
Ultra-filtration of human serum for improved quantitative analysis of low molecular weight biomarkers using ATR-IR spectroscopy†
Franck
Bonnier
*a,
Hélène
Blasco
bc,
Clément
Wasselet
a,
Guillaume
Brachet
d,
Renaud
Respaud
e,
Luis Felipe C. S.
Carvalho
f,
Dominique
Bertrand
g,
Matthew J.
Baker
h,
Hugh J.
Byrne
i and
Igor
Chourpa
a
aUniversité François-Rabelais de Tours, Faculté de Pharmacie, EA 6295 Nanomédicaments et Nanosondes, 31 avenue Monge, 37200 Tours, France. E-mail: franck.bonnier@univ-tours.fr
bCHRU de Tours, Laboratoire de Biochimie et de Biologie Moléculaire, Tours, France
cINSERM, UMR U930 “Imagerie et Cerveau”, Université François Rabelais, Tours, France
dUniversité François Rabelais de Tours, UMR CNRS 7292 Génétique, Immunothérapie, Chimie et Cancer, Faculté de Médecine, 10 Bd Tonnellé, 37032 Tours Cedex, France
eUniversité François-Rabelais de Tours, UMR 1100, CHRU de Tours, Service de Pharmacie, F-37032 Tours, France
fUniversidade do Vale do Paraiba, Laboratory of Biomedical Vibrational Spectroscopy, Sao José dos Campos, Brazil
gData Frame, Nantes, France
hWestCHEM, Technology and Innovation Centre, Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow G1 1XL, UK
iFOCAS Research Institute, Dublin Institute of Technology (DIT), Camden Row, Dublin 8, Ireland
First published on 8th December 2016
Abstract
Infrared spectroscopy is a reliable, rapid and cost effective characterisation technique, delivering a molecular finger print of the sample. It is expected that its sensitivity would enable detection of small chemical variations in biological samples associated with disease. ATR-IR is particularly suitable for liquid sample analysis and, although air drying is commonly performed before data collection, just a drop of human serum is enough for screening and early diagnosis. However, the dynamic range of constituent biochemical concentrations in the serum composition remains a limiting factor to the reliability of the technique. Using glucose as a model spike in human serum, it has been demonstrated in the present study that fractionating the serum prior to spectroscopic analysis can considerably improve the precision and accuracy of quantitative models based on the partial least squares regression algorithm. By depleting the abundant high molecular weight proteins, which otherwise dominate the spectral signatures collected, the ability to monitor changes in the concentrations of the low molecular weight constituents is enhanced. The Root Mean Square Error for the Validation set (RMSEV) has been improved by a factor of 5 following human serum processing with an average relative error in the predictive values below 1% being achieved. Moreover, the approach is easily transferable to different bodily fluids, which would support the development of more efficient and suitable clinical protocols for exploration of vibrational spectroscopy based ex vivo diagnostic tools.
1. Introduction
Bodily fluids have become a much investigated source of samples for the development of rapid and cost effective diagnostic methods.1–4 Reducing the invasiveness of the current approaches to get molecular information linked to pathological events occurring in different organs affected by various diseases is naturally one of main challenges to improve the patients’ comfort. Therefore, performing analysis directly on a few microliters of serum, plasma, saliva or urine has attracted great interest in the medical field, especially due to the facile specimen collection. Blood analysis still holds much promise for accurate ex vivo diagnostics and, compared to deep tissue biopsies, often requiring extreme interventions, the reduced invasiveness of the syringe remains a quite acceptable alternative.Human serum (or plasma) is a vast reservoir for biochemical products collected and accumulated while perfusing the different organs of the body,5 ultimately reflecting the physiological status of a patient. It is expected that modifications in its overall composition could indicate the presence of disease and consequently deliver a diagnostic based on specific molecular signatures.6 It is well accepted that proteins secreted and shed from cells and tissues, such as prostate-specific antigen (PSA) and CA125, can be identified routinely and act as biomarkers of disease.7 In addition, proteolysis within the tissue or deregulated post-translational events participate in fragmentation of proteins produced in tumours that diffuse into the circulation.6 This is further supported by recent associations of pathological conditions with small protein and peptide profiles in serum, notably for diabetes,8 cardiovascular or infectious diseases.9 The number of studies related to serum proteomics, peptidomics or metabolomics has literally exploded in the literature, complex analytical techniques such as chromatography and/or mass spectroscopy being the reference tools.10,11 Quite naturally, vibrational spectroscopy, which has been widely employed in biomedical analysis, from tissue sections12,13 to single cells14,15 with a well demonstrated potential for diagnosis,16,17 has the capability to become the next generation gold standard tool for serum based patient screening.18–20 The advent of imaging technologies coupled to the rapid data collection offered by FTIR systems has contributed considerably to the attraction of infrared absorption spectroscopy for biomedical applications.21–23 However, other methods such as ATR-FTIR are highly suitable for analysis of liquid samples, such as body fluids, allowing delivery of chemical fingerprints from micro-deposition of the samples directly on the Attenuated Total Reflection (ATR) crystal.24,25 The IR spectrum contains rich and specific information about the molecular composition of the human serum which, coupled to advanced multivariate analysis tools, could deliver accurate diagnostics. Recent systematic studies have demonstrated the potential of IR serum based diagnosis with different data mining approaches such as Principal Components Analysis (PCA), PCA coupled to Linear Discriminant Analysis (PCA-LDA), Random Forest or Support Vector Machine (SVM).26–30 However, the development of spectroscopic technologies with medical perspectives needs to align with clinical requirements to enable clinical translation of these promising technologies.31 Although infrared and Raman spectroscopy have great potential for serum based detection of disease biomarkers,32,33 in many cases, detection of the presence of a biomarker is not sufficient, but rather quantification within a physiologically relevant range is required, a common example being glucose levels in blood.34
Partial Least Squares Regression analysis (PLSR) remains one of most used analysis methods for quantitative models35 either in different body fluids such as urine,36 saliva,37,38 serum39 or to evaluate physiological responses to drugs in cells.40 However, the serum composition is highly complex, with up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 different proteins in an overall concentration ranging from 60 to 80 mg mL−1. Moreover, other circulating molecular species such as sugars, lipids, peptides, metabolites are present, adding to the complexity of the mixture and consequently making quantitative analysis of variations in individual constituents a challenging task. Human serum is also characterized by the dynamic range of concentrations observed between the abundant, high molecular weight (HMW) and the sparse, low molecular weight (LMW) molecules. For example, human serum albumin (HSA) (57–71%) and globulins (8–26%) are two abundant HMW proteins completely dominating the composition of the serum quantitatively, potentially impacting the detection and monitoring small variations in the features related to the presence of the informative low molecular weight proteins/peptides/metabolites.41 The analytical capabilities of traditional proteomic methods are limited, given this large dynamic range of concentrations in the serum. Therefore, depletion of the highly abundant proteins is now recognized as the first step, yet to be optimized, in the analysis of the serum composition.42 ATR-FTIR suffers from similar limitations to those encountered with chromatography and/or mass spectroscopy based approaches, which can be overcome by means of sample fractionation prior to data recording.24 Including a separation step prior to IR analysis in order to enhance the specificity and sensitivity has been proposed through coupling with chromatography technologies such as LC-IR and GC-IR.43 Those hyphenated approaches have been reported in chemistry, pharmaceutical and food sciences applications.44–46 However, the development of cryogenic temperature control methods during the sample preparation remains the most promising aspect for possible emergence of such technologies in biomedical applications and human body fluids analysis.47 This present study however investigates the benefits of an alternative approach combining centrifugal filtration/fractionation with ATR-IR spectroscopy for the analysis of human serum as a proof of principle for monitoring and quantified potential low molecular weight biomarkers. Glucose has been used as a model to illustrate the strategy to isolate the relevant fraction of the sample to minimize the influence of the HMW proteins and improve the precision and accuracy of the quantitative models built using the PLSR algorithm. Initially, human serum spiked with systematically varying, physiologically relevant, concentrations of glucose will be used to optimise the measurement protocol, including a study of how the volume deposited on the ATR crystal influences the relevancy of the data collected. PLSR will be employed to build a predictive model over the concentration range to demonstrate the principle of the predictive capacity of the technique and the improved precision afforded by serum fractionation. In the second part of the study, the technique will be demonstrated in patient samples of known varying glucose levels, validating the potential for glucose level monitoring.
000 different proteins in an overall concentration ranging from 60 to 80 mg mL−1. Moreover, other circulating molecular species such as sugars, lipids, peptides, metabolites are present, adding to the complexity of the mixture and consequently making quantitative analysis of variations in individual constituents a challenging task. Human serum is also characterized by the dynamic range of concentrations observed between the abundant, high molecular weight (HMW) and the sparse, low molecular weight (LMW) molecules. For example, human serum albumin (HSA) (57–71%) and globulins (8–26%) are two abundant HMW proteins completely dominating the composition of the serum quantitatively, potentially impacting the detection and monitoring small variations in the features related to the presence of the informative low molecular weight proteins/peptides/metabolites.41 The analytical capabilities of traditional proteomic methods are limited, given this large dynamic range of concentrations in the serum. Therefore, depletion of the highly abundant proteins is now recognized as the first step, yet to be optimized, in the analysis of the serum composition.42 ATR-FTIR suffers from similar limitations to those encountered with chromatography and/or mass spectroscopy based approaches, which can be overcome by means of sample fractionation prior to data recording.24 Including a separation step prior to IR analysis in order to enhance the specificity and sensitivity has been proposed through coupling with chromatography technologies such as LC-IR and GC-IR.43 Those hyphenated approaches have been reported in chemistry, pharmaceutical and food sciences applications.44–46 However, the development of cryogenic temperature control methods during the sample preparation remains the most promising aspect for possible emergence of such technologies in biomedical applications and human body fluids analysis.47 This present study however investigates the benefits of an alternative approach combining centrifugal filtration/fractionation with ATR-IR spectroscopy for the analysis of human serum as a proof of principle for monitoring and quantified potential low molecular weight biomarkers. Glucose has been used as a model to illustrate the strategy to isolate the relevant fraction of the sample to minimize the influence of the HMW proteins and improve the precision and accuracy of the quantitative models built using the PLSR algorithm. Initially, human serum spiked with systematically varying, physiologically relevant, concentrations of glucose will be used to optimise the measurement protocol, including a study of how the volume deposited on the ATR crystal influences the relevancy of the data collected. PLSR will be employed to build a predictive model over the concentration range to demonstrate the principle of the predictive capacity of the technique and the improved precision afforded by serum fractionation. In the second part of the study, the technique will be demonstrated in patient samples of known varying glucose levels, validating the potential for glucose level monitoring.
2. Experimental
2.1 Materials and methods
Sterile, filtered human serum from normal mixed pool (off the clot) was purchased from TCS Biosciences (UK) for the in vitro model prepared from spiked solutions, while patient serum samples were donated by the University Hospital CHU Bretonneau de Tours (France), following the institutional ethical procedures. Initially, the samples were collected during routine blood check-ups, 1 mL of the vial remains being provided for further spectroscopic analysis. Commercially available, centrifugal filtering devices, Amicon Ultra-0.5 ml (Millipore – Merck, Germany), with cut-off points at 10 kDa, were employed to fractionate the serum samples, in both cases. As a result, for each sample, 2 fractions were obtained following filtration; the first representing serum constituents with a molecular weight higher than the cut-off point of the filter used (concentrate); the second corresponding to the fraction passed by the membrane and collected in the vial (filtrate). 0.5 mL of the serum was placed in the centrifugal filter for spinning. The procedure for washing the centrifugal devices prior to serum processing was adapted from,48 based on manufacturer's guidelines, and performed as follows: the Amicon Ultra-0.5 ml filter was spun thrice with a solution of NaOH (0.1 M), followed by 3 rinses with Milli-Q water (Millipore Elix S). For both washing and rinsing, 0.5 mL of the respective liquid was added to the filters and the centrifugation was applied for 10 min at 14000g followed by a spinning with the devices upside down at 1000g for 2 min in order to remove any residual solution contained in the filter. The aim of the study was to illustrate the potential of ATR-IR spectroscopy to detect and screen a potential biomarker in the LMWF. Therefore, based on previous experience demonstrating that 100% of the LMWF is recovered with the 10 kDa, only this cut-off has been included in the present work.24
Additionally, D-glucose (Fisher scientific, UK) was analysed as reference chemical compound for the quantitative analysis on whole and filtered human serum. Notably, glucose has been selected because it is routinely screened in clinics allowing determination of exact blood levels for each patient sample tested.
2.2 Glucose spiked human serum model
The commercial whole human serum was supplemented with known concentrations of glucose; 0.0 mg dL−1 (control), 20 mg dL−1, 60 mg dL−1, 100 mg dL−1, 140 mg dL−1, 180 mg dL−1, 220 mg dL−1, in order to explore the dynamic range and sensitivities of the ATR-IR measurement of unprocessed and filtered samples. The concentrations have been selected to cover a wide range of physiological relevance to simulate hypoglycaemia (<60 mg dL−1), normal level (70–110 mg dL−1) and hyperglycaemia (>120 mg dL−1), in order to optimise the protocols for clinically relevant human serum monitoring using ATR-IR. The serum stock solution has been used as a reference to build the regression models and evaluate the sensitivity of the techniques.2.3 Patient samples blood glucose levels
A total of 15 patient samples have been included in the present study. Glucose levels have been measured at the CHU de Tours using a Cobas analyser following the in house guidelines for routine biochemical analysis. The principle of the test is based on the enzymatic reference method with hexokinase, which catalyses the phosphorylation of glucose to glucose-6-phosphate by ATP.49,50 Subsequently glucose-6-phosphate is oxidized by glucose-6-phosphate dehydrogenase, in the presence of NADP, to gluconate-6-phosphate. This reaction is specific, with no other carbohydrate being oxidized. The rate of NADPH formation during the reaction is directly proportional to the glucose concentration and is measured photometrically in the UV.Glucose level is one of the most common tests performed during blood check-ups and is therefore a perfect model to develop a proof of principle for quantification of low molecular weight biomarkers in human serum using ATR-IR spectroscopy. The instrumentation available at CHU de Tours has a standard deviation of 0.04 mmol L−1 (0.721 mg dL−1), and can therefore be considered as the gold standard. Measured glucose concentrations were provided with the patient samples and have been used as target values for the PLSR models. A summary of information extracted from patient histories is given in Table 1 (see below).
| Sample number | Glucose blood levels | Gender | Age | |
|---|---|---|---|---|
| mmol L−1 (±0.04) | mg dL−1 (±0.721) | |||
| 1 | 3.4 | 61.25 | F | 33 |
| 2 | 3.5 | 63.05 | F | 36 |
| 3 | 3.6 | 64.86 | F | 64 |
| 4 | 3.7 | 66.66 | M | 31 |
| 5 | 3.7 | 66.66 | F | 30 |
| 6 | 3.7 | 66.66 | F | 87 |
| 7 | 3.8 | 68.46 | F | 26 |
| 8 | 6.4 | 115.3 | M | 73 |
| 9 | 6.5 | 117.10 | M | 65 |
| 10 | 6.7 | 120.70 | F | 58 |
| 11 | 7.0 | 126.11 | M | 75 |
| 12 | 7.3 | 131.51 | M | 53 |
| 13 | 9.5 | 171.15 | M | 79 |
| 14 | 10.0 | 180.16 | F | 63 |
| 15 | 11.6 | 208.98 | M | 63 |
All patient samples have been processed with the 10 kDa centrifugal devices in order to carry out HMWF proteins depletion prior to IR analysis. The reproducibility being a critical point to ensure reliability of the results, a set of 10 independent samples (5 unprocessed and 5 filtered mixed pool human serum samples) has been tested on the Cobas analyser before and after ultrafiltration. Ultimately, it has been demonstrated that the concentration of glucose was 4.0 ± 0.1 mmol L−1 in both unprocessed and filtered samples, demonstrating that glucose can freely pass through the membrane of the filters and be quantified in the filtrates recovered. This observation correlates with previous testing of reproducibility highlighting the repeatability of the process.24
2.4 Data collection using the ATR-IR
ATR-IR spectra were recorded using a Bruker Vector 22 equipped with a single reflection golden gate ATR accessory (Specac, UK). A diamond top plate with a 45 °C incident angle was preferred for this study. Penetration depth of the evanescent wave into the sample is both wavenumber and sample dependent, but is typically on the order of 1 μm. Spectral data were the result of 32 scans, with a spectral resolution of 4 cm−1 covering the spectral window 4000–600 cm−1. A background spectrum was also recorded in air (32 scans) and automatically ratioed with the sample spectrum by the software. A built-in quality control is automatically performed by the operating system (OPUS software) every day upon system start up, ensuring the ATR accessory is delivering adequate data and no malfunction of the instrument is detected. Liquid human serum solutions: the spectroscopic analysis of samples was performed directly after deposition of a drop on the crystal (0.1 μl, 0.2 μl, 0.5 μL, 1 μL and/or 2 μl), following air drying. The drying time necessary is directly related to the volume deposited and can be affected by external parameters such as room temperature and humidity, but it generally comprised between 3–5 min. Moreover, the effectiveness of the air drying has been confirmed in real time following the evolution of the main water band in the range 4000–2500 cm−1 directly on screen. The broad water band, with a maximum absorbance between 3270–3340 cm−1, is a reliable indicator of the dehydration of the sample deposited. The intensity of the band gradually decreases as the sample dries, until an equilibrium is reached, at which point no further spectral evolution is observed. When the signal has been found to be stable for at least 60 seconds, the drop is considered dry and the spectra were collected. Although not yet automated and requiring the constant presence of the operator to monitor the screen, this approach has been found particularly efficient and the complete drying can be unambiguously confirmed in the data collected, as illustrated with the raw spectrum in Fig. 1B.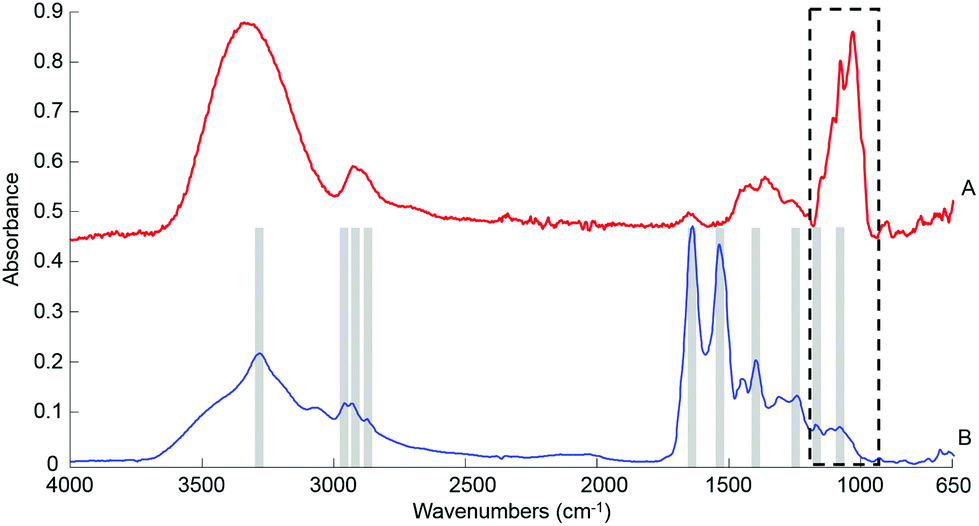 | ||
| Fig. 1 Typical ATR IR spectra collected from a 2 μL drop of 10 g dL−1 pure glucose solution deposited on the ATR crystal (A) and human serum (B). Both spectra have been recorded after air drying. No pre-processing has been applied. The spectra have been offset for clarity and the main features of interest in the serum spectrum are highlighted. | ||
At least 5 drops have been measured per sample and sets of 5 spectra have been collected for each drop in order to take into account both instrumental and inter-drop variability In order to reduce the inter-drop size and shape variability, the deposition has been made on clean and dry crystal. It has been observed that the top diamond plate is quite hydrophobic, preventing the drop from spreading out after deposition and maintaining the round shape of the liquid sample. All drops have been administered with micropipettes, perpendicular to the surface, which ultimately results in reproducible circular shaped samples centred on the crystal after air drying. Finally, only 20 spectra (4 drops) have be used for each sample, selecting the data sets with similar raw maximum absorbance indicating the solutions have been deposited similarly on the crystal. Ultimately, a total of 140 spectra were included in the model study with spiked human serum, while 300 spectra were analysed for the 15 patient samples. 2 μL has been found to be the maximum realistically usable volume, considering the drying time and the large number of samples tested.
2.5 Data pre-processing and analysis
The different pre-processing and data analysis steps were performed using Matlab (Mathworks, USA). Partial Least Squares Regression (PLSR) analysis was exploited as an approach to quantify the spectral variability generated by either the addition of known concentrations of glucose to the human serum or estimate blood glucose levels in patient samples.The analysis of the spectra collected has been restricted to the finger print region, in which the sugar contribution occurs in IR spectra. The spectra collected from spiked human serum have been either min–max normalized (MMN) at the 1637 cm−1 peak corresponding to the amide I band or processed using baseline correction (rubber-band) followed by vector normalization (VN). The IR data collected being highly reproducible with no sign of strong distortion linked to physical effects such as Mie scattering, the baseline correction has been mainly employed to correct the slight offset observed in the spectra between drops. For instance, the rubber-band algorithm employed has been adapted for use on spectral data.51 In the present case, only 2 nodes have been defined at 1800 cm−1 and 900 cm−1 for the baseline correction and this consistently for all spectra. To better appreciate the minimal correction applied to the data sets, the process is illustrated in Fig. S1.† It should be noted that, following filtration, the spectral signature is quite different, due to the depletion of the abundant proteins. Consequently, the typical amide I and II bands are not observed in the filtered serum, but are replaced by a strong peak at 1591 cm−1 most likely assigned to conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C. For consistency, this band, which is the most intense observed in the spectra of the filtered samples, and is also remote from the glucose bands, has been selected for the min–max normalisation.
C. For consistency, this band, which is the most intense observed in the spectra of the filtered samples, and is also remote from the glucose bands, has been selected for the min–max normalisation.
In an attempt to preserve the information related to raw absorbance intensity, a different method has been employed for the data collected from patient samples. Due to the fact that none of the spectral features can be assumed to be consistent between patients, and thus cannot be used as internal standard, the min–max normalisation on the amide I band was found to be insufficient (data not shown). However, in order to minimise the slight drifts observed the spectral background, it has been preferred to normalised the data at 1780 cm−1, located in a region away from any IR bands of the serum. This approach was evaluated in this study to compensate from the offsets observed in the raw data, restoring a common baseline for the spectra without losing the relation between absorbance and the concentrations of different molecular species. The PLSR model has been built from the pre-processed data sets. The algorithm works in a supervised fashion, whereby all different concentrations are known before running the analysis. Ultimately, the output gives an estimation of the model precision (Root Mean Square Error – RMSE) and linearity between the experimental and predicted concentrations (R2). In order to validate the robustness of the models, a 20 fold – cross validation loop has been included in the routine. For each iteration, 50% of the data is randomly selected to constitute the calibration set, while the remaining 50% are used as a validation set for the quantitative predictions. In the present study, particular attention has been accorded to exploit the entire outcome of the PLSR models. For this, for each iteration of the cross validation the RMSE, R2 and predicted concentrations have been extracted and compiled to calculate mean and standard deviations values. It is also important to note that, PLSR being a supervised algorithm, spectral variations not directly related to the target variable of glucose concentration should have little to no impact on the precision estimated. Also the air drying has been carefully monitored for each sample, and any residual water variations in the data would not be expressed in the dimensions selected for the construction of the quantitative model.
Finally, the 2 main notions developed throughout the study are the precision, which is the variability in the measurements realised, and the accuracy corresponding to the how closely the result of an experiment agrees with the “true” or expected result. While the former can be given by mean of the RMSEV from the PLSR models and different calculation of standard deviation, the latter has been particularly used for the patient samples by comparing the relative error (%) between the reference concentrations results provided by the clinicians (gold standard) and the predicted concentrations estimated by PLSR.
3. Results and discussion
3.1 Quantification of glucose levels in spiked human serum
O stretch of COO–), 1242 cm−1 (asymmetric PO2 stretch), 1171 cm−1 (ester C–O asymmetric stretch) and 1080 cm−1 (C–O stretch).54–56 The spectrum from whole human serum is clearly dominated by the abundant proteins contribution such as albumin and globulins24 which swamp the contribution of less represented biomolecules. In comparison, the glucose signature exhibits fewer features, the main peaks being located in the spectral range 1280–800 cm−1, resulting from both the ν(C–O) and ν(C–O–C) vibrational modes,57 as identified in Fig. 1A. In the present work, the limited spectral window of 1800–900 cm−1 was chosen to avoid the regions of strong water absorption. Although the spectral response has stabilised, contributions of water are still visible as a broad background in the region of ∼3300 cm−1.
The volume deposited can have an influence on the data collected, depending on the concentration of the solutions analysed and the coverage of the crystal achieved. The so-called “coffee ring effect” has been documented extensively in literature54,58 and describes the uneven distribution of the constituents following air drying. Commonly, a higher concentration is obtained on the edge of the deposited material which can result in some spectral variability. In complex mixtures such as human serum, different chemical constituents are deposited at different rates, as described by Vroman,59 resulting in spatially inhomogeneous chemical composition in the dried deposit. Fig. 2 presents the dependence of the area under the curve observed in raw data (AUC – between baseline and peak maximum), calculated for the band 1180–955 cm−1, depending on deposited volume, for two different concentrations of glucose solution. The 10 g dL−1 solution does not exhibit any noticeable decrease in the AUC between 0.5 μL and 2 μL, explained by a full coverage of the crystal coupled to a saturation of the signal.24 The notion of saturation is defined by the loss of linearity between the absorbance and the concentrations measured. In contrast to transmission IR spectroscopy, which can deliver absorbance up to 3 with a saturation manifesting itself as highly noisy spectra coupled to plateau effects, ATR-IR spectra intensities are limited by the depth of penetration of the evanescent wave. In the present study and considering the instrumentation used, it has been observed that the maximum absorbance that can possibly be recorded from protein rich samples such as human serum would never exceed 0.45. Following air drying, the Beer–Lambert type dependence of the absorbance on initial sample concentration and length of the optical path can both be partially lost and the critical parameter is the amount of matter deposited. In other words, the deposited sample thickness, and therefore absorbance, depends on the concentration of the droplet applied, but if the sample thickness exceeds the evanescent wave penetration depth, no further increase in the absorbance can be observed. However, when reducing the volume below 0.5 μL, the deposit thickness becomes less than the sampling depth, and the AUC consequently decreases. The AUC pattern for the 0.1 g dL−1 is rather different, a maximum intensity being found with 0.2 μL. This is a result of the coffee ring effect, as the glucose is accumulated in the edges of the drop, outside the field of data collection for larger volumes (Fig. 2, 0.5 μL–1 μL–2 μL), thus delivering poor signals, while the 0.2 μL deposit is completely contained on the crystal and entirely recorded. The decrease observed for the 0.1 μL sample confirms that the drops are smaller than the ATR crystal area. These observations clearly demonstrate the difficulty to accurately estimate the maximum and minimum glucose concentrations that can be measured with IR, due to the different behaviours at low and high concentrations. However, as demonstrated in the ESI (Fig. S2†), based on the amount of glucose deposited, such limits can be estimated. Also, as the highest concentrations measurable are much greater than any relevant range for clinical applications, with values between 1210 mg dL−1 and 24205 mg dL−1 for respectively 2 μL and 0.1 μL drops, the minimum concentrations are indeed of far more importance. While a 0.1 μL drop would be associated with a limit of detection around 15 mg dL−1, increasing the volume deposited would gradually decrease the minimum concentrations measurable to 7.5 mg dL−1, 3 mg dL−1, 1.5 mg dL−1 and 0.75 mg dL−1 for respectively 0.2 μL, 0.5 μL, 1 μL and 2 μL. Such observation implies that larger volumes should deliver more accurate outcomes from the analysis, however further investigations of human serum have been performed with both 2 μL and 0.2 μL. Although 0.1 μL would have been optimum to ensure the volume deposited is smaller than the crystal size, due to the viscosity of the serum, the smallest volume that could be deposited as drops was 0.2 μL.
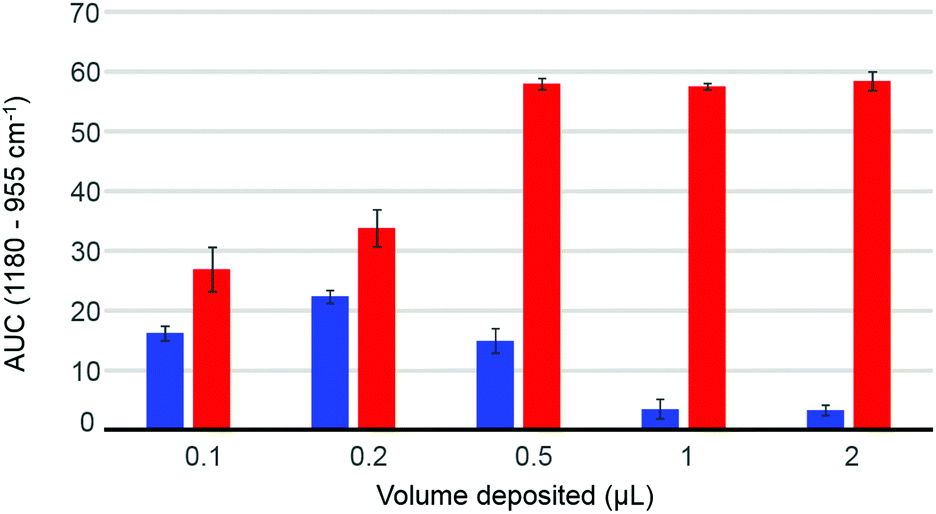 | ||
| Fig. 2 Evolution of Area Under the Curve (AUC) of the band at 1180–955 cm−1 as a function of volume of glucose solution deposited. Red: 10 g dL−1 glucose solution; blue: 0.1 g dL−1 glucose solution. Intensity of the blues bars have been multiplied by 5 for better visualization on the graph. Error bars are the results of 5 independent measurements. | ||
The first step of the study was to build a model accurately mirroring clinically relevant blood variations of glucose levels using human serum spiked with D-glucose. For this, different amounts of pure glucose have been added to the human serum stock solution in order to achieve variable concentrations in the physiologically relevant range of 20 mg dL−1–220 mg dL−1. The normal glucose concentration being between 70 mg dL−1 and 110 mg dL−1, simulation for hypoglycaemia and hyperglycaemia have been deliberately included in the set of samples prepared and tested. Fig. 3A presents mean ATR-IR spectra collected following air drying from the different solutions. The (1637 cm−1) min–max normalized data exhibit similar profiles, only the region attributed to sugar being affected by the increase of glucose concentration. In Fig. 3B, it can be observed how the 1140–950 cm−1 spectral region evolves from the serum stock solution (red) to the highest concentration prepared (cyan), the main features at 1105 cm−1, 1078 cm−1, 1032 cm−1 and 989 cm−1 systematically increasing with glucose concentration.
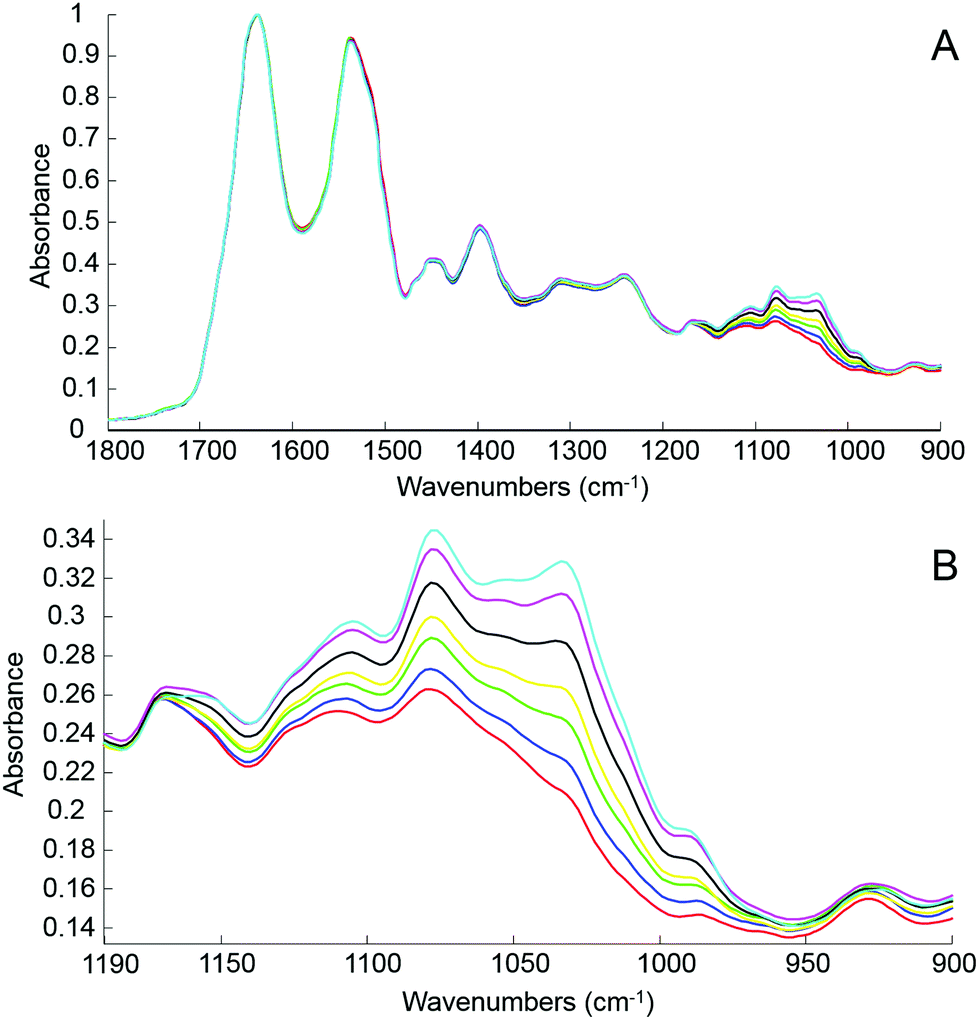 | ||
| Fig. 3 Mean ATR-IR spectra collected from unprocessed whole human serum (red) and supplemented with 20 mg dL−1 (blue), 60 mg dL−1 (green), 100 mg dL−1 (yellow), 140 mg dL−1 (black), 180 mg dL−1 (magenta) and 220 mg dL−1 (cyan) of glucose respectively. A: Finger print region 1800–900 cm−1; B: Glucose region 1190–900 cm−1. Min–max normalized spectra on the amide I band (1637 cm−1) used for illustration. | ||
Pre-processed spectra have been analyzed using the PLSR algorithm to determine the relationship between spectral variations and glucose concentrations. The method being supervised, the different concentrations are taken into account during the calculations. The first step generates a scatter plot, (not shown). Fig. 4 presents the first two weighting vectors A and B, confirming the discrimination of the data is based on glucose features, which, as demonstrated in Fig. 2 and 3, mainly occur in the 1190–950 cm−1 window. The similarities between the 2 weighting vectors support the fact that more than 1 dimension is required to fully describe the spectral variability due to glucose concentrations. Plotting the Root Mean Square Error from the validation set (RMSEV) as displayed in Fig. 5 is commonly used to guide the operator in choosing the optimal number of dimensions necessary to reach the best model. In the present case, a 20 fold cross validation has been preferred, leading to the creation of 20 independent calibration/validation sets and thus 20 predictive models. In order to simplify the illustration, the error bars in Fig. 5 illustrate the standard deviation calculated between each iteration of the cross validation. As expected, a strong decrease in the RMSEV is observed within the 3 first dimensions, which is normal behaviour for well discriminated data, followed by a stabilisation of the values observed without any further improvement of the model precision. Considering the min–max normalised spectra used for illustration in Fig. 5, a minimum is found at 2.078 ± 0.252 mg dL−1, corresponding to 8 dimensions. For consistency between data sets, the minimum found has always been selected to build the predictive models. It should be noted that similar plots can be obtained from the RMSE for the calibration sets (not shown).
 | ||
| Fig. 4 First (A) and second (B) PLS weighting vector corresponding respectively to dimension 1 and 2 of the scatter plot. Min–max normalized spectra on the amide I band (1637 cm−1) used for illustration. | ||
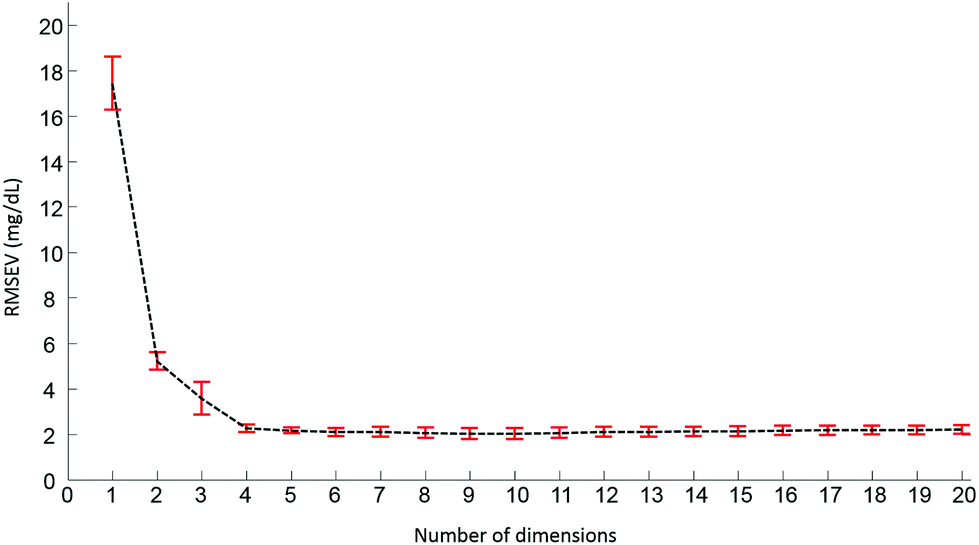 | ||
| Fig. 5 Evolution of the root mean square error on the validation set (RMSEV) according to the number of dimensions selected in the PLS model. Value are average calculated from the 20 iteration of the cross validation associated to corresponding error bar illustrating the standard deviation. Min–max normalized spectra on the amide I band (1637 cm−1) used for illustration. | ||
Ultimately, after selection of the optimal number of dimensions for the data set analysed, a predictive model can be built from the PLSR scatter plot (Fig. 4), to compare the observations corresponding to the known concentrations of glucose in the samples with the estimated concentrations from the spectral data sets. In the example presented in Fig. 6, a really good linearity was reached, with a R2 value of 0.9992. The standard deviation (2.172e−4) is indicative of good repeatability between the 20 iterations of the cross validation. However, the standard deviation of the RMSEV (0.2526 mg dL−1) is equal to about 10% of the mean value, but remains acceptable considering the precision of the model is below the mg dL−1 range. Moreover, the error bars display no overlapping of data, indicating that each concentration can be unambiguously identified. This approach has been replicated for all data sets collected from the human serum samples spiked with glucose, either unprocessed (whole serum) or following centrifugal filtration with a 10 kDa device. The summary of the PLSR results is presented in Tables 2 and 3.
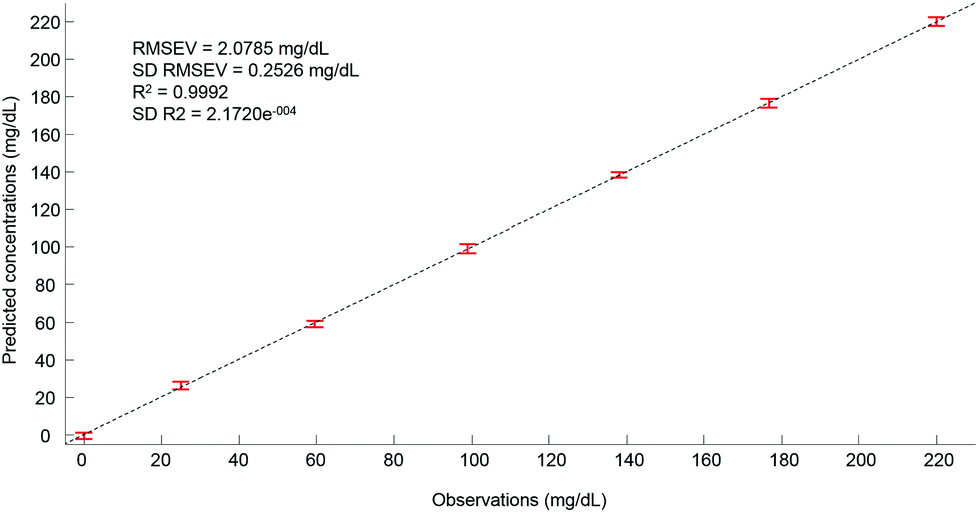 | ||
| Fig. 6 Predictive model build from the PLS analysis. For each concentrations the value displayed is an average of the concentration predicted with the corresponding standard deviation calculated from the 20 iterations of the cross validation. Mean RMSEV and R2 values are given on the plot both also with their respective standard deviation. Min–max normalized spectra on the amide I band (1637 cm−1) used for illustration. | ||
| Deposit type | Drop – 2 μL | Drop – 0.2 μL | |||||
|---|---|---|---|---|---|---|---|
| RMSEV | STD | R 2 | RMSEV | STD | R 2 | ||
| MMN: min–max normalised at 1637 cm−1; VN: baseline and vector normalised, STD: standard deviation, GLU: spectra cut to the range 1190–950 cm−1. | |||||||
| MMN | FP | 2.078 | 0.252 | 0.999 | 12.347 | 1.852 | 0.974 |
| GLU | 2.388 | 0.126 | 0.999 | 15.419 | 0.753 | 0.96 | |
| VN | FP | 2.137 | 0.181 | 0.999 | 12.512 | 1.36 | 0.973 |
| GLU | 2.806 | 0.362 | 0.999 | 15.942 | 2.058 | 0.956 | |
| Deposit type | Drop – 2 μL | Drop – 0.2 μL | |||||
|---|---|---|---|---|---|---|---|
| RMSEV | STD | R 2 | RMSEV | STD | R 2 | ||
| MMN: min–max normalised at 1591 cm−1; VN: baseline and vector normalised, STD: standard deviation, GLU: spectra cut in range 1190–950 cm−1. | |||||||
| MMN | FP | 2.199 | 0.250 | 0.999 | 5.337 | 0.551 | 0.995 |
| GLU | 3.465 | 0.317 | 0.998 | 5.506 | 0.417 | 0.995 | |
| VN | FP | 2.679 | 0.234 | 0.998 | 5.357 | 0.396 | 0.995 |
| GLU | 4.001 | 0.337 | 0.997 | 8.746 | 0.539 | 0.986 | |
A direct comparison of the predictive results obtained from 2 μL and 0.2 μL (Table 2) drops of whole human serum unambiguously demonstrates that the precision of the PLSR models is strongly affected by the size of the drops. The best RMSEV (2.078 ± 0.252 mg dL−1) was achieved with the Min–Max Normalised (MMN) ATR-IR spectra collected from the 2 μL drops, while this value is up to 6 times higher for the 0.2 μL deposits (12.347 ± 1.852 mg dL−1). It should be noted that neither reducing the spectral range of the glucose windows (1190–950 cm−1) (see Table 2 GLU) nor using vector normalisation (VN) improves the precision of the predictive models (see Table 2 VN). This observation is rather encouraging and supports the notion that, although the samples exhibit a heterogeneous distribution of the different molecular species following air drying, the centre of the drop still delivers quantitative information that can be linearly captured by the PLSR models.
Interestingly, the 0.2 μL does not improve the PLSR quantitative model, which suggests that attempting to ensure that the entire sample is deposited on the ATR crystal, with a coverage less than the recording area, is not the best strategy when performing ATR analysis. An experimental limitation of ATR-IR spectroscopy is the absence of a bright field imaging system coupled to the spectrometer, making the visualisation of the drop positions quite difficult, especially with volumes less than 1 μL. Therefore, it is simply impossible to ensure the 0.2 μL samples have been positioned identically on the ATR crystal, explaining the high RMSEV values.
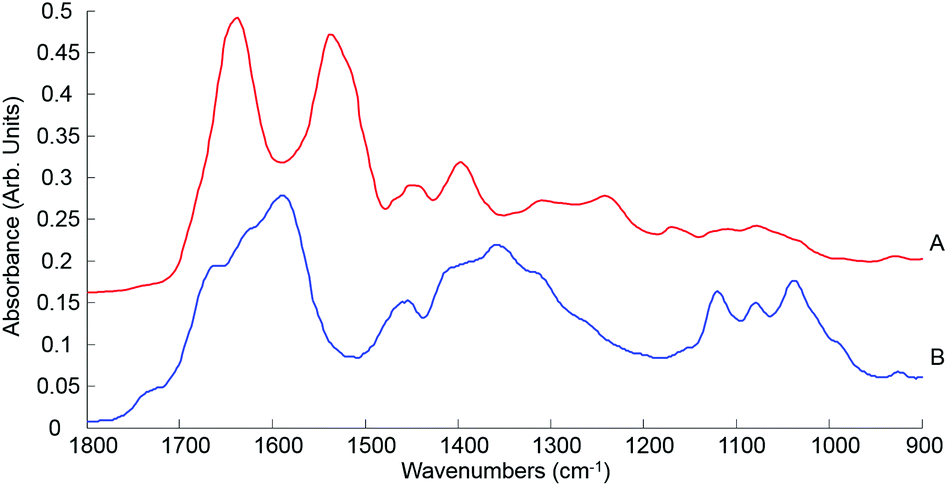 | ||
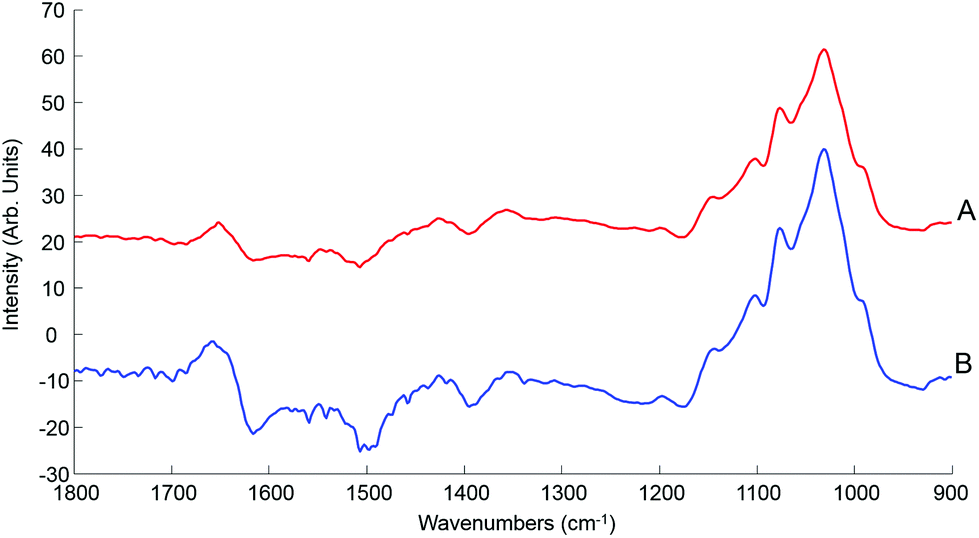
| Fig. 7 Mean ATR-IR spectra collected from human serum stock solution unprocessed whole (A) and 10 kDa (B) in the finger print region 1800–900 cm−1. Spectra offset for clarity. | ||
This initial step of the study is rather reassuring, indicating that centrifugal filtration does not affect the sample integrity, which remains representative of the concentrations of the different serum physiological constituents. The main concern was regarding the drying pattern of biological samples characterised by low concentrations, as illustrated in Fig. 2, which are more affected by the coffee ring effect which could result in a drastic decrease in the IR spectral intensity due to accumulation of molecules in the edge of the dried drop, with little or no contribution at the centre of it. However, despite the considerably reduced concentration in filtered serum, this effect remains limited and the recordings performed on the 2 μL drops are perfectly relevant, delivering good predictive models by means of PLSR analysis. Following those observation, the use of 0.2 μL samples can be disregarded for analysis of patient samples, in favour of the 2 μL. A comparison with a PLSR model constructed from aqueous glucose solutions indicates that the coffee ring effect is limited in filtered serum. In pure glucose solutions, it is observed that the RMSEV tends to be slightly higher than those obtained in Table 3 (see Fig. S4†). The small drop in the R2 value (0.9987) illustrates the difficulty to get a linear correlation between spectral variations and concentrations for pure glucose solutions. It also supports the fact that the residual serum constituents present in the 10 kDa filtrates play a key role in maintaining a more consistent drying pattern while reducing the coffee ring effect. Thus, it is important to deplete the human serum and remove the HMW fraction, but preserving the whole LMW fraction seems to be a better approach than trying to further separate or extract glucose from the samples as the surrounding matrix clearly impacts the precision of the measurements done.
3.2 Glucose level quantification in patient samples
Although the quantitative capabilities of ATR-IR spectroscopy can be quite easily demonstrated with model human serum spiked with glucose, both commercially available, the analysis of patient samples can be considered a more delicate matter, the main reason being the multi-parametrical variability observed in clinical applications. When spiking human serum with glucose, only one physiological constituent is affected, while all others remain at similar levels. In that situation, any peak away from the glucose spectral window can be used as an internal standard for calibration of the data, explaining the good results obtained with a MMN to the amide I band. The overall protein content being the same between spiked samples, the multivariate analysis of the spectra ultimately highlights the linear evolution of the band ratio between the proteins and glucose. However, samples harvested from patients can display an intrinsic variability, directly reflecting their physiological state on the day. In real conditions, there is no real control, as each individual has its own metabolism, characterised by different ground blood levels for all the serum constituents. Uric acid, blood urea nitrogen (BUN), creatinine, total proteins (albumin, globulin), total bilirubin, alkaline phosphatase, GGTP, LDH, SGOT (also called AST) are some of the most screened serum constituents, although only a few are routinely tested. Glucose is indeed one of them, used for monitoring either hypoglycaemia or hyperglycaemia, particularly important for the detection of diabetes. In addition to the fact that glucose can freely diffuse through the centrifugal device membranes and be fully collected in the filtrate, the choice of the glucose was also motivated because of its physiological relevance and normal levels in human serum. With concentrations ranging between 70–100 mg dL−1, it can be detected in whole unprocessed serum, which was the condition required to be able to build a comparative model between patients samples before and after fractionation.A total of 15 patient samples have been analysed using ATR-IR spectroscopy, selected to cover a wide range of glucose concentrations mirroring cases of hypoglycaemia and hyperglycaemia (Table 1). The initial measurements have been performed from the unprocessed samples, as provided from the clinician, from 2 μL air dried drops. Similarly to the spiked samples, the spectra, min–max normalised at 1780 cm−1, have been analysed with the PLSR algorithm and the results are presented in Fig. 9A. With a RMSEV of 11.87 ± 0.88 mg dL−1, the precision of the predictive model has strongly decreased in comparison to the observation made from spiked serum. Furthermore, the R2 reflects the lack of linearity between the spectral variations and the glucose levels, thus the poor quality of the analysis performed. As shown in Fig. 8B, although the first (a) and second (b) PLSR weighting vectors contain contributions which can be associated with glucose (Fig. 1), there are also significant other contributions associated with serum proteins in the region 1650–1350 cm−1.
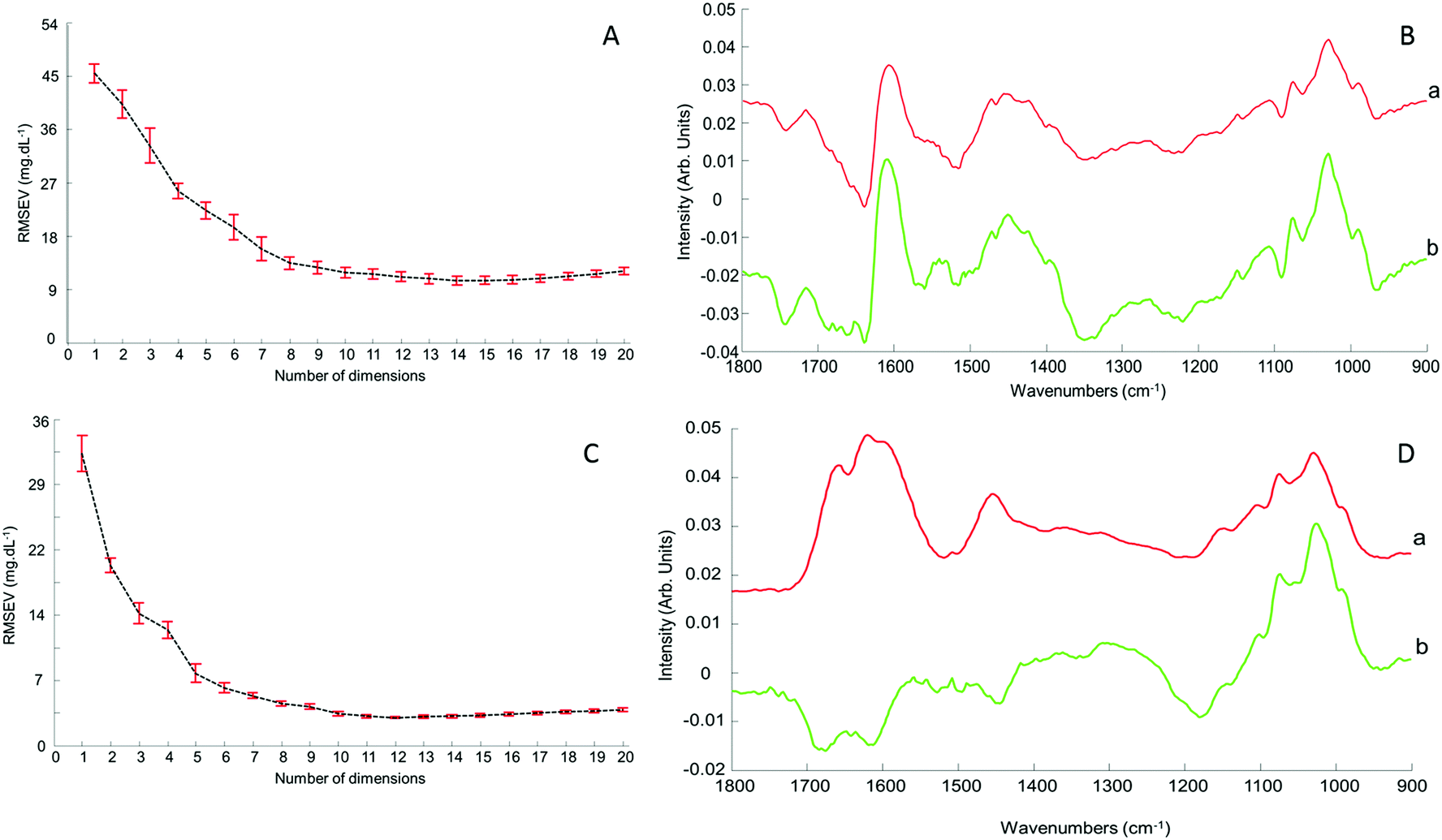 | ||
| Fig. 8 Evolution of the root mean square error on the validation set (RMSEV) according to the number of dimensions selected in the PLS model with corresponding first (a) and second (b) PLS weighting vector corresponding respectively to dimension 1 and 2 of PLSR model. A: Unprocessed serum; B: 10 kDa filtered serum. | ||
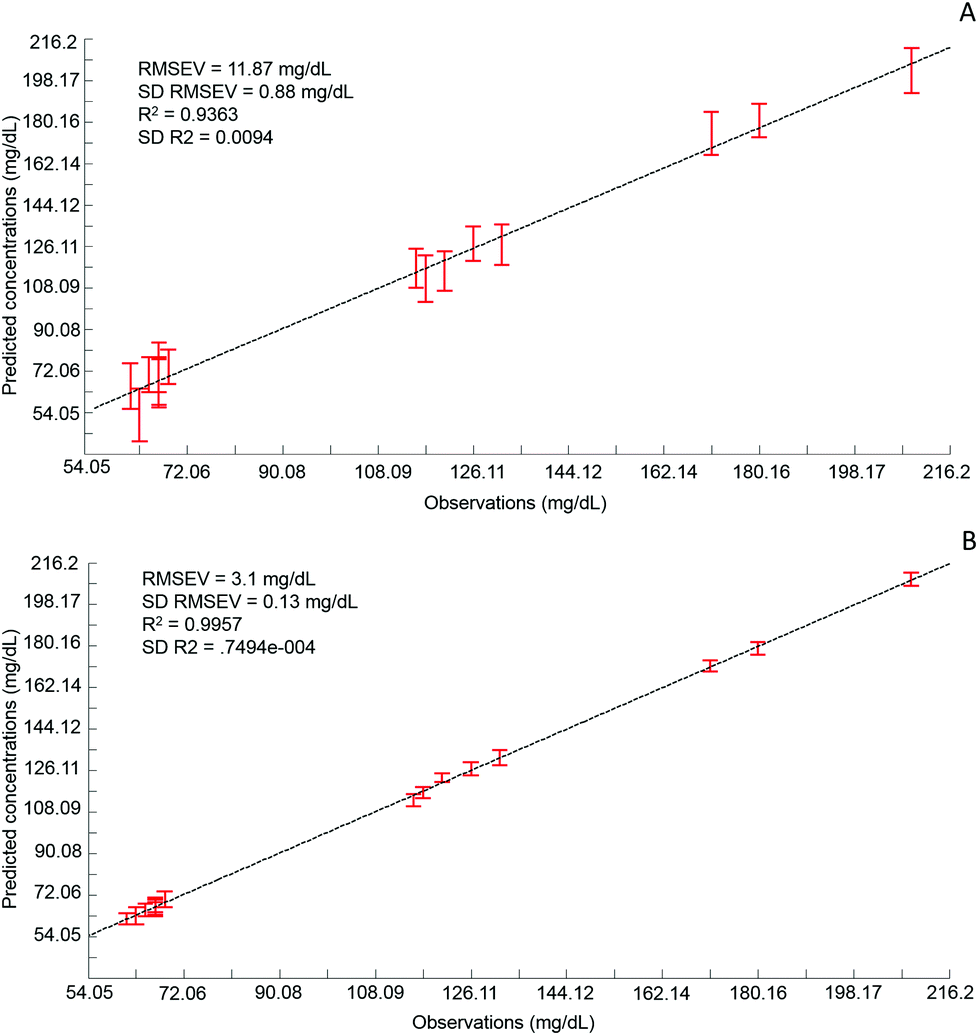 | ||
| Fig. 9 A: Predictive model built from the PLS analysis of patient unprocessed samples; B: Predictive model built from the PLS analysis of patient 10 kDa filtered samples. For each concentrations the value displayed is an average of the concentration predicted with the corresponding standard deviation calculated from the 20 iterations of the cross validation. Mean RMSEV and R2 values are given on the plot both also with their respective standard deviation. Min–max normalized at 1780 cm−1. | ||
Following the first set of measurements, the same patient samples have been processed with a 10 kDa centrifugal device in order to fractionate the serum by depletion of the abundant proteins. From the resulting filtrates, ATR-IR spectra have been recorded, also min–max normalised at the 1780 cm−1 and analysed in identical conditions for comparison purposes. The PLSR model is presented in Fig. 9B, displaying a much better RMSEV value (3.1 ± 0.13 mg dL−1). A comparison of both plots unambiguously supports the improvement after centrifugal filtration that can be achieved, confirmed by the reduced standard deviation associated with each concentration tested. Consequently, the linearity of the predictive model is restored to an acceptable value for R2 of 0.9957. The first (a) PLSR weighting vector, shown in Fig. 8D, still contains significant contributions associated with serum proteins in the region 1650–1350 cm−1, although the spectrum (b) of the second is now dominated by glucose.
Table 4 gives an overview of the different predicted concentrations for each patient for both unprocessed and fractionised samples. The mean values from the 20 cross validations iterations are given in mg mL−1 with the corresponding standard deviation and finally the relative error compared to the real glucose concentration, as given by the clinician, has been expressed in percentage. Ranging from 16.39% to 0.25%, the relative error for the unprocessed serum clearly highlights the difficulties to estimate the glucose levels in whole serum. Indeed some of the samples display acceptable mean predictive values because of the supervised fashion of the PLSR algorithm. As for any calibration curve, the trend line illustrates the best fit between data point, meaning few samples are located close to it. In comparison, the filtered serum delivers higher accuracy, ranging from 2.51% to 0% mean relative error. Looking at the samples independently, it can be seen that the majority of them are below 1% relative error, clearly supporting the increased accuracy of the model with fractionated samples.
| Patient | Concentrations (mg dL−1) | Unprocessed serum | 10 kDa filtered serum | ||||
|---|---|---|---|---|---|---|---|
| Predicted concentrations (mg dL−1) | |||||||
| Mean | STD | % | Mean | STD | % | ||
| STD: standard deviation; %: relative error between reference and mean predicted value expressed in percent. | |||||||
| 1 | 61.25 | 65.39 | 9.84 | 6.75 | 61.68 | 2.45 | 0.71 |
| 2 | 63.05 | 52.71 | 11.46 | 16.39 | 63.05 | 3.74 | 0.00 |
| 3 | 64.86 | 70.34 | 7.59 | 8.45 | 65.37 | 2.73 | 0.78 |
| 4 | 66.66 | 73.44 | 10.69 | 10.17 | 65.66 | 3.04 | 1.51 |
| 5 | 66.66 | 66.94 | 10.98 | 0.42 | 67.16 | 2.68 | 0.76 |
| 6 | 66.66 | 67.04 | 9.90 | 0.58 | 67.02 | 3.66 | 0.55 |
| 7 | 68.46 | 73.71 | 7.62 | 7.67 | 70.18 | 3.33 | 2.51 |
| 8 | 115.3 | 116.54 | 8.49 | 1.08 | 112.92 | 2.65 | 2.06 |
| 9 | 117.1 | 112.04 | 10.04 | 4.32 | 116.30 | 2.42 | 0.69 |
| 10 | 120.7 | 115.32 | 8.68 | 4.46 | 122.65 | 1.90 | 1.61 |
| 11 | 126.11 | 127.20 | 7.44 | 0.86 | 126.64 | 2.89 | 0.42 |
| 12 | 131.51 | 126.72 | 8.91 | 3.64 | 131.43 | 3.39 | 0.06 |
| 13 | 171.15 | 175.12 | 9.48 | 2.32 | 171.44 | 2.50 | 0.17 |
| 14 | 180.16 | 180.61 | 7.16 | 0.25 | 179.03 | 2.77 | 0.63 |
| 15 | 208.98 | 202.37 | 9.66 | 3.16 | 209.01 | 3.00 | 0.01 |
| Mean % = 4.7 | Mean % = 0.84 | ||||||
Considering the complexity of human serum, concerns can be raised towards the monitoring of low molecular weight physiological constituents potentially relevant for diagnosis. The proof of principle presented throughout this study demonstrates that glucose levels, a well-known biomarker for diabetes, can be monitored by means of infrared spectroscopy. As highlighted in Fig. 1, specific spectral features can be identified for the detection of glucose in complex biological mixtures. Moreover, using multivariate analysis tools such as PLSR, a linear relationship between glucose levels and the intensity of glucose spectral features can be obtained. The interference presented by the HMWF of the human serum is illustrated in the case of patient samples (Fig. 9), indicating that the presence of the abundant proteins can be identified as a limiting factor when focusing on the spectral analysis of potential low molecular weight biomarkers. Similarly to glucose, the constituents of the LMWF of the serum can exhibit weak contributions in the IR data collected directly impacting on the precision and accuracy of the quantitative models developed. The concept of human serum fractionation proposed in the present work, implies that separating the fractions of interest to perform spectral characterisation independently would ultimately lead to better accuracy. This statement can be further supported by the presence of 3 different patients with similar glucose concentrations as given by the clinicians. Although patients 4, 5 and 6 all have glucose levels found at 66.6 mg dL−1, all individuals tested would logically present different overall serum composition, notably with consequent variations in the protein contents. While the predictive model built from the whole serum delivers glucose concentrations of respectively 73.44 mg dL−1, 66.94 mg dL−1and 67.04 mg dL−1, after depletion of the abundant proteins, those values are found to be 65.66 mg dL−1, 67.16 mg dL−1and 67.02 mg dL−1. Although the analysis is not able to generate exact glucose concentrations for those 3 patients due to the precision of the model, it is noticeable that the inter-individual variability is considerably reduced, leading to improved accuracy. Instrumentation available in clinics present high performance towards glucose level monitoring, and with a standard deviation of 0.72 mg dL−1 (0.04 mmol L−1), it is hardly conceivable that IR can deliver better results. Although, with a RMSEV of 3.1 ± 0.13 mg dL−1, fractionating the human serum before analysis places the ATR-IR approach in a clinically relevant range of concentrations allowing the identification of patients with abnormal glucose levels (either hypo- or hyper-glycaemia), the aim of the work proposed was to further demonstrate the potential for body fluids screening towards disease diagnosis, in general. However, the precision of the model expressed by the RMSEV indicates the lowest variations in glucose concentration that can be considered statistically relevant for the discrimination of 2 patients with close results. More importantly, the precision and accuracy play key roles in identifying patients at risk, according to blood glucose (BG) falling in the range corresponding to hypoglycaemia or hyperglycaemia. For this reason, the detection and monitoring of biomarkers are subject to requirements specifically defined by the clinical context. For instance, when considering blood glucose monitoring, the concept of the Parkes error grid has been implemented in 1994.60 This model defines performance zones for the results collected, aiming to assess the clinical accuracy of BG monitoring devices. Notably, the Parkes error grid has been introduced as an accepted evaluation tool according to the ISO15197:2013 guideline “In vitro diagnostic test systems – requirements for blood-glucose monitoring systems for self-testing in managing diabetes mellitus”. Example of Parkes error grids constructed from the PLSR analysis performed from whole and filtered patient serum are given in Fig. 10. Each estimated value from both PLSR models (3000 predicted concentrations) has been plotted in order to better visualize their distribution across the risk zones (it should be noted that the model is based on Diabetes type I). While for the whole serum, a majority of the values are found in zone A, defined as the zone of “clinical accurate measurements with no effect on clinical action”, it can also be observed that, for the lowest concentrations, many are located in zone B. A general consensus would support that, based on the definition of the risk boundaries, a clinically accurate BG meter should show at least 95% of its data points in zone A of the Parkes error grid. Although the number of patients in the present study remains limited, it is nevertheless interesting to observe the significant improvement obtained after depletion of the abundant proteins by ultrafiltration and how all the predicted concentrations from the PLSR model are now gathered in zone A, unambiguously removing any doubts about the data interpretation (Fig. 10B). This further supports the increased clinical relevancy of the quantitative measurement performed after isolation of the LMW fraction. Recently, an increasing numbers of publications promote the capabilities of IR and Raman spectroscopy for serum analysis in both animal and human models from cardiovascular disease to cancers diagnosis.61–63 However even facing the difficulties to accurately perform specific discrimination or diagnosis,2,53 resorting to pre-analytical sample preparation procedures is rarely or never considered. Ultimately, the benefits of serum fractionation using centrifugal filtration demonstrated in the case of glucose levels can be easily transferable to any other low molecular weight biomarkers with swapped contribution in the IR data by the HMWF. Indeed, the transferability into clinics remains an open discussion and further in depth investigation is needed to better evaluate the place of centrifugal filtration in medical routines. However, separation techniques such as those based on chromatography hold a pivotal position in diagnosis based on proteomics, peptidomics or metabolomics. Considering the cost to equip a service with an operational LC/MS systems, the labour intensive procedure for sample preparation and analysis and the running costs attached to maintenance of the instrumentation, the IR approach coupled to centrifugal filtration remains a competitive and cost effective alternative, especially in cancer detection. The centrifugal filters can generate enough sample for analysis in less than a minute and, although they can represent an additional cost to patient testing, compared to protocols involving electrophoresis and/or immunoassays, the 3 euros per device (around 3 dollars) could appear to be insignificant. However, this study remains a proof of concept and the emergence of new technologies for automated sample fractionation and protein depletion based on similar membrane based principle may be necessary (e.g. microfluidic based) for translation of such an approach to the clinical environment. Therefore such optimisations would contribute to further broaden the range of applications of vibrational spectroscopic techniques.
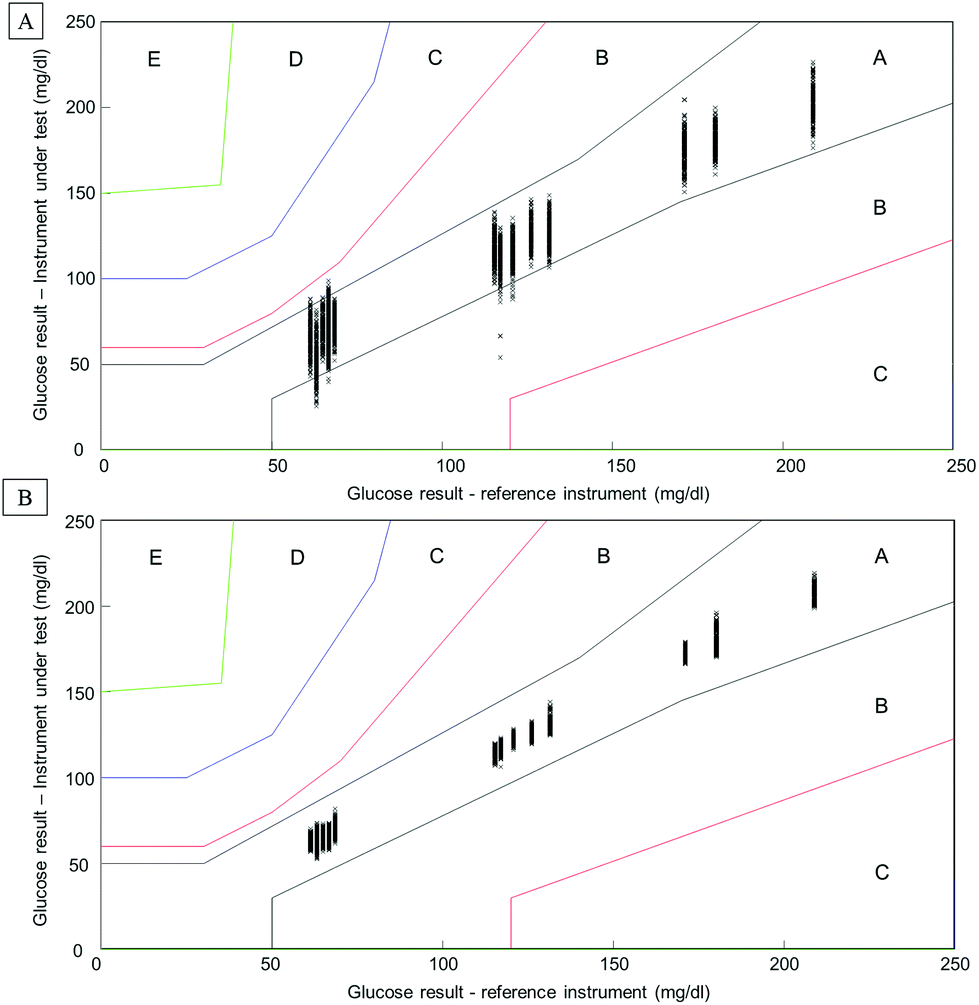 | ||
| Fig. 10 Parkes error grid for patient serum samples. A: Predicted values obtained from the PLSR analysis performed on the whole human serum from patients. B: Predicted values obtained from filtered human serum. Labels A–E define the risks zone restricted to the range 0–250 mg dL−1. | ||
4. Conclusion
Screening of human serum, and by extension all body fluids, is still an emerging field of application and in order to strengthen the position of infrared spectroscopy as a potential clinical tool, numerous questions need to be considered and addressed by the community. Firstly, the technique should be competitive with other approaches currently used, such as mass spectroscopy, in terms of sensitivity and specificity, and, secondly, it should be able to deliver information relevant for clinical diagnosis. Infrared spectroscopy holds immense promise for the implementation of new quantitative analytical techniques in clinical routines. The label free/reagent free argument is often used to further support the relevancy to develop such techniques. However, in order to comply with the high requirements associated with body fluids based diagnostics, a number of improvements of the experimental protocol can be proposed. Notably, delivering quantitative information of the different serum constituents is crucial to validate the approach as a potential clinical tool. Centrifugal filtration of human serum has been proposed to specifically isolate relevant fractions of the samples for more accurate spectroscopic analysis. Using glucose as an example, it has been clearly illustrated that the depletion of the abundant proteins has greatly reduced the spectral variability and consequently significantly improved the precision and accuracy of the quantitative models for potential low molecular weight biomarkers built from the PLSR analysis. In the present study, it has been highlighted that the patient samples are characterised by a normal variability, reflecting the physiological state of the individuals tested, but also that such effects can be considerably reduced by means of easy and rapid pre-analytical samples preparation steps. Furthermore, as the sample processing is also applicable to a wider range of body fluids, the methodology presented in the present work will certainly be beneficial for the field and lead to drastic improvements in the strategies oriented toward their implementation as the next generation of diagnostics techniques clinical tools.Acknowledgements
This work was funded in part by the Science Foundation Ireland, Principle Investigator Award 11/PI/1108.References
- M. Arellano, J. Jiang, X. Zhou, L. Zhang, H. Ye, D. T. Wong and S. Hu, Front. Biosci., Scholar Ed., 2009, 1, 296–303 CrossRef.
- M. J. Baker, S. R. Hussain, L. Lovergne, V. Untereiner, C. Hughes, R. A. Lukaszewski, G. Thiefin and G. D. Sockalingum, Chem. Soc. Rev., 2016, 45, 1803–1818 RSC.
- S. M. Hanash, C. S. Baik and O. Kallioniemi, Nat. Rev. Clin Oncol., 2011, 8, 142–150 CrossRef PubMed.
- C. E. Thomas, W. Sexton, K. Benson, R. Sutphen and J. Koomen, Cancer Epidemiol. Biomarkers Prev., 2010, 19, 953–959 CrossRef CAS PubMed.
- D. W. Greening and R. J. Simpson, J. Proteomics, 2010, 73, 637–648 CrossRef CAS PubMed.
- A. Tessitore, A. Gaggiano, G. Cicciarelli, D. Verzella, D. Capece, M. Fischietti, F. Zazzeroni and E. Alesse, Int. J. Proteomics, 2013, 2013, 125858 Search PubMed.
- R. S. Tirumalai, K. C. Chan, D. A. Prieto, H. J. Issaq, T. P. Conrads and T. D. Veenstra, Mol. Cell. Proteomics, 2003, 2, 1096–1103 CAS.
- D. Basso, A. Valerio, R. Seraglia, S. Mazza, M. G. Piva, E. Greco, P. Fogar, N. Gallo, S. Pedrazzoli, A. Tiengo and M. Plebani, Pancreas, 2002, 24, 8–14 CrossRef PubMed.
- R. B. Rubin and M. Merchant, Am. Clin. Lab., 2000, 19, 28–29 CAS.
- T. Kimhofer, H. Fye, S. Taylor-Robinson, M. Thursz and E. Holmes, Br. J. Cancer, 2015, 112, 1141–1156 CrossRef CAS PubMed.
- D. B. Liesenfeld, N. Habermann, R. W. Owen, A. Scalbert and C. M. Ulrich, Cancer Epidemiol. Biomarkers Prev., 2013, 22, 2182–2201 CrossRef CAS PubMed.
- S. M. Ali, F. Bonnier, K. Ptasinski, H. Lambkin, K. Flynn, F. M. Lyng and H. J. Byrne, Analyst, 2013, 138, 3946–3956 RSC.
- L. M. Fullwood, D. Griffiths, K. Ashton, T. Dawson, R. W. Lea, C. Davis, F. Bonnier, H. J. Byrne and M. J. Baker, Analyst, 2014, 139, 446–454 RSC.
- J. Dorney, F. Bonnier, A. Garcia, A. Casey, G. Chambers and H. J. Byrne, Analyst, 2012, 137, 1111–1119 RSC.
- Z. Farhane, F. Bonnier, A. Casey, A. Maguire, L. O'Neill and H. J. Byrne, Analyst, 2015, 140, 5908–5919 RSC.
- L. F. Carvalho, F. Bonnier, K. O'Callaghan, J. O'Sullivan, S. Flint, H. J. Byrne and F. M. Lyng, Exp. Mol. Pathol., 2015, 98, 502–509 CrossRef CAS PubMed.
- F. M. Lyng, D. Traynor, I. R. Ramos, F. Bonnier and H. J. Byrne, Anal. Bioanal. Chem., 2015, 407, 8279–8289 CrossRef CAS PubMed.
- I. Taleb, G. Thiefin, C. Gobinet, V. Untereiner, B. Bernard-Chabert, A. Heurgue, C. Truntzer, P. Hillon, M. Manfait, P. Ducoroy and G. D. Sockalingum, Analyst, 2013, 138, 4006–4014 RSC.
- J. R. Hands, G. Clemens, R. Stables, K. Ashton, A. Brodbelt, C. Davis, T. P. Dawson, M. D. Jenkinson, R. W. Lea, C. Walker and M. J. Baker, J. Neuro-Oncol., 2016, 127, 463–472 CrossRef PubMed.
- C. Hughes, G. Clemens, B. Bird, T. Dawson, K. M. Ashton, M. D. Jenkinson, A. Brodbelt, M. Weida, E. Fotheringham, M. Barre, J. Rowlette and M. J. Baker, Sci. Rep., 2016, 6, 20173 CrossRef CAS PubMed.
- G. Bellisola and C. Sorio, Am. J. Cancer Res., 2012, 2, 1–21 CAS.
- B. R. Wood, M. Kiupel and D. McNaughton, Vet. Pathol., 2014, 51, 224–237 CrossRef CAS PubMed.
- M. Pilling and P. Gardner, Chem. Soc. Rev., 2016, 45, 1935–1957 RSC.
- F. Bonnier, G. Brachet, R. Duong, T. Sojinrin, R. Respaud, N. Aubrey, M. J. Baker, H. J. Byrne and I. Chourpa, J. Biophotonics, 2016, 9(10), 1085–1097 CrossRef CAS PubMed.
- C. M. Orphanou, L. Walton-Williams, H. Mountain and J. Cassella, Forensic Sci. Int., 2015, 252, e10–e16 CrossRef CAS PubMed.
- E. Staniszewska-Slezak, A. Fedorowicz, K. Kramkowski, A. Leszczynska, S. Chlopicki, M. Baranska and K. Malek, Analyst, 2015, 140, 2273–2279 RSC.
- G. L. Owens, K. Gajjar, J. Trevisan, S. W. Fogarty, S. E. Taylor, B. Da Gama-Rose, P. L. Martin-Hirsch and F. L. Martin, J. Biophotonics, 2014, 7, 200–209 CrossRef CAS PubMed.
- J. Ollesch, S. L. Drees, H. M. Heise, T. Behrens, T. Bruning and K. Gerwert, Analyst, 2013, 138, 4092–4102 RSC.
- D. Perez-Guaita, J. Kuligowski, S. Garrigues, G. Quintas and B. R. Wood, Analyst, 2015, 140, 2422–2427 RSC.
- H. J. Byrne, P. Knief, M. E. Keating and F. Bonnier, Chem. Soc. Rev., 2016, 45, 1865 RSC.
- H. J. Byrne, M. Baranska, G. J. Puppels, N. Stone, B. Wood, K. M. Gough, P. Lasch, P. Heraud, J. Sule-Suso and G. D. Sockalingum, Analyst, 2015, 140, 2066–2073 RSC.
- K. Kong, C. Kendall, N. Stone and I. Notingher, Adv. Drug Delivery Rev., 2015, 89, 121–134 CrossRef CAS PubMed.
- C. Lacombe, V. Untereiner, C. Gobinet, M. Zater, G. D. Sockalingum and R. Garnotel, Analyst, 2015, 140, 2280–2286 RSC.
- S. Yotsukura and H. Mamitsuka, Crit. Rev. Oncol. Hematol., 2015, 93, 103–115 CrossRef PubMed.
- S. Wold, M. Sjostrom and L. Eriksson, Chemom. Intell. Lab. Syst., 2001, 109–130 CrossRef CAS.
- H. M. Heise, G. Voigt, P. Lampen, L. Küpper, S. Rudloff and G. Werner, Appl. Spectrosc., 2001, 55, 434–443 CrossRef CAS.
- A. R. Shaw and H. H. Mantsch, Infrared Spectroscopy in Clinical and Diagnostic Analysis, John Wiley & Sons Ltd, Chichester, 2006 Search PubMed.
- S. Khaustova, M. Shkurnikov, E. Tonevitsky, V. Artyushenko and A. Tonevitsky, Analyst, 2010, 135, 3183–3192 RSC.
- I. Elsohaby, J. T. McClure, C. B. Riley, R. A. Shaw and G. P. Keefe, J. Vet. Diagn. Invest., 2016, 28, 30–37 CrossRef PubMed.
- H. Nawaz, F. Bonnier, A. D. Meade, F. M. Lyng and H. J. Byrne, Analyst, 2011, 136, 2450–2463 RSC.
- F. Di Girolamo, J. Alessandroni, P. Somma and F. Guadagni, J. Proteomics, 2010, 73, 667–677 CrossRef CAS PubMed.
- J. L. Luque-Garcia and T. A. Neubert, J. Chromatogr., A, 2007, 1153, 259–276 CrossRef CAS PubMed.
- K. N. Patel, J. K. Patel, M. P. Patel, G. C. Rajput and H. A. Patel, Pharm Methods, 2010, 1, 2–13 CrossRef PubMed.
- A. Ioannou and C. Varotsis, J. Phys. Chem. Biophys., 2016, 6, 210 Search PubMed.
- J. Kuligowski, M. Cascant, S. Garrigues and M. de la Guardia, Talanta, 2012, 99, 660–667 CrossRef CAS PubMed.
- A. Edelmann, J. Diewok, J. R. Baena and B. Lendl, Anal. Bioanal. Chem., 2003, 376, 92–97 CrossRef CAS PubMed.
- S. V. Patil and S. D. Barhate, World J. Pharm. Res., 2014, 4, 214–225 Search PubMed.
- F. Bonnier, M. J. Baker and H. J. Byrne, Anal. Methods, 2014, 6(14), 5155–5160 RSC.
- A. Kunst, B. Draeger and J. Ziegenhorn, in Methods of Enzymatic Analysis, ed. H. U. Bergmeyer, 3rd edn, 1984, vol. VI, pp. 163–172 Search PubMed.
- A. H. B. Wu, Tietz Clinical Guide to Laboratory Tests, WB Saunders Company, Philadelphia, 4th edn, 2006 Search PubMed.
- K. Peter, Interactions of Carbon Nanotubes with Human Lung Epithelial Cells In Vitro, Assessed by Raman Spectroscopy, Dublin Institute of technology, 2010 Search PubMed.
- K. M. Dorling and M. J. Baker, Trends Biotechnol., 2013, 31, 327–328 CrossRef CAS PubMed.
- C. Hughes, M. Brown, G. Clemens, A. Henderson, G. Monjardez, N. W. Clarke and P. Gardner, J. Biophotonics, 2014, 7, 180–188 CrossRef CAS PubMed.
- F. Bonnier, F. Petitjean, M. J. Baker and H. J. Byrne, J. Biophotonics, 2014, 7, 167–179 CrossRef CAS PubMed.
- J. R. Hands, P. Abel, K. Ashton, T. Dawson, C. Davis, R. W. Lea, A. J. McIntosh and M. J. Baker, Anal. Bioanal. Chem., 2013, 405, 7347–7355 CrossRef CAS PubMed.
- J. R. Hands, K. M. Dorling, P. Abel, K. M. Ashton, A. Brodbelt, C. Davis, T. Dawson, M. D. Jenkinson, R. W. Lea, C. Walker and M. J. Baker, J. Biophotonics, 2014, 7, 189–199 CrossRef CAS PubMed.
- C. Petibois, A. M. Melin, A. Perromat, G. Cazorla and G. Deleris, J. Lab. Clin. Med., 2000, 135, 210–215 CrossRef CAS PubMed.
- L. Lovergne, G. Clemens, V. Untereiner, R. A. Lukaszweski, G. D. Sockalingum and M. J. Baker, Anal. Methods, 2015, 7, 7140–7149 RSC.
- L. Vroman, A. L. Adams, G. C. Fischer and P. C. Munoz, Blood, 1980, 55, 156–159 CAS.
- J. L. Parkes, S. L. Slatin, S. Pardo and B. H. Ginsberg, Diabetes Care, 2000, 23, 1143–1148 CrossRef CAS PubMed.
- A. Sahu, S. Sawant, H. Mamgain and C. M. Krishna, Analyst, 2013, 138, 4161–4174 RSC.
- S. L. Haas, R. Muller, A. Fernandes, K. Dzeyk-Boycheva, S. Wurl, J. Hohmann, S. Hemberger, E. Elmas, M. Bruckmann, P. Bugert and J. Backhaus, Appl. Spectrosc., 2010, 64, 262–267 CrossRef CAS PubMed.
- J. Backhausa, R. Muellera, N. Formanskia, N. Szlamaa, H. G. Meerpohlb, M. Eidtb and P. Bugertc, Vib. Spectrosc., 2010, 52, 173–177 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6an01888b |
| This journal is © The Royal Society of Chemistry 2017 |