is controlled by an Autolab PGSTAT 100 potentiostat through a PC interface. The cyclic voltammograms are recorded with a sweep rate of 300 mV s−1, in dried acetonitrile (Sigma-Aldrich, HPLC grade). The concentration of the complexes is 8 × 10−4 mol L−1, with 0.1 mol L−1 tetrabutylammonium perchlorate as the supporting electrolyte. Before each measurement, the samples are purged by nitrogen.
Synthetic procedures and characterization of “elbow-shaped” ligands and respective bpy-based Ru(II) complexes
Synthetic procedures and characterization of bpz-based Ru(II) complexes
Acknowledgements
Q. D. and B. E. gratefully acknowledge the Université catholique de Louvain, the Fonds National pour la Recherche Scientifique (F.R.S.-F.N.R.S.), the Région Wallonne, and the Prix Pierre et Colette Bauchau for financial support. L. M. thanks the Belgian American Educational Foundation (BAEF) for the Cabeaux-Jacobs Fellowship. F. L. thanks the Labex Arcane, France (ANR-11-LABX-0003-01) and the chemistry platform NanoBio campus in Grenoble for luminescence lifetime measurement facilities. Q. D. also warmly thanks Christophe Rubay for his scientific help.References
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Garland Science, New York, 5th edition, 2007 Search PubMed.
- R. R. Iyer, A. Pluciennik, V. Burdett and P. L. Modrich, Chem. Rev., 2006, 106, 302–323 Search PubMed.
- P. Modrich, J. Biol. Chem., 2006, 281, 30305–30309 Search PubMed.
- T. Carell, Angew. Chem., Int. Ed., 2015, 54, 15330–15333 Search PubMed.
- J. H. Hoeijmakers, Nature, 2001, 411, 366–374 Search PubMed.
- J. Jiricny, Nat. Rev. Mol. Cell Biol., 2006, 7, 335–346 Search PubMed.
- P. Liu, Y. Li, H. Wang, Z. Wang and X. Hu, Tetrahedron Lett., 2012, 53, 6654–6656 Search PubMed.
- Y. Chen, K. Li, M. Zhao, Y. Li and B. Chen, Tetrahedron Lett., 2013, 54, 1627–1630 Search PubMed.
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 Search PubMed.
- A. W. McKinley, P. Lincoln and E. M. Tuite, Coord. Chem. Rev., 2011, 255, 2676–2692 Search PubMed.
- A. C. Komor and J. K. Barton, Chem. Commun., 2013, 49, 3617–3630 Search PubMed.
- B. Elias, L. Herman, C. Moucheron and A. Kirsch-De Mesmaeker, Inorg. Chem., 2007, 46, 4979–4988 Search PubMed.
- L. Herman, B. Elias, F. Pierard, C. Moucheron and A. Kirsch-De Mesmaeker, J. Phys. Chem. A, 2007, 111, 9756–9763 Search PubMed.
- K. Kobayashi, H. Ohtsu, K. Nozaki, S. Kitagawa and K. Tanaka, Inorg. Chem., 2016, 55, 2076–2084 Search PubMed.
- I. Ortmans, B. Elias, J. M. Kelly, C. Moucheron and A. Kirsch-DeMesmaeker, Dalton Trans., 2004, 4, 668–676 RSC.
- J. A. Smith, M. W. George and J. M. Kelly, Coord. Chem. Rev., 2011, 255, 2666–2675 CrossRef CAS.
- P. M. Keane, F. E. Poynton, J. P. Hall, I. V. Sazanovich, M. Towrie, T. Gunnlaugsson, S. J. Quinn, C. J. Cardin and J. M. Kelly, Angew. Chem., Int. Ed., 2015, 54, 8364–8368 Search PubMed.
- S. M. Cloonan, R. B. Elmes, M. Erby, S. A. Bright, F. E. Poynton, D. E. Nolan, S. J. Quinn, T. Gunnlaugsson and D. C. Williams, J. Med. Chem., 2015, 58, 4494–4505 Search PubMed.
- B. Elias, C. Creely, G. W. Doorley, M. M. Feeney, C. Moucheron, A. Kirsch-DeMesmaeker, J. Dyer, D. C. Grills, M. W. George, P. Matousek, A. W. Parker, M. Towrie and J. M. Kelly, Chem. – Eur. J., 2008, 14, 369–375 Search PubMed.
- C. Sentagne, J.-C. Chambron, J.-P. Sauvage and N. Paillous, J. Photochem. Photobiol., B, 1994, 26, 165–174 Search PubMed.
- P. Vicendo, S. Mouysset and N. Paillous, Photochem. Photobiol., 1997, 65, 647–655 Search PubMed.
- M. H. Lim, H. Song, E. D. Olmon, E. E. Dervan and J. K. Barton, Inorg. Chem., 2009, 48, 5392–5397 Search PubMed.
- E. Ruba, J. R. Hart and J. K. Barton, Inorg. Chem., 2004, 43, 4570–4578 Search PubMed.
- A. J. McConnell, M. H. Lim, E. D. Olmon, H. Song, E. E. Dervan and J. K. Barton, Inorg. Chem., 2012, 51, 12511–12520 Search PubMed.
- Q. Deraedt, L. Marcélis, T. Auvray, G. S. Hanan, F. Loiseau and B. Elias, Eur. J. Inorg. Chem., 2016, 22, 3649–3658 Search PubMed.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 Search PubMed.
- E. Amouyal, A. Homsi, J.-C. Chambron and J.-P. Sauvage, J. Chem. Soc., Dalton Trans., 1990, 6, 1841–1845 Search PubMed.
- R. J. Crutchley, A. B. P. Lever and A. Poggi, Inorg. Chem., 1983, 22, 2647–2650 Search PubMed.
- C. Sentagne, J.-C. Chambron, J.-P. Sauvage and N. Paillous, J. Photochem. Photobiol., B, 1994, 26, 165–174 Search PubMed.
- J. Olofsson, L. M. Wilhelmsson and P. Lincoln, J. Am. Chem. Soc., 2004, 126, 15458–15465 Search PubMed.
- G. Pourtois, D. Beljonne, C. Moucheron, S. Schumm, A. Kirsch-De Mesmaeker, R. Lazzaroni and J. L. Bredas, J. Am. Chem. Soc., 2004, 126, 683–692 Search PubMed.
- J. Olofsson, B. Önfelt and P. Lincoln, J. Phys. Chem. A, 2004, 108, 4391–4398 Search PubMed.
- E. Gicquel, J. P. Souchard, F. Magnusson, J. Chemaly, P. Calsou and P. Vicendo, Photochem. Photobiol. Sci., 2013, 12, 1517–1526 CAS.
- G. D. Hager and G. A. Crosby, J. Am. Chem. Soc., 1975, 97, 7031–7037 Search PubMed.
- J. V. Caspar, E. M. Kober, B. P. Sullivan and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 630–632 Search PubMed.
- J. V. Caspar and T. J. Meyer, J. Am. Chem. Soc., 1983, 105, 5583–5590 Search PubMed.
- C. G. Coates, L. Jacquet, J. J. McGarvey, S. E. J. Bell, A. H. R. Al-Obaidi and J. M. Kelly, J. Am. Chem. Soc., 1997, 119, 7130–7136 Search PubMed.
- R. M. Hartshorn and J. K. Barton, J. Am. Chem. Soc., 1992, 114, 5919–5925 Search PubMed.
- A. C. Bhasikuttan, M. Suzuki, S. Nakashima and T. Okada, J. Am. Chem. Soc., 2002, 124, 8398–8405 Search PubMed.
- A. Cannizzo, F. van Mourik, W. Gawelda, G. Zgrablic, C. Bressler and M. Chergui, Angew. Chem., Int. Ed., 2006, 45, 3174–3176 Search PubMed.
- E. J. C. Olson, D. Hu, A. Hörmann, A. M. Jonkman, M. R. Arkin, E. D. A. Stemp, J. K. Barton and P. F. Barbara, J. Am. Chem. Soc., 1997, 119, 11458–11467 Search PubMed.
- P. Lincoln, A. Broo and B. Nordèn, J. Am. Chem. Soc., 1996, 118, 2644–2653 Search PubMed.
- Y. Sun, L. E. Joyce, N. M. Dickson and C. Turro, Chem. Commun., 2010, 46, 2426–2428 RSC.
- L. Marcélis, J. Ghesquière, K. Garnir, A. Kirsch-De Mesmaeker and C. Moucheron, Coord. Chem. Rev., 2012, 256, 1569–1582 Search PubMed.
- L. Marcelis, C. Moucheron and A. Kirsch-De Mesmaeker, Philos. Trans. R. Soc. London, Ser. A, 2013, 371, 20120131 Search PubMed.
- C. Turro, S. H. Bossmann, Y. Jenkins, J. K. Barton and N. J. Turro, J. Am. Chem. Soc., 1995, 117, 9026–9032 Search PubMed.
- B. Onfelt, P. Lincoln, B. Norden, J. S. Baskin and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5708–5713 Search PubMed.
- C. Moucheron, A. Kirsch-De Mesmaeker and S. Choua, Inorg. Chem., 1997, 36, 584–592 Search PubMed.
- J. D. McGhee and P. H. von Hippel, J. Mol. Biol., 1974, 86, 469–489 Search PubMed.
- A. Srishailam, Y. P. Kumar, P. Venkat Reddy, N. Nambigari, U. Vuruputuri, S. S. Singh and S. Satyanarayana, J. Photochem. Photobiol., B, 2014, 132, 111–123 Search PubMed.
- A. N. Boynton, L. Marcélis and J. K. Barton, J. Am. Chem. Soc., 2016, 138, 5020–5023 Search PubMed.
- B. A. Jackson and J. K. Barton, J. Am. Chem. Soc., 1997, 119, 12986–12987 Search PubMed.
- B. A. Jackson, V. Y. Alekseyev and J. K. Barton, Biochemistry, 1999, 38, 4655–4662 CrossRef CAS PubMed.
- B. A. Jackson and J. K. Barton, Biochemistry, 2000, 39, 6176–6182 Search PubMed.
- S. Steenken and S. V. Jovanovic, J. Am. Chem. Soc., 1997, 119, 617–618 Search PubMed.
- B. M. Zeglis and J. K. Barton, Nat. Protoc., 2007, 2, 357–371 CrossRef CAS PubMed.
- K. M. Boyle and J. K. Barton, Inorg. Chim. Acta, 2016, 452, 3–11 Search PubMed.
- A. G. Weidmann, A. C. Komor and J. K. Barton, Philos. Trans. R. Soc. London, Ser. A, 2013, 371, 20120117 Search PubMed.
- J. SantaLucia and D. Hicks, Annu. Rev. Biophys. Biomol. Struct., 2004, 33, 415–440 Search PubMed.
- E. C. Riesgo, X. Jin and R. P. Thummel, J. Org. Chem., 1996, 61, 3017–3022 Search PubMed.
- N. B. Vicker, H. V. Bailey, W. Heaton, J. M. Day, A. Purohit and B. V. L. Potter, WO2009/066072A2, 2009 Search PubMed.
- S. Villa, G. Cignarella, D. Barlocco, M. Gervasoni, G. Carcassola, L. Giannino and P. Mantegazza, Il Farmaco, 2003, 58, 929–937 Search PubMed.
- W. Travis, C. E. Knapp, C. N. Savory, A. M. Ganose, P. Kafourou, X. Song, Z. Sharif, J. K. Cockcroft, D. O. Scanlon, H. Bronstein and R. G. Palgrave, Inorg. Chem., 2016, 55, 3393–3400 Search PubMed.
- E. M. Castanheira, M. S. Carvalho, A. R. Rodrigues, R. C. Calhelha and M. J. Queiroz, Nanoscale Res. Lett., 2011, 6, 379 Search PubMed.
- R. B. Dixit, T. S. Patel, S. F. Vanparia, A. P. Kunjadiya, H. R. Keharia and B. C. Dixit, Sci. Pharm., 2011, 79, 293–308 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qi00223d |
| ‡ Present address: Trasis sa, Rue Gilles Magnée 90, 4430 Ans, Belgium. |
| This journal is © the Partner Organisations 2017 |