
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel reaction of 3,4-dibromofuran with azo diesters to give tetrahydropyridazinones†
Kati M.
Aitken
,
R. Alan
Aitken
* and
Alexandra M. Z.
Slawin
EaStCHEM, School of Chemistry, University of St. Andrews, North Haugh, St. Andrews, Fife, UK KY16 9ST. E-mail: raa@st-and.ac.uk; Fax: +44 1334 463808; Tel: +44 1334 463865
First published on 23rd February 2016
Abstract
Cycloaddition of 3,4-dibromofuran with azo diesters proceeds by a Diels–Alder reaction followed by a novel rearrangement to give 3,5-dibromotetrahydropyridazin-4-ones. Variable-temperature NMR spectroscopy shows four separate conformations at low temperature attributed to restricted rotation about the carbamate functions. The ethyl compound decomposes upon storage with loss of the carbamate groups and aromatisation to give a simple bromohydroxypyridazinium salt.
We recently described the generation and spectroscopic characterisation of 1,4-oxazine, the first parent fully unsaturated six-membered ring heterocycle containing one group 15 and one group 16 atom.1 Although the successful method ultimately involved pyrolytic deprotection of the N-Boc derivative, we also investigated several other approaches including a cycloaddition – double bond functionalisation – cycloreversion route that led to the discovery of a novel and unexpected heterocyclic rearrangement described here.
The approach involves Diels–Alder cycloaddition with an azo diester, functionalisation of the newly formed double bond to give a cyclopropane, epoxide or aziridine 1, hydrolysis of the esters and decarboxylation followed by mild oxidation to give the azo compound, and finally spontaneous extrusion of N2 to give the target heterocycle 2 (Scheme 1). This strategy has previously been successful in forming 1,4-cyclohexadiene (X = Y = CH2),2 1,4-dihydropyridines (X = CH2, Y = NR),3–5 and 4H-pyran (X = CH2, Y = O),6 but to our knowledge has not been attempted with X = O, although a related strategy involving extrusion of maleic anhydride rather than N2 was used to produce 1,4-dioxin 2 (X = Y = O).7 The Diels–Alder cycloaddition between furan and diethyl azodicarboxylate is well known, having been first reported over 50 years ago,8,9 and it has also been extended to other azo diesters such as dibenzyl azodicarboxylate,10 although caution has to be exercised in altering the dienophile since azodibenzoyl (PhCO–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–COPh) instead gives the apparent [2 + 2]-cycloadduct with furan11 Since we experienced problems in forming an aziridine directly from the double bond of the furan – diethyl azodicarboxylate adduct, we decided to prepare the adduct of a 3,4-difunctionalised furan which, following hydrogenation, could be converted into an aziridine in a stepwise manner.
N–COPh) instead gives the apparent [2 + 2]-cycloadduct with furan11 Since we experienced problems in forming an aziridine directly from the double bond of the furan – diethyl azodicarboxylate adduct, we decided to prepare the adduct of a 3,4-difunctionalised furan which, following hydrogenation, could be converted into an aziridine in a stepwise manner.
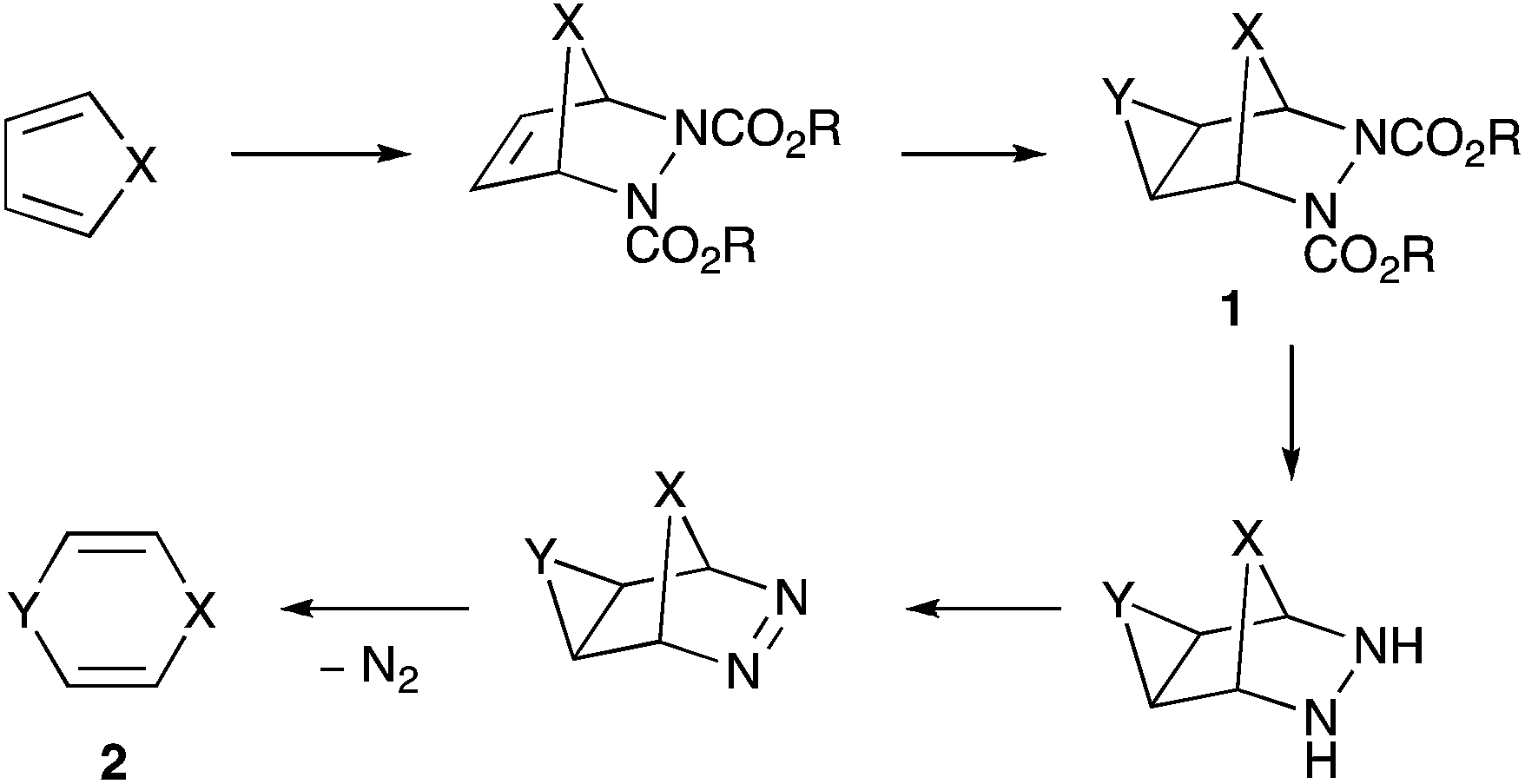 | ||
| Scheme 1 | ||
3,4-Dibromofuran 3 was readily prepared in moderate yield by oxidative cyclodehydration of (E)-2,3-dibromobut-2-en-1,4-diol,12,13 The only previous reports of cycloadditions involving 3 are [4 + 2]-cycloaddition with benzyne14 and [4 + 3]-cycloaddition with allyl cation equivalents.15 When 3 was allowed to react with either diethyl or diisopropyl azodicarboxylate at room temperature for 7 days a new single product was formed in each case which was isolated in pure form by column chromatography. The structures of these products were initially obscured by the occurrence of a dynamic process leading to extremely broad NMR spectra at room temperature. However at 55 °C, sharper spectra were obtained,‡ and these were incompatible with either the [4 + 2]- or [2 + 2]-cycloadducts 4 or 5 (Scheme 2). In particular, although the compounds had the expected molecular formulae, they were unsymmetrical and there was clear evidence for the presence of a ketone carbonyl group in addition to the carbamate esters. The matter was finally resolved by a single crystal X-ray diffraction study on the isopropyl compound (Fig. 1) which clearly showed the 3,5-dibromotetrahydropyridazin-4-one structure 6.§ Although the formation of pyridazin-4-ones has been reported in the cycloaddition of azo diesters with a range of functionalised dienes,16–18 none of these involve furans and the current process is clearly quite different, requiring a fundamental rearrangement. We propose that the expected [4 + 2]-adduct 4 is formed initially but this undergoes a spontaneous rearrangement by one of the two mechanisms shown in Scheme 3.
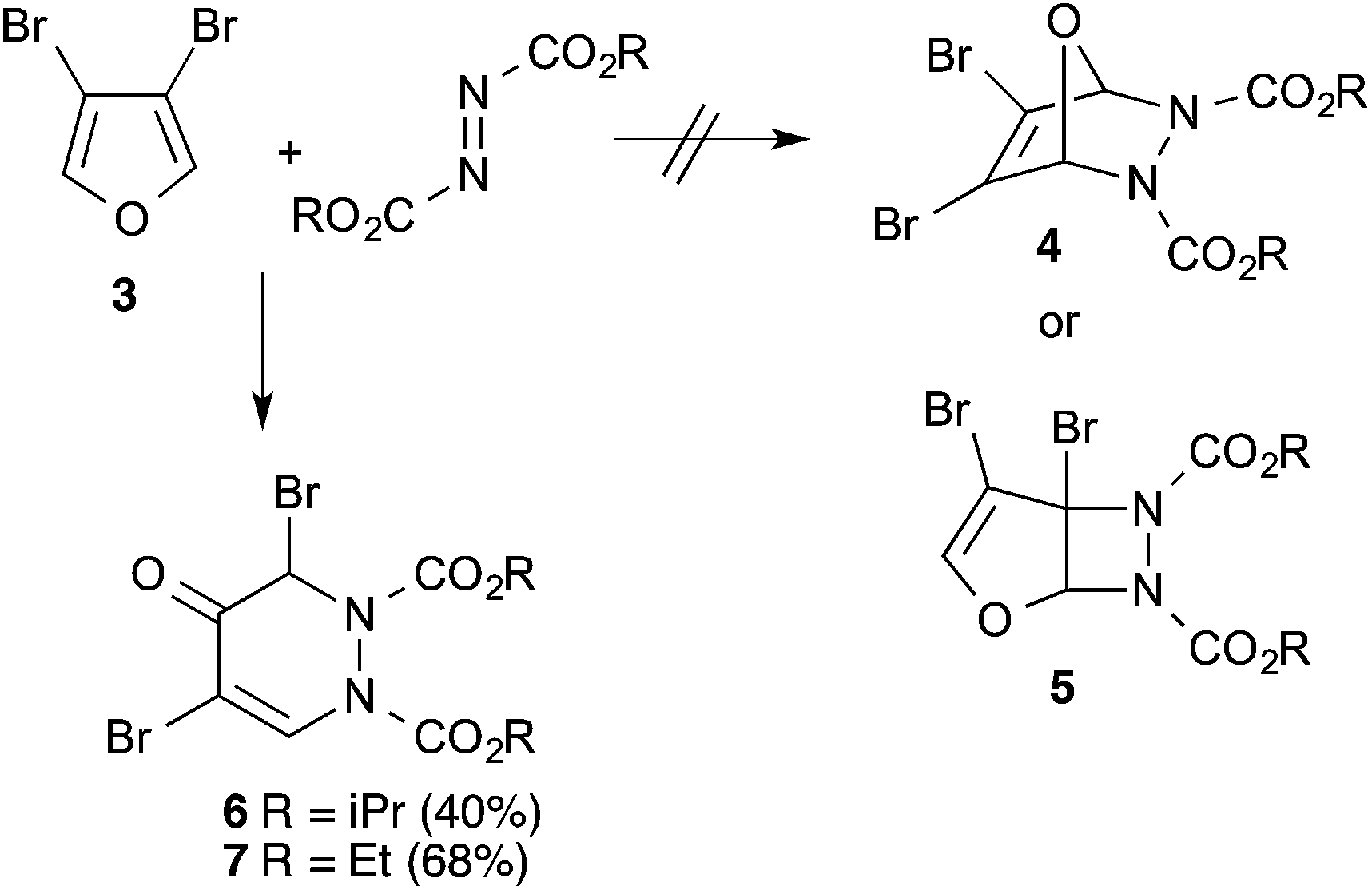 | ||
| Scheme 2 | ||
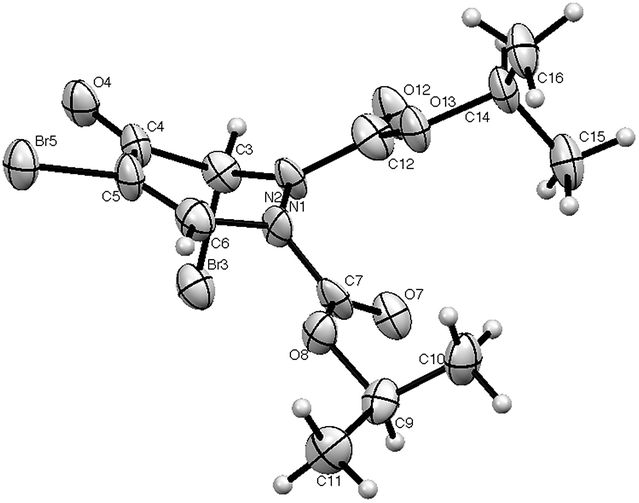 | ||
| Fig. 1 X-ray structure of 6 showing numbering scheme used. | ||
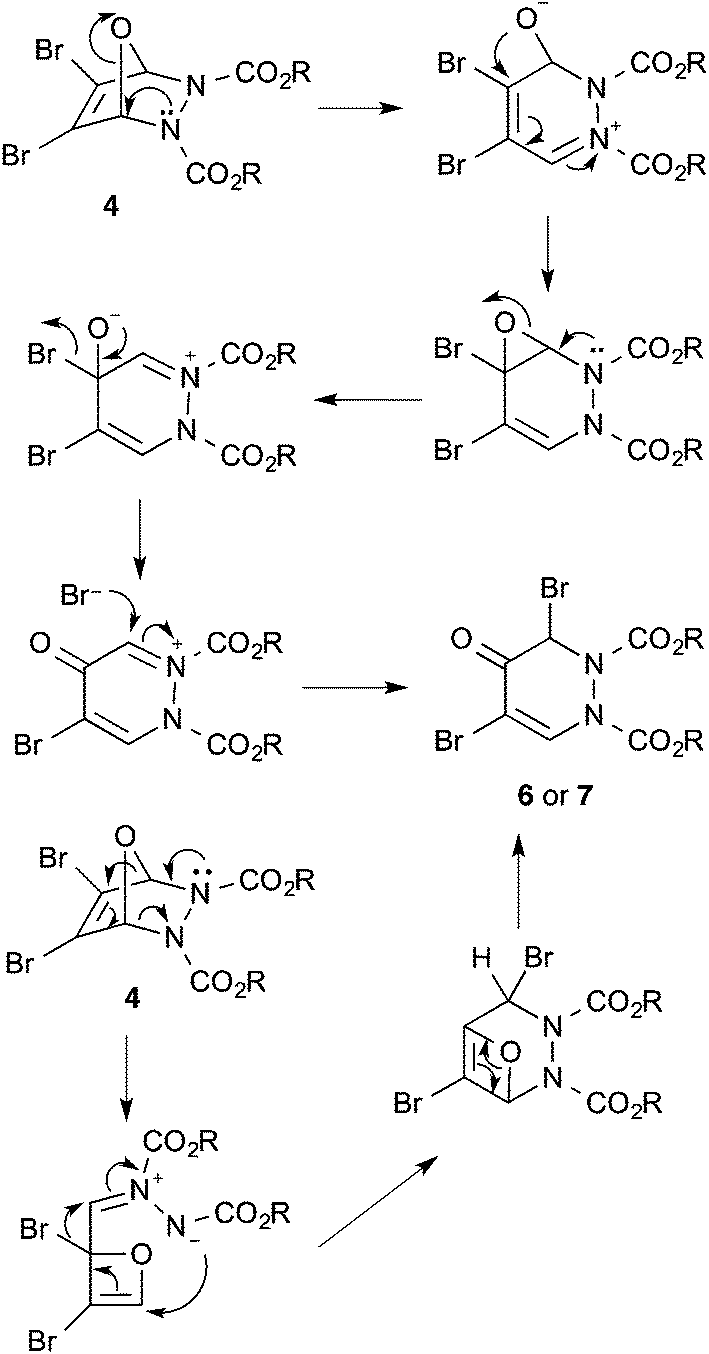 | ||
| Scheme 3 | ||
The occurrence of dynamic processes in hydrogenated pyridazine carbamate systems is a well-studied area that has elicited some controversy in the past. There are three main phenomena that can occur: restricted rotation about the carbamate N–C(O) bond, inversion at the pyramidal carbamate N, and ring inversion in half chair forms, and the occurrence of each of these in various 1,2-dialkoxycarbonyl-1,2,3,6-tetrahydropyridazines has been described in detail.19,20 While these earlier studies were based on 1H NMR, additional insight was provided by a detailed variable temperature 13C NMR study,21 which was also backed up by X-ray structure determinations of the compounds involved, including 8–10 (Scheme 4).22 As compared to these model compounds, 6 is devoid of symmetry and so shows separate 1H and 13C NMR signals for each atom at high temperature. The spectra at −30 °C show a full set of signals for four separate forms in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 ratio.¶ Upon warming these coalesce first into two forms, which then further coalesce into one, with all the free energies of activation in the range 57–61 kJ mol−1 (13.7–14.5 kcal mol−1). A significant difference between 6 and the model systems 8–10 in the solid state is its high degree of planarity. The values of the angle sum at N shown in Scheme 4 show that one nitrogen (N2) is completely planar and the other (N1) nearly so, in stark contrast to compounds 8 and 9 which adopt half chair conformations with distinctly pyramidal N atoms, but rather similar to the dibromo compound 10 which exists in a chair conformation with axial bromine atoms and one nearly and one completely planar nitrogen.22
:1:1:1 ratio.¶ Upon warming these coalesce first into two forms, which then further coalesce into one, with all the free energies of activation in the range 57–61 kJ mol−1 (13.7–14.5 kcal mol−1). A significant difference between 6 and the model systems 8–10 in the solid state is its high degree of planarity. The values of the angle sum at N shown in Scheme 4 show that one nitrogen (N2) is completely planar and the other (N1) nearly so, in stark contrast to compounds 8 and 9 which adopt half chair conformations with distinctly pyramidal N atoms, but rather similar to the dibromo compound 10 which exists in a chair conformation with axial bromine atoms and one nearly and one completely planar nitrogen.22
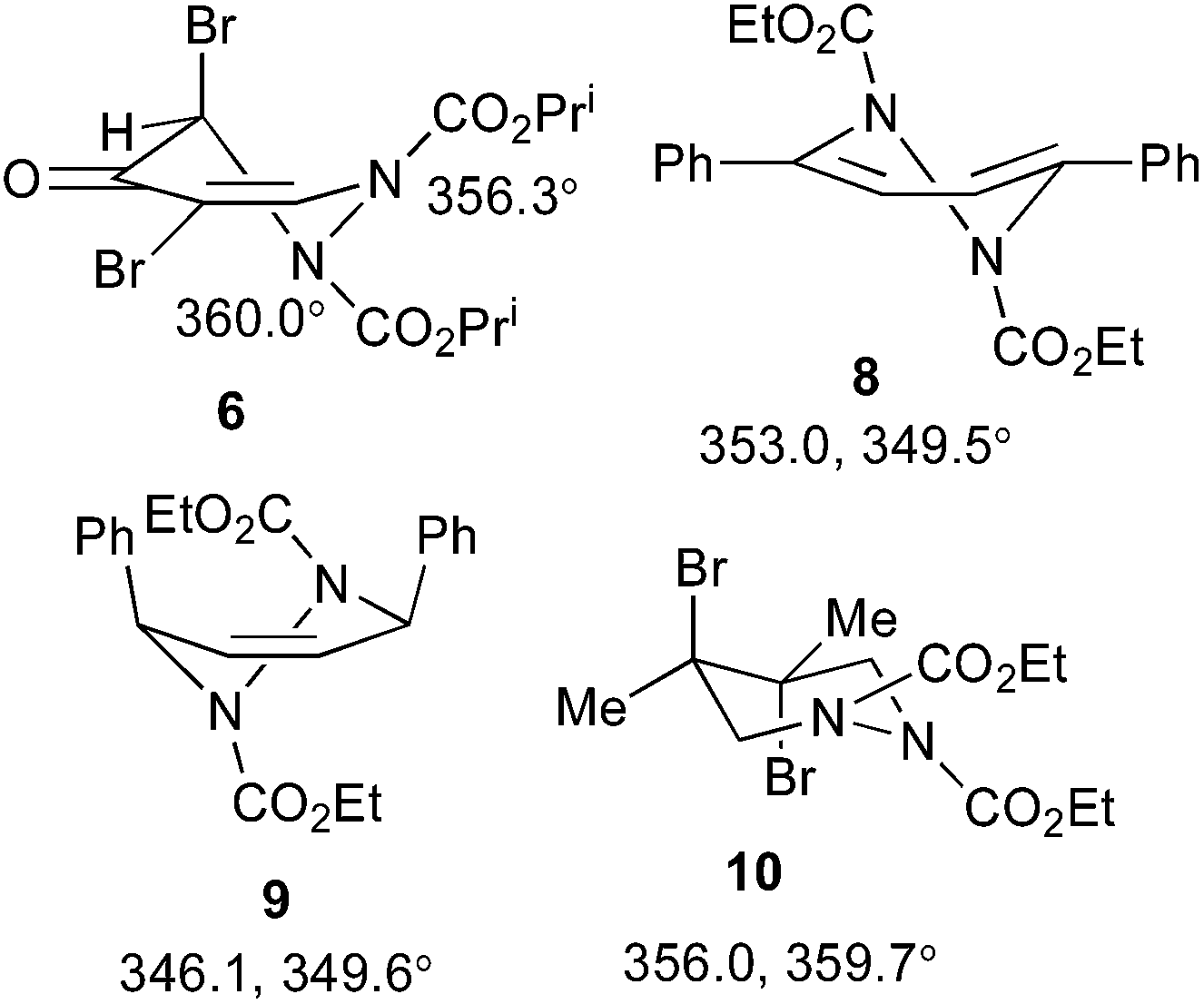 | ||
| Scheme 4 X-ray structures of 6 and 8–10 showing angle sums at N. | ||
Taking all the evidence into account, we believe that the NMR behaviour of compound 6 is most likely explained by restricted rotation about the carbamate N–C(O) bonds with the four interconverting forms as shown in Scheme 5. Ring inversion seems less likely than in the related ring systems where it has been previously observed, nitrogen inversion seems unlikely with such planar nitrogens in the solid state, and the free energy barriers to interconversion of the different forms are in the same range as for previously described systems involving carbamate rotation.
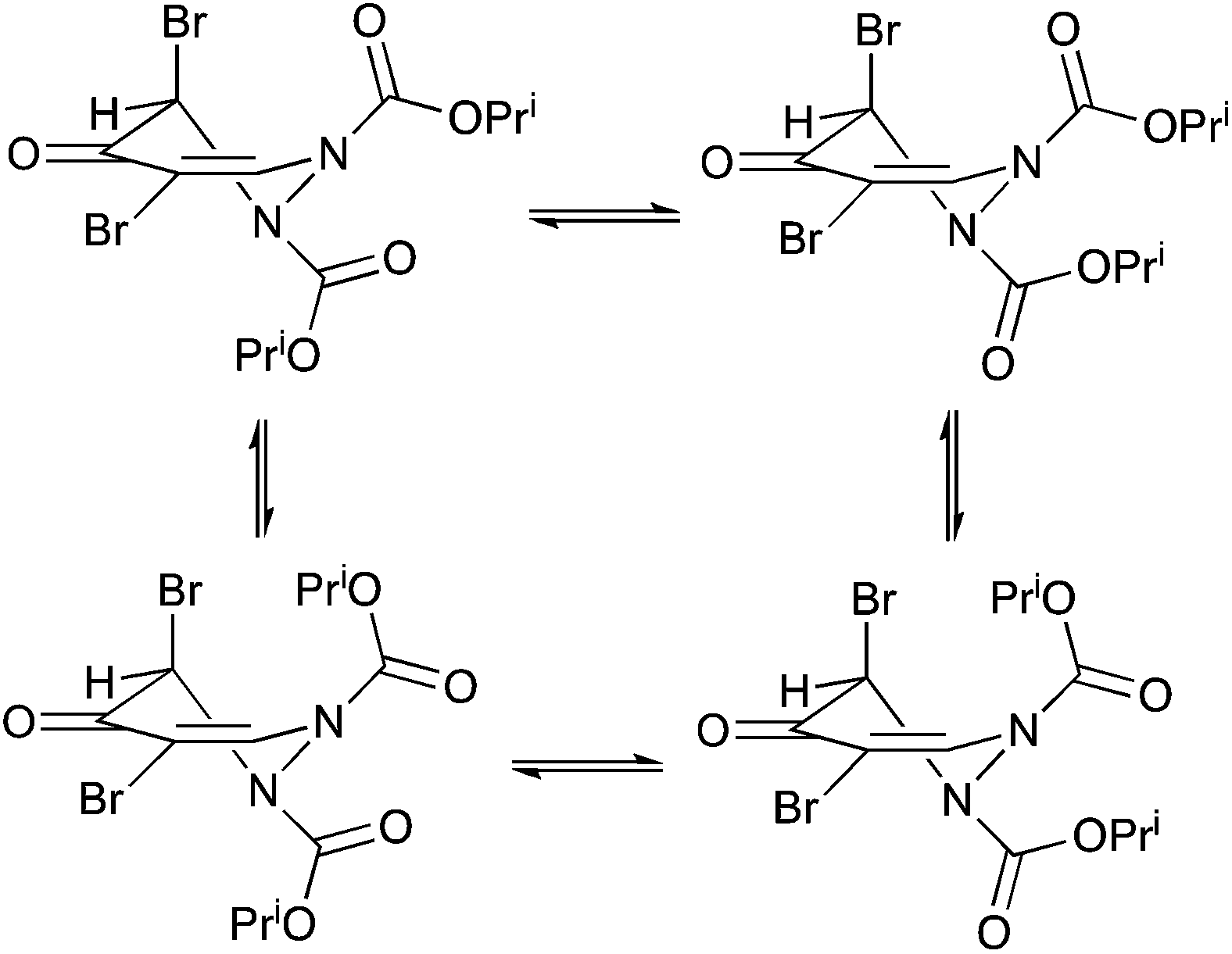 | ||
| Scheme 5 | ||
The diethyl compound 7 was observed to be significantly less stable than 6 and, after storage for a period of weeks, it was found to have decomposed with formation of a completely new compound with unexpected spectroscopic properties.|| This was found by X-ray diffraction to be the monohydrate of 5-bromo-4-hydroxypyridazinium bromide 11.** The structure (Fig. 2) features a hydrogen bonded network containing water molecules, with each water molecule being bonded by oxygen to the OH of 11 and by hydrogen to two bromide ions, while each bromide is bonded to two waters and to the NH of 11.
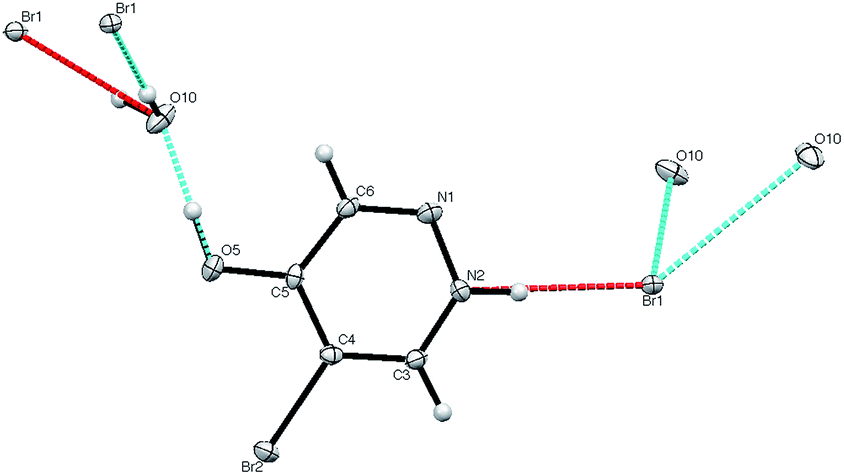 | ||
| Fig. 2 X-ray structure of 11. | ||
We propose that this is formed by HBr catalysed dealkylation and decarboxylation followed by aromatisation as shown in Scheme 6.
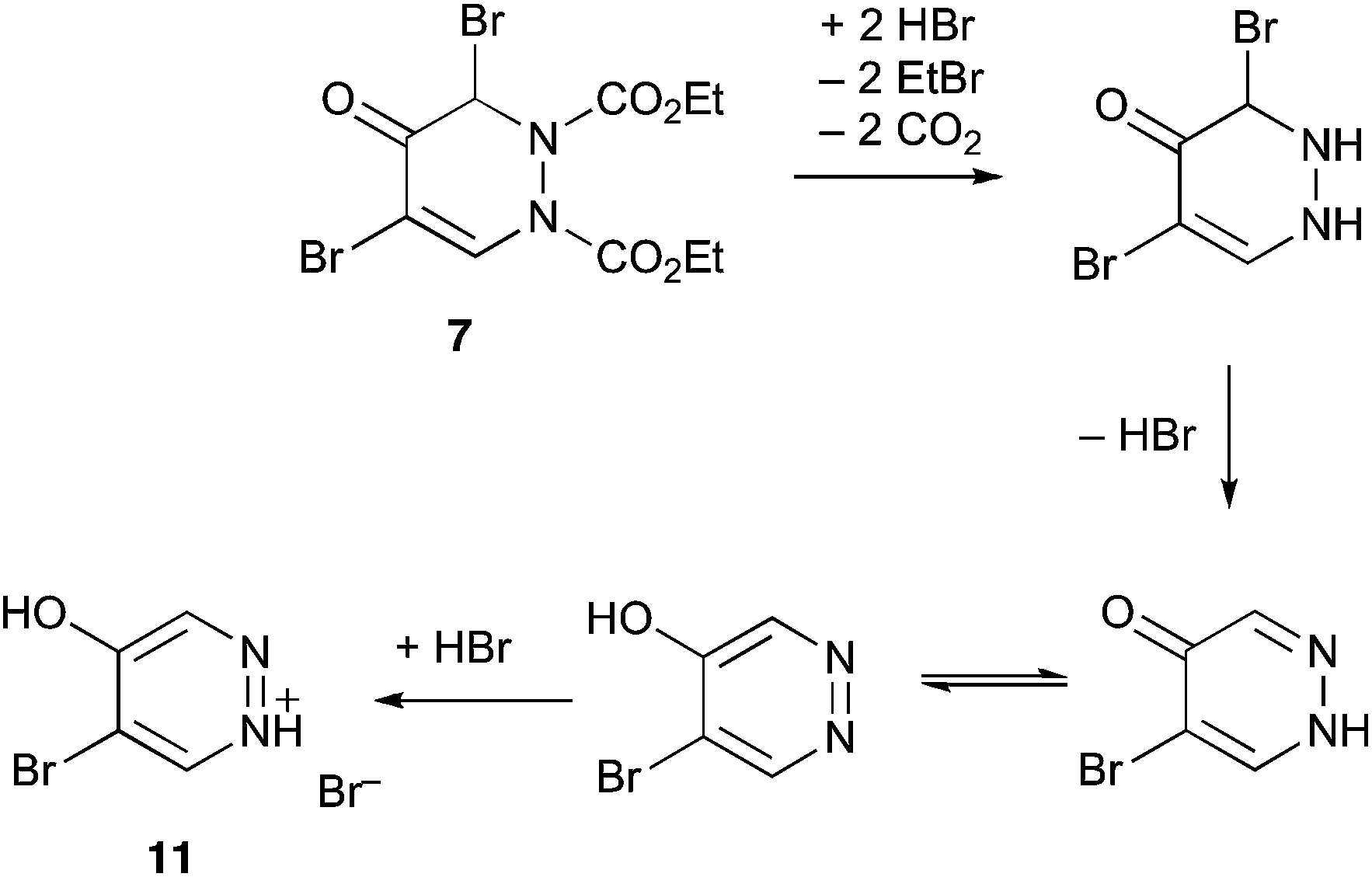 | ||
| Scheme 6 | ||
Although 3,4-dibromofuran has not been widely investigated as a 1,3-diene before, the reactions reported here show that it has the potential to provide access to highly functionalised pyridazines by reaction with readily available azo diesters. Its reactions with other types of dienophile may lead to useful functionalised products and this is currently under investigation.
Notes and references
- R. A. Aitken, K. M. Aitken, P. G. Carruthers, M.-A. Jean and A. M. Z. Slawin, Chem. Commun., 2013, 49, 11367–11369 RSC.
- E. L. Allred, J. C. Hinshaw and A. L. Johnson, J. Am. Chem. Soc., 1969, 91, 3382–3383 CrossRef CAS.
- D. C. Horwell and J. Deyrup, J. Chem. Soc., Chem. Commun., 1972, 485–486 RSC.
- A. I. Meyers, D. M. Stout and T. Takaya, J. Chem. Soc., Chem. Commun., 1972, 1260–1261 RSC.
- D. M. Stout, T. Takaya and A. I. Meyers, J. Org. Chem., 1975, 40, 563–569 CrossRef CAS.
- W. Adam and A. Berkessel, Chem. Ber., 1985, 118, 5018–5023 CrossRef CAS.
- R. A. Aitken, J. I. G. Cadogan and I. Gosney, J. Chem. Soc., Perkin Trans. 1, 1994, 927–931 RSC.
- Yu. K. Yur'ev and N. S. Zefirov, Russ. J. Gen. Chem., 1959, 29, 2916–2921 Search PubMed.
- C. W. Huffman, US Pat., 2 957 874 ( 1960).
- B. K. Bandlish, J. N. Brown, J. W. Timberlake and L. M. Trefonas, J. Org. Chem., 1973, 33, 1102–1105 CrossRef.
- N. S. Zefirov and A. A. Dudinov, Zh. Org. Khim., 1967, 3, 428 ( J. Org. Chem. USSR , 1967 , 3 , 411 ) CAS.
- M. Gorzynski and D. Rewicki, Liebigs Ann. Chem., 1986, 625–637 CrossRef CAS.
- S. P. H. Mee, V. Lee, J. E. Baldwin and A. Cowley, Tetrahedron, 2004, 60, 3695–3712 CrossRef CAS.
- W. D. Neudorff, N. Schulte, D. Lentz and A. D. Schlüter, Org. Lett., 2001, 3, 3115–3118 CrossRef CAS PubMed.
- H. Beck, C. B. W. Stark and H. M. R. Hoffmann, Org. Lett., 2000, 2, 883–886 CrossRef CAS PubMed.
- W. Ried and U. Reiher, Chem. Ber., 1987, 120, 1597–1599 CrossRef CAS.
- M. T. Aranda, M. C. Aversa, A. Barattucci, P. Bonaccorsi, M. C. Carreño, M. B. Cid and J. L. García Ruano, Tetrahedron: Asymmetry, 2000, 11, 1217–1225 CrossRef CAS.
- R. S. Vartanyan, A. L. Gyul'budagyan, A. K. Khanamiryan, E. N. Mkrtumyan and Z. V. Kazaryan, Khim. Geterotsikl. Soedin., 1991, 27, 783–785 ( Chem. Heterocycl. Compd. , 1991 , 27 , 613–616 ) Search PubMed.
- J. E. Anderson and J. M. Lehn, Tetrahedron, 1968, 24, 137–149 CrossRef CAS.
- E. W. Bittner and J. T. Gerig, J. Am. Chem. Soc., 1972, 94, 913–922 CrossRef CAS.
- T. H. Fisher, J. C. Crook and S. Chang, Tetrahedron, 1987, 43, 2443–2450 CrossRef CAS.
- M. Kaftory, T. H. Fisher and S. M. Dershem, J. Chem. Soc., Perkin Trans. 2, 1989, 1887–1891 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, hydrogen bonding parameters for 11 and copies of 1H and 13C NMR spectra for compounds 6, 7 and 11. CCDC 1443587 and 1443588. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra02735k |
| ‡ Compound 6: colourless plates, mp 104–106 °C (found: C, 33.8; H, 3.7; N, 6.3; m/z 428.9484. C12H16Br2N2O5 requires C, 33.7; H, 3.8; N, 6.5%; 79Br, 81Br − M + H, 428.9484); νmax/cm−1 1746, 1695 and 1590; δH (CDCl3, 55 °C) 8.38 (1H, br s, CH), 6.80 (1H, br s, CHBr), 5.13 (1H, septet, J 6.3), 5.04 (1H, septet, J 6.3), 1.37 (6H, d, J 6.3), 1.33 (3H, d, J 6.3) and 1.32 (3H, d, J 6.3); δC (55 °C) 175.4, 150.0 (br, N–CO), 141.6 ( CH), 98.9 (br, CBr), 74.4 and 73.9 (CHMe2), 57.2 (CHBr), 21.79, 21.70, 21.68 and 21.60 (CHMe2). |
| § Crystal data for 6, C12H16Br2N2O5, Mr = 428.08, colourless prism, monoclinic, space group P21/c, a = 11.676(5), b = 9.708(4), c = 14.448(7) Å, β = 92.350(11)°, V = 1636.3(13) Å3, Z = 4, Dc 1.738 Mg m−3, T = 93(2) K, R1 = 0.1080 and wR2 = 0.2634 for 2091 reflections [I > 2σ(I)] and 194 parameters. |
| ¶ δH (CDCl3, −30 °C) 8.60, 8.51, 8.41, 8.33 (1H, 4 × s, CH), 7.00, 6.95, 6.85, 6.79 (1H, 4 × s, CHBr), 5.25–5.00 (2H, m) and 1.49–1.27 (12H, m); δC (−30 °C) 176.0, 175.8, 175.7, 175.6, 150.4, 149.8, 149.4, 149.1, 148.9, 148.6, 147.5, 147.2, 142.3, 142.1, 141.8, 141.1, 99.6, 98.4, 98.1, 97.7, 74.8, 74.7, 74.4, 74.2, 74.1, 74.0, 73.8, 73.6, 56.9, 56.8, 56.4, 56.2, 21.8, 21.7, 21.63, 21.59, 21.5 and 21.4. |
| || Compound 11: colourless needles, mp 176–178 °C; δH (CDCl3) 9.00 (1H, s), 7.91 (1H, s) and 6.36 (1H, br s); δH (CD3SOCD3) 8.84 (1H, s) and 7.86 (1H, s); δC (CD3SOCD3) 166.2 (C–OH), 146.7 (CH), 142.4 (CH) and 112.5 (C–Br). |
| ** Crystal data for 11·H2O, C4H6Br2N2O2, Mr = 273.91, colourless prism, orthorhombic, space group Pbca, a = 13.4208(15), b = 6.0747(7), c = 19.193(2) Å, V = 1564.8(3) Å3, Z = 8, Dc 2.325 Mg m−3, T = 93(2) K, R1 = 0.0326 and wR2 = 0.0756 for 1253 reflections [I > 2σ(I)] and 107 parameters. Data were recorded using a Rigaku mercury 70, MoKα radiation (confocal optic, λ 0.71070 Å) and Saturn detector. The structures were solved by direct methods and refined using full-matrix least-squares methods. |
| This journal is © The Royal Society of Chemistry 2016 |